

Abstract
The aim of this study was to develop novel hydrogel-based beads and characterize their potential to deliver and release a drug exhibiting pH-dependent solubility into distal parts of gastrointestinal (GI) tract. Oxycellulose beads containing diclofenac sodium as a model drug were prepared by the ionotropic external gelation technique using calcium chloride solution as the cross-linking medium. Resulting beads were characterized in terms of particle shape and size, encapsulation efficacy, swelling ability and in vitro drug release. Also, potential drug–polymer interactions were evaluated using Fourier transform infrared spectroscopy. The particle size was found to be 0.92–0.96 mm for inactive (oxycellulose only) and 1.47–1.60 mm for active (oxycellulose–diclofenac sodium) beads, respectively. In all cases, the sphericity factor was between 0.70 and 0.81 with higher values observed for samples containing higher polymer and drug concentrations. The swelling of inactive beads was found to be strongly influenced by the pH and composition (i.e. Na+ concentration) of the selected media (simulated gastric fluid vs. phosphate buffer pH 6.8). The encapsulation efficiency of the prepared particles ranged from 58% to 65%. Results of dissolution tests showed that the drug loading inside of the particles influenced the rate of its release. In general, prepared particles were able to release the drug within 12–16 h after a lag time of 4 h. Fickian diffusion was found as the predominant drug release mechanism. Thus, this novel particulate system showed a good potential to deliver drugs specifically to the distal parts of the human GI tract.
Key words: controlled release, external gelation, lag time, microencapsulation, oxidized cellulose
INTRODUCTION
In the past two decades, significant advances in controlled release technology have been reached, and numerous approaches have been investigated to achieve the desired drug release behaviour (1). The carriers for controlled drug delivery should ensure that the active substance is released at the demanded site, in the appropriate dose and for the required time. Furthermore, they should also be biocompatible or biodegradable. Recent approaches suggested the use of drug delivery vehicles which are non-toxic natural or synthetic polymers that will be eliminated harmlessly from the body (2). Oxycellulose (OC) (Fig. 1a) is a semi-synthetic derivative of naturally occurring cellulose which undergoes chemical depolymerization and enzymatic hydrolysis under in vivo conditions (3,4). Being a non-toxic, biodegradable, biocompatible and bioresorbable polymer (5,6), OC is a well-established surgical material commercially available as a knitted fabric or staple fibre used in humans to stop bleeding and prevent the undesirable tissue adhesion occurring during post-operative healing processes (7,8). OC is practically insoluble in water and common organic solvents (9) and can form different salts, which differ in their physicochemical properties.
Fig. 1.

Comparison of chemical structure of oxycellulose (a) and alginate (b)
These salts can be produced by simple reactions of OC with inorganic basic agents (10). It is difficult to achieve the sustained drug release during the transition through the upper part of gastrointestinal tract (GIT) from particles based on soluble OC salts (Na+, K+). On the other hand, multivalent cations, such as calcium, magnesium, zinc, cobalt, or aluminium, form insoluble OC salts. The conversion of soluble salts to insoluble ones during the microparticles preparation is the way to ensure a controlled drug release. For instance, divalent calcium cations have been reported to induce cross-linking between OC chains leading to the formation of a stronger, more water resistant gel (11–13).
In general, this method is known as “ionotropic gelation” or “external gelation technique” and leads to the formation of microspheres. Active substances (in a liquid or solid form) can be either evenly dispersed or entrapped within this matrix during gelation or be adsorbed on the surface of preformed particles (14). The variety of manufacturing techniques together with a wide range of carrier materials offer many possibilities to control the particles characteristics which can be tailor-made to meet the required therapeutic action. In some cases, the matrix of these controlled drug delivery devices remains behind in the body and must be surgically removed after the drug is exhausted (15).
In this study, diclofenac sodium (DS) was used as a model drug. It is a good candidate for incorporation into dosage forms with delayed and prolonged release due to its properties, such as short biological half-life (1–2 h) and its efficiency in chronic diseases (16). DS is a potent anti-inflammatory drug, marketed since 1970 (17); it is widely used in the treatment of rheumatoid disorders, other chronic inflammatory diseases (18,19) and also in suppression of colorectal tumorigenesis due to increased colon cell apoptosis (1,20). When administered, DS is characterized by rapid systemic clearance and thus a necessity of repeated daily dosing. Therefore, therapy with DS warrants the use of a sustained release formulation for prolonged action and improved patient compliance (21). DS is the sodium salt of a phenylacetic acid (pKa ∼ 4) (22) exhibiting a pH-dependent solubility. Moreover, its solubility is significantly dependent on the ionic strength and composition of dissolution medium used (e.g. presence of common ions). It is slightly soluble in water, sparingly soluble in phosphate buffer (PB) solutions of pH 6.8 (DS solubility 670 μg/ml) and 7.2. Because DS is practically insoluble in hydrochloric acid of pH 1.2 (23,24), OC particles are expected to by-pass the gastric environment and start to release the drug in the intestine where the pH of the intestinal content is more favourable. Thus, undesirable side effects of DS on gastric mucosa are minimized (25).
The main objective of our study was to investigate the feasibility of the external gelation technique to produce drug-loaded particles for extended drug release. OC beads were prepared using sodium salt of oxidized cellulose (NaOC) obtained by the C6 oxidation of cellulose anhydroglucose units. Calcium chloride solution was used as a cross-linking agent in order to retard the drug release rate.
