

Abstract
The study reports on the drug release behavior of a potent synthetic somatostatin analogue, octreotide acetate, from biocompatible and biodegradable microspheres composed of poly-lactic-co-glycolic acid (PLGA) following a single intramuscular depot injection. The serum octreotide levels of three Oakwood Laboratories formulations and one Sandostatin LAR® formulation were compared. Three formulations of octreotide acetate-loaded PLGA microspheres were prepared by a solvent extraction and evaporation procedure using PLGA polymers with different molecular weights. The in vivo drug release study was conducted in male Sprague–Dawley rats. Blood samples were taken at predetermined time points for up to 70 days. Drug serum concentrations were quantified using a radioimmunoassay procedure consisting of radiolabeled octreotide. The three octreotide PLGA microsphere formulations and Sandostatin LAR® all showed a two-phase drug release profile (i.e., bimodal). The peak serum drug concentration of octreotide was reached in 30 min for all formulations followed by a decline after 6 h. Following this initial burst and decline, a second-release phase occurred after 3 days. This second-release phase exhibited sustained-release behavior, as the drug serum levels were discernible between days 7 and 42. Using pharmacokinetic computer simulations, it was estimated that the steady-state octreotide serum drug levels would be predicted to fall in the range of 40–130 pg/10 μL and 20–100 pg/10 μL following repeat dosing of the Oakwood formulations and Sandostatin LAR® every 28 days and every 42 days at a dose of 3 mg/rat, respectively.
Key words: in vivo drug release, pharmacokinetic simulation, PLGA microspheres, polypeptide/protein drug delivery, single depot injection
INTRODUCTION
Poly(d,l-lactide-co-glycolide) (PLGA), consists of one or more of three different hydroxy acid monomers, d-lactic, l-lactic, and/or glycolic acids, and the polymer can be made to be highly crystalline [e.g., poly(l-lactic acid), i.e., PLA], or completely amorphous [e.g., poly(d,l-lactic-co-glycolic acid)] (1,2). Many studies indicate that PLA and PLGA formulations containing therapeutic molecules are biocompatible and when used in therapeutic applications in vivo do not exhibit untoward reactions either locally or systemically (3,4). In recent years, the interest in biodegradable and biocompatible polymers, which control and prolong the action of therapeutic agents, has grown considerably in importance because delivery systems composed of biodegradable and biocompatible polymers do not require removal from the body at the end of the treatment period due to their degradation into physiologically occurring compounds that can be readily excreted from the body (5). Moreover, encapsulation of a drug provides an opportunity to avoid degradation of the drug in the biological milieu and can improve the bioavailability of the drug (6,7). Injectable PLGAs are an important advanced delivery system for week-to-month controlled release delivery of small molecular weight molecules and larger molecular weight molecules, e.g., peptides, proteins, and DNA/RNA (8–16). Moreover, PLGA microspheres have been used successfully to deliver therapeutic peptides and proteins in a sustained-release manner, and many commercial long-acting release (LAR®) drug formulations of microspheres are available on the market (e.g., Lupron Depot®, Zoladex®, Decapeptyl®, Eligard®, Enantone®, Trenantone®, Risperdal Consta® and Profact®) (17–27). Injectable sustained-release formulations possess many important advantages over conventional formulations, such as reduced dosing frequency, reduced overall total dose administered, reduced side effects, which all give rise to improved patient adherence. While oral drug delivery continues to be the primary route of administration for a variety of reasons, the parenteral route does offer important advantages when oral administration is difficult or not suitable. Inter-subject variability caused by differences in oral absorption (i.e., due to illness and extensive gastrointestinal metabolism) may be minimized by the parenteral route of administration. Finally, extended drug release can be achieved by parenteral sustained-release formulations over days up to several months by advanced injectible depot drug delivery systems.