MATERIALS AND METHODS
Materials
Diclofenac sodium (Amoli Organics, Mumbai, India) was the model drug, oxidized cellulose (Synthesia a.s., Pardubice, Semtín, Czech Republic) was used as a polymer carrier and calcium chloride dihydrate (Kirsch Pharma, Salzgitter, Germany) as a cross-linking agent. Simulated gastric fluid pH 1.2 (35% hydrochloric acid, sodium chloride—both Merck KGaA, Darmstadt, Germany) and phosphate buffer pH 6.8 (dodecahydrate sodium hydrogen phosphate, potassium dihydrogen phosphate—both Merck KGaA, Darmstadt, Germany) were applied as dissolution media. All materials were of Ph. Eur. quality; OC corresponded to USP XXVI monography.
Methods
Bead Preparation
Inactive calcium cross-linked OC beads (samples S01–S03) were prepared by an ionotropic gelation. NaOC dispersions (8%, 10% and 12%) were prepared by dispersing 8, 10 and 12 g of NaOC in purified water, respectively. Dispersions were heated up to 60°C to swell faster and continuously mixed using a magnetic stirrer Ikamag (RT5 Power, IKA-Werke, Staufen, Germany) at 600 rpm for 10 min. Subsequently, dispersions were homogenized using an Ultra-Turrax (T25 basic, IKA-Werke, Staufen, Germany) at 13,000 rpm for 5 min. The volume of each dispersion was finally adjusted to 100 ml with purified water. Dispersions (for composition, see Table I) were extruded through a needle with an internal diameter of 0.4 mm at a dropping rate of 0.175 ml/min into 120 ml of 1 M CaCl2 aqueous solution. The distance between the edge of the needle and the surface of the solution was adjusted to 0.5 cm. Beads were formed immediately and were left in the cross-linking solution for 1 h to harden while gently mixed.
Table I.
Composition of the Prepared Dispersions
| Sample | Dispersion composition (%, w/w) | |
|---|---|---|
| OC | DS | |
| S01 | 8 | – |
| S02 | 10 | – |
| S03 | 12 | – |
| S1 | 10 | 2 |
| S2 | 10 | 4 |
| S3 | 12 | 2 |
| S4 | 12 | 4 |
Particles were subsequently washed three times with purified water and dried at 30°C in a cabinet drier (HORO-048B, Dr. Hofmann GmbH, Germany) for 24 h before further testing. Drug-loaded beads (samples S1–S4) were prepared using the same procedure. The drug (2% or 4%, w/w—see Table I) was suspended into the OC dispersions (10% and 12%, w/w) prior to high-shear homogenization.
Viscosity Measurements
To investigate the influence of the viscosity on the characteristics of the prepared microparticles, rheological measurements of OC dispersions S01–S03 were performed (viscometer Stabinger SVM 3000, Antonpaar, Graz, Austria). The results are presented as the mean of three measurements of single point viscosity ± standard deviations (SD).
Morphological Properties
All bead samples were examined using scanning electron microscopy (SEM) Hitachi S-4300 (Hitachi Scientific Instruments, Japan). The samples were mounted directly onto the SEM sample holder using a double-side sticking tape and were gold spray-coated in a sputter coater (SCD 005, BalTec Inc., Switzerland) and then analysed at 8.0 kV acceleration voltages.
Particle size of beads was measured using a stereoscopic microscope (STM 902 Opting, Czech Republic) equipped with a charge-coupled device camera (Alphaphot, Nicon, Japan). Beads were visualized under ×7 magnification. The obtained images of 200 randomly chosen beads were stored and subsequently processed using the Ia32 software (Leco Corporation, USA).
Swelling Properties
Swelling properties of inactive OC beads (S01–S03) were evaluated in two selected environments—simulated gastric juice of pH 1.2 simulating the stomach environment and phosphate buffer of pH 6.8 corresponding to the pH of the proximal part of small intestine. Certain quantities of OC beads (100 mg) were placed into dissolution baskets and accurately weighed. Baskets were immersed into a beaker containing 100 ml of both media. At predefined time intervals (15, 30, 60, 120, 180, 240, 300 and 360 min), each basket was withdrawn, blotted to remove the excess of water and immediately weighed. The swelling of the beads was expressed as the relative weight gain/loss of the beads vs. time according to the following equation (26):
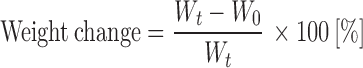 |
1 |
where Wt is the weight of wet beads at the time t and W0 is the initial weight at the time zero. The experiments were carried out in triplicate for each sample ± standard deviations.
Determination of Drug Content
The drug content was determined by dissolving appropriate amount of particles in purified water under sonication using an ultrasonic bath Sonorex (RK 52H, Bandelin, Germany) for at least 1 h. Once the particles were completely disintegrated, the samples were filtered and analysed spectrophotometrically at 276 nm (Lambda 25, Perkin Elmer, Wellesley, MA, USA) for DS content.
Drug-loading efficiency was calculated from the obtained values by using the following equation (26):
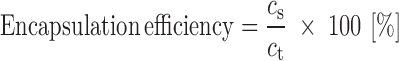 |
2 |
where cs corresponds to the actual DS content and ct corresponds to the theoretical drug load. The assay was carried out in triplicate.
Fourier Transform Infrared Spectroscopy
A Nicolet FTIR Impact 410 Spectrometer (Nicolet Instrument Co., Madison, WI, USA) equipped with a deuterated triglycine sulphate detector and diamond attenuated total reflectance (ATR) accessory (DuraSample IR-Technologies USA) was employed for ATR Fourier transform infrared (FTIR) experiments. Samples were placed in contact with the crystal using the same pressure. Each spectrum comprised 64 co-added scans measured at a spectral resolution of 4 cm−1 in the 4,000–600-cm−1 range. Spectral data were obtained with Omnic E.S.P. software version 5.2 (Nicolet instruments Co.)
In Vitro Release Studies
Drug release profiles of OC beads containing DS (S1–S4) were obtained using basket method in an automatic dissolution apparatus (SOTAX AT 7 On-Line System—Donau Lab, Switzerland) at 100 rpm. The dissolution medium volume used was 500 ml either of simulated gastric juice of pH 1.2 or phosphate buffer of pH 6.8 both kept at 37.0 ± 0.5°C. The bead samples were weighted with respect to their encapsulation efficiency: Thus, each sample contained 10 mg of diclofenac sodium. Samples, withdrawn at times as follows: 0.5, 1, 2 and then each 2 h, were analysed for the released drug amount using an UV spectrophotometer (Lambda 25, Perkin Elmer, Wellesley, MA, USA) at 276 nm. In order to propose the drug release mechanism from matrix beads, the experimental data were treated according to the following equations (27):
- Zero-order equation

3 - First-order equation
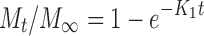
4 - Square root-time kinetics (Higuchi model)
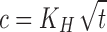
5 - Korsmeyer–Peppas equation
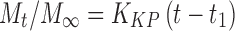
6 - Hixson–Crowell model
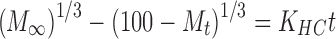
7 - Baker–Lonsdale model

8
where Mt is the amount of drug released at time t; M∞ is the absolute cumulative amount of drug released at infinitive time; K0, K1, KH, KHC, KBL are the zero-order, first-order, Higuchi, Hixson–Crowell and Baker–Lonsdale, release constants, respectively, and KKP is the release constant comprising the structural and geometrical characteristics. Release exponent n characterizes the mechanism of drug release, in particular, n = 0.5 corresponds to a Fickian diffusion release, 0.5 < n < 1.0 to an anomalous transport, n = 1.0 to a zero-order release kinetics and n > 1.0 to a super case II transport (27,28).
With regards to inconstant variance of some measured data, the weighted least squares method was applied to dissolution data. The release constant average value of each model with confidential regions (at significant level p ≤ 0.05) and determination coefficient R2 of regression analysis were calculated. Observing that R2 is only an orientational measure for regression function suitability, the autocorrelation test of residua deviations (Durbin–Watson test) was determined. Regression diagnostics were calculated by means of QC.Expert™ 2.5 software.
RESULTS AND DISCUSSION
Inactive Oxycellulose Beads
Particle Size and Morphological Properties
Particle size and sphericity factor of obtained wet and dry inactive OC beads are shown in Table II together with the viscosity of prepared OC dispersions. Dispersions S01, S02 and S03 contained 8%, 10% and 12% of OC polymer, respectively. With increasing polymer concentration, higher viscosities of the dispersions were reached, i.e. 0.08, 0.18 and 0.55 mPa s, respectively. Concentrations higher than 12% of OC formed highly viscous dispersions which were not possible to extrude because of the needle obstruction. Therefore, the preparation of beads using such concentrations was not possible.
Table II.
Some Characteristics of OC Dispersion, Dry and Wet Inactive Beads (Mean ± SD)
| Sample | Dispersion viscosity (Pa s) | Wet beads | Dry beads | ||
|---|---|---|---|---|---|
| Diameter (mm) | SF | Diameter (mm) | SF | ||
| S01 | 0.08 ± 0.02 | 1.89 ± 0.11 | 0.73 ± 0.02 | 0.92 ± 0.05 | 0.70 ± 0.02 |
| S02 | 0.18 ± 0.02 | 2.01 ± 0.10 | 0.80 ± 0.02 | 0.94 ± 0.05 | 0.80 ± 0.01 |
| S03 | 0.55 ± 0.02 | 2.15 ± 0.11 | 0.81 ± 0.02 | 0.96 ± 0.09 | 0.81 ± 0.02 |
All beads were of comparable size in the range of 1.89–2.15 mm for wet beads and from 0.92 to 0.96 mm for dry beads. During drying, the beads shrank significantly owing to the evaporation of water (29), and this affected the particle size (−51% to –55%) and slightly influenced sphericity factor (SF) in the sample S01 (−4.1%). The viscosity of polymer dispersion had a significant effect on the size of the resultant beads as well. The more viscous the polymer dispersion, the more difficult is the formation of smaller droplets (30); therefore, bigger particles were obtained (see Table II).
Sphericity values of dry matrix beads ranged from 0.70 to 0.81. Generally, the theoretical value of this parameter is 1.0, and value around and above 0.80 was reported for particles indicating good sphericity (31). As it can be seen from the results, the samples S02 and S03 prepared from more concentrated OC dispersions showed good sphericity (0.80 and 0.81, respectively), while the lowest OC concentration, i.e. 8% (sample S01), led to the formation of beads with lower sphericity value (0.70), and hence, this sample was excluded from further testing. Scanning electron micrographs of inactive dried beads showed that the beads exhibited rough surface (Fig. 2).
Fig. 2.