Octreotide is a cyclic octapeptide and a potent synthetic somatostatin analogue that has become the mainstay of medical therapy for tumor control in neuroendocrine disorders such as acromegaly and gastroenteropancreatic neuroendocrine tumors (28–30). Octreotide is an effective first-line agent for a large group of acromegalic patients (independent of initial tumor extension). In addition, octreotide is safe and effective in the treatment of postoperative chylothorax in children with congenital heart disease. It is a useful adjunctive therapy to the conventional treatment of this complication (31). Despite its many potential uses, the clinical applications of octreotide are limited by its short half-life, and frequent injections are required to ensure an adequate control of the disease. Therefore, in order to obtain a long-term and constant therapeutic effect, a sustained-release depot formulation is required in patients who have constant daily pain (32). Two types of commercially available octreotide formulations are on market as pharmaceutical products. One commercially available product is an injectable form (lactate buffer, pH 4.2) for subcutaneous administration 3 times daily. The other marketed product is a sustained-release formulation (PLGA microspheres) for intramuscular administration on a monthly basis. It has been reported that the monthly sustained-release intramuscular formulation may be a useful substitute for the daily subcutaneous octreotide injection in the management of chronic therapy. It is useful to develop an optimal depot formulation of octreotide with ideal release parameters and optimal drug stability within the dosage form, since chronic therapy over decades is required in the chronic acromegalic patient. For the management of chronic diseases, reduced frequent dosing provides valuable benefits in enhancing the quality of life in patients. Moreover, recent results have confirmed the antiproliferative effect of octreotide sustained-release formulation in patients with well-differentiated metastatic gastroenteropancreatic neuroendocrine tumors of the midgut.
Computer simulation is a useful tool to estimate the pharmacokinetic parameters after repeated dosing of long-acting formulations. It can be difficult to examine the repeated dosing study of long-acting dosage forms in animals because it is a time and financially consuming process. In addition, aging of animals and the ethical aspects of animal experimentation have to be considered in the repeated dosing study. The time to reach the steady-state concentrations (Tss), the minimum drug–serum concentration (Cmin), the maximum drug–serum concentration (Cmax) and the drug–serum concentration range (Rmin–max) after repeated dosing can be calculated and estimated by simulation. These calculated parameters (Tss, Cmin, Cmax, Rmin–max) can be used for the evaluation of low, ineffective drug concentrations or unnecessarily high drug concentrations which may be poorly tolerated.
Many factors have to be considered to explain the in vivo drug absorption process such as drug release from the microspheres, volumes of injection, distribution of blood vessels, mobility of subjects, and area of injection sites. Sometimes, physiological factors may critically affect drug absorption because the rate and extent of intramuscular drug absorption are very erratic and variable (33,34). In this work, we evaluate the pharmacokinetic properties of octreotide from microspheres and investigate the pharmacokinetic parameters and repeated serum–drug concentrations from the point of view of the in vivo drug absorption. Therefore, the objectives of this study were the following: (1) to measure and assess the in vivo drug absorption behavior of octreotide for 70 days from heteropolymeric microspheres composed of the diblock co-polymer, poly-lactic-co-glycolic acid (PLGA) following a single depot intramuscular injection; (2) to compare the serum octreotide levels of three Oakwood Laboratories formulations and one Sandostatin LAR®, a commercial product of long-acting octreotide for intramuscular (IM); and (3) to compare in vivo pharmacokinetic drug absorption behavior with computer simulated pharmacokinetic drug release.
EXPERIMENTAL
Materials and Animals
Octreotide acetate (H2N-D-Phe-Cys-Phe-D-Trp-Lys-Thr-Cys-Thr-ol; MW = 1,018.4 g/mol) and octreotide-RIA kits were purchased from Bachem Inc (Torrance, CA). PLGA were from Boehringer Ingelheim (Ingelheim, Germany), and Sandostatin LAR® kits were purchased commercially. All other chemicals used were of analytical HPLC-reagent-grade.
Male Sprague–Dawley rats were used for the in vivo drug release study. Following University Institutional Animal Care and Use Committee (IACUC) approval, male rats weighing about 300 gm were injected with four different formulations. Six rats (per group) were randomly assigned to each PLGA microsphere group and seven rats (per group) were randomly assigned to Sandostatin LAR® group according to IACUC approval.
Microsphere Formulations
Three formulations of octreotide acetate-loaded PLGA microspheres were prepared by a solvent extraction and evaporation procedure with three different molecular weights (58,000, 70,000, and 76,400 Da) PLGA. Each formulations was administered in Group 1 (58,000 Da), Group 2 (70,000 Da), or Group 3 (76,400 Da), and Sandostatin LAR® 20 mg was administered in Group 4. Each formulation contained 20 mg of octreotide per vial. The marketed product, Sandostatin LAR® 20 mg was used for comparison. Six (Group 1, 2 and 3) or seven (Group 4) rats were used per group for the Octreotide product.