SEM photographs of the surface of inactive OC beads: sample S02 (a, b) and sample S03 (c, d). Magnification ×215 (a, c), ×1,204 (b, d). The bar corresponds to 400 μm (a, c) and 100 μm (b, d)
Bead Swelling
Figure 3 shows the swelling behaviour, i.e. the water uptake, expressed as the percentage weight gain for the inactive OC beads of samples S02 and S03 in simulated gastric fluid (SGF) of pH 1.2 and the PB of pH 6.8. From the obtained data, it is apparent that the swelling of particles was influenced by OC concentration. In both dissolution media, the water uptake slightly increased as the OC concentration rose from 10.0% to 12.0%. This result is in agreement with published literature data (32).
Fig. 3.

Water uptake of inactive OC beads in SGF and PB (SD values were lower than 4%)
It is obvious that OC beads exhibited different swelling properties in the different dissolution media. It was observed that beads’ swelling is significantly higher in PB compared to SGF. This effect could probably be due to the difference in the concentration of sodium ions in both media. Higher water uptake was observed in PB which contains higher concentration of Na+ ions. NaOC is a hydrophilic and water soluble anionic polysaccharide, but the Ca2+ ions inducing cross-linking of OC led to creation of insoluble calcium salt. Thus, prepared beads are sufficiently stable in the aqueous media. The ion-exchange process between Na+ and Ca2+ ions bound to COO− groups of anhydroglucose chain is supposed to be responsible for the swelling. As a result, an electrostatic repulsion between the negatively charged COO− groups increases and enhances gel swelling. Exchanged Ca2+ ions precipitated in the form of insoluble calcium phosphate causing a slight turbidity of the swelling medium (29). Maximum weight gain of the sample S02 was 505.67% while that of sample S03 was 595.79% in PB after 60 min. After attaining the maximum water uptake, the beads began to lose their weight. Lower water uptake was observed in SGF, where sample S02 showed weight gain of 73.75% and sample S03 of 106.77%, respectively. The swelling behaviour of the particles in this medium was almost the same during the entire period of 360 min. These results suggest the possibility of slight swelling of the prepared beads in the stomach and subsequently, large water uptake after the transfer to small intestine region and later on erosion and drug release at intended site of action (33).
Drug-Loaded Beads
Particle Size and Morphological Properties
Incorporation of DS into OC beads influenced the shape of the final particles (Fig. 4). Wet DS loaded particles (Table III) were more spherical than inactive wet OC particles (see Table II). The wet DS beads exhibited sphericity in the range of 0.81–0.86 while dry DS beads showed values from 0.78 to 0.81. The decrease of SF values within drying process for samples S1, S2, S3 and S4 was calculated 3.7%, 4.8%, 4.8% and 5.8%, respectively. The mean particle size of obtained DS–OC beads was bigger than that of inactive beads (see Table II), ranging from 2.36 to 2.88 mm for the wet particles and 1.47 to 1.60 mm for the dry ones.
Fig. 4.

Stereomicrographs of DS–OC beads a wet S1, b wet S2, c wet S3, d wet S4, e dry S1, f dry S2, g dry S3 and h dry S4. Magnification ×7. The bar corresponds to 1,000 μm
Table III.
Some Characteristics of Wet and Dry DS–OC Beads (Mean ± SD)
| Sample | Wet beads | Dry beads | EE (%) | Theoretical content (%) | Practical content (mg/100 mg particles) | ||
|---|---|---|---|---|---|---|---|
| Diameter (mm) | SF | Diameter (mm) | SF | ||||
| S1 | 2.36 ± 0.11 | 0.81 ± 0.01 | 1.47 ± 0.131 | 0.78 ± 0.02 | 65.8 | 16.66 | 10.97 ± 0.27 |
| S2 | 2.59 ± 0.11 | 0.83 ± 0.02 | 1.52 ± 0.109 | 0.79 ± 0.02 | 60.7 | 28.57 | 17.33 ± 0.60 |
| S3 | 2.67 ± 0.10 | 0.84 ± 0.02 | 1.56 ± 0.127 | 0.80 ± 0.02 | 58.2 | 14.28 | 8.32 ± 0.66 |
| S4 | 2.88 ± 0.12 | 0.86 ± 0.02 | 1.60 ± 0.138 | 0.81 ± 0.02 | 58.6 | 25 | 14.65 ± 0.34 |
The results showed that the particle size of the DS–OC beads increased with increasing polymer concentration and DS amount in the dispersion. This could be explained in terms of increased viscosity and subsequent larger droplets formation.
Scanning electron micrographs of DS–OC particles and their surface morphology are shown in Fig. 5. SEM photographs revealed small acicular crystals on the surface. These particles are most probably crystals of DS and were perhaps formed as a result of DS migration along with water to the surface during drying and its subsequent recrystallization (32).
Fig. 5.

SEM photographs of surface of DS–OC beads a, b sample S3 and c, d sample S4. Magnification ×215 (a, c) and ×1,204 (b, d). Higher magnification showed acicular crystals on the surface of the beads. The bar corresponds to 400 μm (a, c) and 100 μm (b, d)
Drug Content, Encapsulation Efficiency
Results of DS practical content ranging from 8.32 to 17.33 mg/100 mg was used for calculation of encapsulation efficiencies (EE) according to formula 2 (Table III). The EE values ranged between 58.2% and 65.8%. It can be observed that the samples with lower polymer concentration (i.e. 10%) (samples S1 and S2) tend to have higher EE than samples with higher one (i.e. 12%). From Table II, it is obvious that with increasing the concentration of the sodium salt of oxycellulose led to the preparation of polymer dispersions with higher viscosity. With increasing viscosity of polymer dispersion, the encapsulation efficiency decreased from 65.8% to 58.2% at the samples S1 and S3 and from the 60.7% to 58.6% at the samples S2 and S4, respectively. Thus, it seems that the encapsulation efficiency was influenced by viscosity of prepared polymer dispersions as published in experimental study by Cheng et al. focused on microspheres based on polycaprolactone (34).
Evaluation of Drug–Polymer Interactions
FTIR analyses were performed to evaluate the possible interactions between NaOC and DS in the prepared beads. FTIR spectra of the initial NaOC powder and drug-free OC beads are compared in Fig. 6. Except for the difference in the intensity of peaks in the regions of 1,250–1,485, 1,520–1,675 and 3,000–3,750 cm−1, the spectra of OC beads appear similar to the spectrum of pure NaOC powder. Peaks in the region from 850 to 1,180 cm−1 in the FTIR spectra of the NaOC powder and OC beads are ascribed to the cellulose backbone. C = O vibration (belonging to the carboxyl groups) and O–H vibration (caused by crystallization water) could be observed as the peak at about 1,600 cm−1 in the spectra of the NaOC powder and OC beads. Owing to the presence of residual carboxyl groups, the peak at 1,730 cm−1 in the sample of NaOC was observed. Disappearance of this peak in the spectra of OC beads could be caused by the reaction of Ca2+ ions from cross-linking medium with free carboxyl groups in a process of ionotropic gelation. When DS–OC beads were compared with drug-free ones (Figs. 6 and 7), the occurrence of peaks at 1,450 and 1,505 cm−1 was observed only in the case of drug-loaded particles. This phenomenon could be explained by the presence of DS. The broad peak in the region 3,000–3,750 cm−1 in the spectra of the pure NaOC powder, drug-free OC beads and DS–OC beads is attributed to the hydrogen bonded O–H stretching vibration (9). These results and also the fact that there were no additional peaks appearing in the FTIR spectra of beads indicate that no change in the solid-state structure of OC occurred during the ionotropic gelation.
Fig. 6.

FTIR spectra of NaOC powder and drug-free beads
Fig. 7.

FTIR spectra of DS powder and beads with DS
In Vitro Release Studies
Dissolution studies were performed in two dissolution media of different pH values (pH 1.2 and 6.8, respectively). Within 24-h dissolution test in acidic medium, only very small amount of DS (less than 4%) was released from all tested samples due to the fact that DS is practically insoluble (∼1.2 μg/ml) (23) under these conditions. Therefore, dissolution data are not presented as the values were close to zero during all the tested period. Matrix particles stayed intact during this test, and it is a good presumption for their successful passage through stomach. The release profiles of DS–OC bead samples in phosphate buffer of pH 6.8 are shown in Fig. 8. Table IV presents standard deviations of average values of released drug to provide better lucidity of the Fig. 8.
Fig. 8.

DS dissolution profiles from beads in pH 6.8
Table IV.
Standard Deviations of the Amount of DS Released from Beads Within the Dissolution Test in the Dissolution Medium with pH Value 6.8
| Sample | Standard deviations (%) at time intervals (min) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 30 min | 60 min | 120 min | 240 min | 360 min | 480 min | 600 min | 720 min | 840 min | 960 min | 1,080 min | |
| S1 | 0.00 | 0.00 | 0.00 | 0.58 | 1.62 | 3.88 | 3.64 | 3.07 | 3.54 | 2.76 | 3.44 |
| S2 | 0.00 | 0.00 | 0.00 | 1.01 | 2.62 | 4.57 | 6.84 | 4.48 | 1.64 | 1.16 | 0.96 |
| S3 | 0.00 | 0.00 | 0.00 | 0.14 | 1.32 | 3.00 | 3.19 | 9.57 | 9.35 | 5.39 | 5.85 |
| S4 | 0.00 | 0.00 | 0.00 | 0.00 | 2.42 | 2.08 | 2.56 | 2.96 | 1.76 | 1.95 | 1.41 |
All samples exhibited an initial 4 h lag time and then prolonged DS release. Observed lag time could be probably caused, due to the relatively stable association of calcium ions with polyguluronate sequences which served as stable cross-linking points within the gels. The same effect was observed in preparation of alginate microparticles (35). During dissolution test, the beads did not disintegrate and their swelling was observed. Exchange of Na+ ions presented in phosphate buffer with Ca2+ ions in the bead structure is probably responsible for swelling and forming gel layer controlling the drug release. Swelling of alginate gels was also described in experimental study by Setty et al. (36).
The studied samples differed slightly from each other in two selected variables—encapsulated drug amount and OC concentration. From the obtained results, it is evident that the samples S2 (t50% = 459 min) and S4 (t50% = 548 min) with higher DS loading in the particles (DS content 17.33 and 14.65 mg, respectively) released the drug faster than samples S1 (t50% = 624 min) and S3 (t50% = 672 min) containing lower DS amount (DS content 10.97 and 8.32 mg, respectively). The faster drug release from the beads with higher drug content could be caused by higher drug concentration gradient between beads and dissolution medium (37). Another aspect is OC concentration. When a bead is placed in PB, OC swells, water penetrates into the matrix forming the pores necessary for drug release. As the polymer level in the formulation is increased, drug diffusion is slowed due to lower porosity and higher tortuosity of the matrix (38,39). Therefore, the fastest DS release from the sample S2 is a result of the highest drug content and the lowest OC concentration. Conversely, the slowest DS release from the sample S3 is caused by the lowest DS content and the highest OC amount. However, the drug release from a polymer matrix is a complicated process, it is influenced by many factors and its mechanism is not easy to determine (39).
Drug Release Mechanism
The dissolution data of OC bead samples were treated according to various kinetic models (Eqs. 3, 4, 5, 6, 7 and 8) to estimate the drug release mechanism and release rate constants. Fittings of DS release data to different kinetic equations are summarized in Table V. Durbin–Watson test was applied to the dissolution data to recognize a risk of apparent regression existence by means of the autocorrelation of residual deviations.
Table V.
Fittings of DS Release Data to Different Kinetic Equations
| Model | Zero order | First order | Higuchi | Korsmeyer–Peppas | Hixson–Crowell | Baker–Lonsdale | |
|---|---|---|---|---|---|---|---|
| Sample | K 0 (h−1) (CI) | K 1 (h−1) (CI) | K H (h−1/2) (CI) | K KP (h−n) (CI) | n (CI) | K HC (h−1/3) (CI) | K BL (CI) |
| R 2 | R 2 | R 2 | R 2 | R 2 | R 2 | ||
| S1 | 8.8 (6.7–11.0) | 0.21a (−0.05–0.47) | 54.0 (45.9–62.0) | 0.031a (0.004–0.227) | 1.4 (0.4–2.4) | 0.009 (0.007–0.011) | 0.016 (0.012–0.020) |
| 0.98 | 0.91 | 0.99 | 0.96 | 0.98 | 0.98 | ||
| S2 | 8.4 (5.9–10.9) | 0.12 (−0.03–0.28) | 48.5 (37.5–59.7) | 0.143 (0.040–0.508) | 0.8 (0.2–1.4) | 0.009 (0.005–0.012) | 0.025 (0.020–0.029) |
| 0.97 | 0.84 | 0.96 | 0.92 | 0.96 | 0.99 | ||
| S3 | 7.4 (6.1–8.7) | 0.19 (0.05–0.33) | 46.9 (43.2–50.5) | 0.036 (0.016–0.083) | 1.3 (0.9–1.7) | 0.007 (0.006–0.008) | 0.015 (0.009–0.026) |
| 0.98 | 0.83 | 0.99 | 0.96 | 0.98 | 0.96 | ||
| S4 | 9.2 (6.5–11.9) | 0.16a (0.02–0.30) | 57.7 (46.9–68.4) | 0.084 (0.037–0.193) | 1.0 (0.6–1.5) | 0.008 (0.006–0.011) | 0.031 (0.019–0.043) |
| 0.97 | 0.92 | 0.99 | 0.98 | 0.98 | 0.97 | ||
aAutocorrelation of residual deviations were found (Durbin–Watson test)
According to R2 values as the best-fit release kinetic model was revealed Higuchi kinetic model (for S1, S3 and S4, R2 = 0.99; for S2, R2 = 0.96) indicating Fickian diffusion-type release mechanism. This finding was also confirmed by fitting the dissolution data with Baker–Lonsdale mathematical model (R2 > 0.96) which assumed that the drug release from the bead samples took place by diffusion through water filled channels (40). Good correlation with semi empirical Hixson–Crowell model (for S1, S3 and S4, R2 = 0.98; for S2, R2 = 0.96) indicated that DS could be partly released from OC beads also via erosion process which is characterized by changes in DS–OC bead surface area and diameter (41). Determination coefficient R2 for Korsmeyer–Peppas kinetic model for samples S1, S2, S3 and S4 took value 0.96, 0.92, 0.96 and 0.98, respectively. Release exponent n characterizes the mechanism of drug release, but n value in our study had a large confidence interval (∆ 0.8–2.0); thus, it was not possible to predict drug release mechanism. Resultant DS release from OC beads was in correspondence with zero-order model (R2 > 0.97). For first-order model, the poor correlation was observed (R2 < 0.92). Thus, it can be concluded that DS was released from OC particles by the combination of diffusion (Higuchi, Baker–Lonsdale) and erosion (Hixson–Crowell) processes.
CONCLUSION
OC beads were successfully prepared using an ionotropic gelation method. The formed OC particles containing DS showed good entrapment and encapsulation efficiencies and spherical shape. FTIR studies did not reveal any significant interactions between the drug and the polymer. The in vitro release profile in pH 6.8 showed lag time of 4 h and then the ability of beads to prolong the drug release for next 12–16 h. The dissolution profile of incorporated DS was influenced by drug loading inside the particles: Faster drug release was determined from particles containing higher DS amount. Fickian diffusion was found as the predominant drug release mechanism.
Acknowledgements
This experimental work was realized by support of IGA VFU Brno Czech Republic, project 44/2011/FaF.
References
- 1.Gonzáles-Rodríguez ML, Maestrelli F, Mura P, Rabasco AM. In vitro release of sodium diclofenac from a central core matrix tablet aimed for colonic drug delivery. Eur J Pharm Sci. 2003;20:125–131. doi: 10.1016/S0928-0987(03)00181-7. [DOI] [PubMed] [Google Scholar]
- 2.Wang Q, Zhang J, Wang A. Preparation and characterization of a novel pH-sensitive chitosan-g-poly (acrylic acid)/attapulgite/sodium alginate composite hydrogel bead for controlled release of diclofenac sodium. Carbohyd Polym. 2009;78:731–737. doi: 10.1016/j.carbpol.2009.06.010. [DOI] [Google Scholar]
- 3.Dimitrijevich SD, Tatarko M, Gracy RW. Biodegradation of oxidized regenerated cellulose. Carbohyd Res. 1990;195:247–256. doi: 10.1016/0008-6215(90)84169-U. [DOI] [PubMed] [Google Scholar]
- 4.Dimitrijevich SD, Tatarko M, Gracy RW, Wise GE, Oakford LX. In vivo degradation of oxidized regenerated cellulose. Carbohyd Res. 1990;198:331–341. doi: 10.1016/0008-6215(90)84303-C. [DOI] [PubMed] [Google Scholar]
- 5.Stillwell RL, Marks MG, Saferstein L, Wiseman DM. Oxidized cellulose: chemistry, processing and medical applications. In: Domb JA, Kost J, Wiseman MD, editors. Handbook of biodegradable polymers. Amsterdam: Harwood Academic; 1997. pp. 291–306. [Google Scholar]
- 6.Kumar V, Yang T. HNO3/H3PO4-NANO2 mediated oxidation of cellulose—preparation and characterization of bioabsorbable oxidized celluloses in high yields and with different levels of oxidation. Carbohyd Polym. 2002;48:403–412. doi: 10.1016/S0144-8617(01)00290-9. [DOI] [Google Scholar]
- 7.Zhu L, Kumar V, Banker GS. Examination of aqueous oxidized cellulose dispersions as a potential drug carrier. I. Preparation and characterization of oxidized cellulose–phenylpropanolamine complexes. AAPS PharmSciTech. 2004;5(4):1–7. doi: 10.1208/pt050451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zimnitski DS, Yurkshtovich TL, Bychkovsky PM. Synthesis and characterization of oxidized cellulose. J Polym Sci A-Polym Chem. 2004;42:4785–4791. doi: 10.1002/pola.20302. [DOI] [Google Scholar]
- 9.Kumar V, Kang J, Yang T. Preparation and characterization of spray-dried oxidized cellulose microparticles. Pharm Dev Technol. 2001;6:449–458. doi: 10.1081/PDT-100002253. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt R, Bogan D, Moore J. EU Patent Office, Pat. No. 1 153 618, 2001.
- 11.Kosaraju SL. Colon targeted delivery systems: review of polysaccharides for encapsulation and delivery. Crit Rev Food Sci Nutr. 2005;45:251–258. doi: 10.1080/10408690490478091. [DOI] [PubMed] [Google Scholar]
- 12.Maestrelli F, Zerrouk N, Cirri M, Mennini N, Mura P. Microspheres for colonic delivery of ketoprofen–hydroxypropyl-β-cyclodextrin complex. Eur J Pharm Sci. 2008;34:1–11. doi: 10.1016/j.ejps.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 13.Jay SM, Saltzman WM. Controlled delivery of VEGF via modulation of alginate microparticle ionic crosslinking. J Control Release. 2008;134:26–34. doi: 10.1016/j.jconrel.2008.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burgesss DJ, Hickey AJ. Microsphere technology and applications. In: Swarbrick J, Boylan JC, editors. Encyclopedia of pharmaceutical technology. New York: Marcel Dekker; 2002. pp. 1783–1793. [Google Scholar]
- 15.Kumar MNV. Nano and microparticles as controlled drug delivery devices. J Pharm Pharm Sci. 2000;3:234–258. [PubMed] [Google Scholar]
- 16.Su SF, Chou ChH, Kung ChF, Huang JD. In vitro and in vivo comparison of two diclofenac sustained release oral formulations. Int J Pharm. 2003;250:39–46. doi: 10.1016/S0378-5173(03)00237-0. [DOI] [PubMed] [Google Scholar]
- 17.Fini A, Fazio G, Rosetti F, Holgado MA, Iruín A, Alvarez-Fuentes J. Diclofenac salts. III. Alkaline and earth alkaline salts. J Pharm Sci. 2005;94:2416–2431. doi: 10.1002/jps.20436. [DOI] [PubMed] [Google Scholar]
- 18.Piyakulawat P, Praphairaksit N, Chantarasiri N, Muangsin N. Preparation and evaluation of chitosan/carrageenan beads for controlled release of sodium diclofenac. AAPS PharmSciTech. 2007;8(4):E1–E11. doi: 10.1208/pt0804097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biju SS, Saisivam S, Rajan MG, Mishra PR. Dual coated erodible microcapsules for modified release of diclofenac sodium. Eur J Pharm Biopharm. 2004;58:61–67. doi: 10.1016/j.ejpb.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 20.Chan TA. Nonsteroidal anti-inflammatory drugs, apoptosis, and colon-cancer chemoprevention. Lancet Oncol. 2002;3:166–174. doi: 10.1016/S1470-2045(02)00680-0. [DOI] [PubMed] [Google Scholar]
- 21.Billa N, Yuen KH, Khader MAA. Gamma-scintigraphic study of the gastrointestinal transit and in vivo dissolution of a controlled release diclofenac sodium formulation in xanthan gum matrices. Int J Pharm. 2000;201:109–120. doi: 10.1016/S0378-5173(00)00399-9. [DOI] [PubMed] [Google Scholar]
- 22.Savaser A, Özkan Y, Ismer A. Preparation and in vitro evaluation of sustained release tablet formulations of diclofenac sodium. Farmaco. 2005;60:171–177. doi: 10.1016/j.farmac.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 23.Kincl M, Vrečer F, Veber M. Characterization of factors affecting the release of low-solubility drug from prolonged release tablets. Anal Chim Acta. 2004;502:107–113. doi: 10.1016/j.aca.2003.09.050. [DOI] [Google Scholar]
- 24.Dvořáčková K, Rabišková M, Masteiková R, Muselík J, Krejčová Soluble filler as a dissolution profile modulator for slightly soluble drugs in matrix tablets. Drug Dev Ind Pharm. 2009;35:930–940. doi: 10.1080/03639040802710243. [DOI] [PubMed] [Google Scholar]
- 25.Gonzáles-Rodríguez ML, Holgado MA, Sánchez-Lafuente C, Rabasco AM, Fini A. Alginate/chitosan particulate systems for sodium diclofenac release. Int J Pharm. 2002;232:225–234. doi: 10.1016/S0378-5173(01)00915-2. [DOI] [PubMed] [Google Scholar]
- 26.Paskaris G, Bouropoulos N. Swelling studies and in vitro release of verapamil from calcium alginate and calcium alginate–chitosan beads. Int J Pharm. 2006;323:34–42. doi: 10.1016/j.ijpharm.2006.05.054. [DOI] [PubMed] [Google Scholar]
- 27.Costa P, Lobo JMS. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13:123–133. doi: 10.1016/S0928-0987(01)00095-1. [DOI] [PubMed] [Google Scholar]
- 28.Samani MS, Montaseri H, Kazemi A. The effect of polymer blends on release profiles of diclofenac sodium from matrices. Eur J Pharm Biopharm. 2003;55:351–355. doi: 10.1016/S0939-6411(03)00030-4. [DOI] [PubMed] [Google Scholar]
- 29.Smrdel P, Bogataj M, Zega A, Planinšek O, Mrhar A. Shape optimization and characterization of polysaccharide beads prepared by ionotropic gelation. J Microencapsul. 2008;25:90–105. doi: 10.1080/02652040701776109. [DOI] [PubMed] [Google Scholar]
- 30.Yang YY, Wan JP, Chung TS, Pallathadka PK, Ng S, Heller J. POE–PEG–POE triblock copolymeric microspheres containing protein I. Preparation and characterization. J Control Release. 2001;75:115–128. doi: 10.1016/S0168-3659(01)00373-X. [DOI] [PubMed] [Google Scholar]
- 31.Deasy PB, Law MFL. Use of extrusion–spheronization to develop an improved oral dosage form of indomethacin. Int J Pharm. 1997;148:201–209. doi: 10.1016/S0378-5173(96)04846-6. [DOI] [Google Scholar]
- 32.Al-Kassas RS, Al-Gohary OMN, Al-Faadhel MM. Controlling of systemic absorption of gliclazide through incorporation into alginate beads. Int J Pharm. 2007;341:230–237. doi: 10.1016/j.ijpharm.2007.03.047. [DOI] [PubMed] [Google Scholar]
- 33.Bajpai SK, Sharma S. Investigation of swelling/degradation behavior of alginate beads crosslinked with Ca2+ and Ba2+ ions. React Funct Polym. 2004;59:129–140. doi: 10.1016/j.reactfunctpolym.2004.01.002. [DOI] [Google Scholar]
- 34.Cheng X, Liu R, He Y. A simple method for the preparation of monodisperse protein-loaded microspheres with high encapsulation efficiencies. Eur J Pharm Biopharm. 2010;76:336–341. doi: 10.1016/j.ejpb.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 35.Pongjanyakul T, Puttipipatkhachorn S. Xanthan–alginate composite gel beads: molecular interactions and in vitro characterization. Int J Pharm. 2007;331:61–71. doi: 10.1016/j.ijpharm.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 36.Setty CM, Sahoo SS, Sa B. Alginate-coated alginate–polyethylenimine beads for prolonged release of furosemide in simulated intestinal fluid. Drug Dev Ind Pharm. 2005;31:435–446. doi: 10.1080/03639040500214647. [DOI] [PubMed] [Google Scholar]
- 37.Barakat NS, Ahmad AAE. Diclofenac sodium loaded-cellulose acetate butyrate: effect of processing variables on microparticles properties, drug release kinetics and ulcerogenic activity. J Microencapsul. 2008;25:31–45. doi: 10.1080/02652040701747928. [DOI] [PubMed] [Google Scholar]
- 38.Mulani HT, Patel B, Shah NJ. Development of pH-independent matrix type sustained release drug delivery system of propranolol hydrochloride. J Appl Pharm Sci. 2011;01:83–92. [Google Scholar]
- 39.Fredenberg S, Wahlgren M, Reslow M, Axelsson A. The mechanism of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems—a review. Int J Pharm. 2011;415:34–52. doi: 10.1016/j.ijpharm.2011.05.049. [DOI] [PubMed] [Google Scholar]
- 40.Sood A, Panchagnula R. Drug release evaluation of diltiazem CR preparations. Int J Pharm. 1998;175:95–107. doi: 10.1016/S0378-5173(98)00268-3. [DOI] [Google Scholar]
- 41.Hixson AW, Crowell JH. Dependence of reaction velocity upon surface and agitation: I—theoretical consideration. Ind Eng Chem. 1931;23:923–931. doi: 10.1021/ie50260a018. [DOI] [Google Scholar]