Drug Administration and Blood Sampling
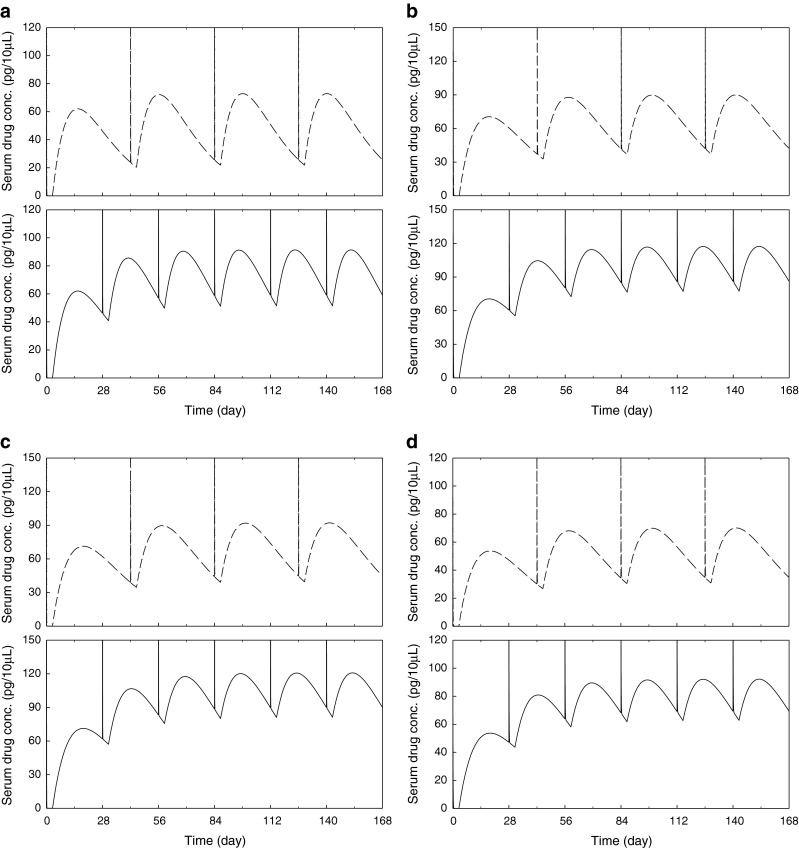
The microsphere formulations were reconstituted with Sterile Water for Injection (SWFI) to create a uniform suspension for administration. Reconstitution procedure of the Sandostatin LAR® 20 mg was performed according to the instructions in the Sandostatin LAR® 20 mg kit. The required volume of suspension to deliver a 3 mg dose was administered via a hypodermic needle intramuscularly into the rat’s quadriceps muscle located on the cranial aspect of the femur. The required volume was calculated using the following equation:
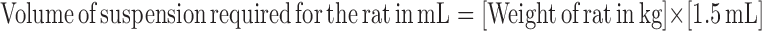 |
(For example, a 300 gm rat received 0.45 mL of suspension)
The injection site was pressed for around 1 min to prevent bleeding and microsphere leaking.
Rat blood samples were collected before drug dosing and thereafter at predetermined time points. After drug administration, samples were collected at 30 min, 1, 2, 6 h, 1, 3, 7, 14, 21, 28, 35, 42, 49, 60, and 70 days after dosing. During the first day, there was frequent sampling to capture the burst release. Sampling schedule was determined based on the previous reports (35). All rats had approximately 0.6–0.8 mL of blood drawn from the tail by a tail-vein nicking method at the specific time points. The blood samples were centrifuged at 10,000 rpm for 5 min and the serum was decanted into 0.5 mL polypropylene tubes followed by storage at −20°C until serum octreotide analysis was performed.
Serum Drug Analysis
The serum concentrations of octreotide were determined using a validated radioimmunoassay (RIA) method (35,36). The commercial octreotide-RIA kit (Bachem Inc., Torrance, CA) allowed for quantification of the peptide within the range of 1–125 pg; and hence, only 10 μL of serum sample was required for each tube in the analysis. The 3-day validated RIA procedure involved the following: On day 1, addition of primary rabbit anti-octreotide antibody to all tubes and incubation 16–24 h at 4°C; On day 2, addition of ([125I]–Tyr0)–Octreotide (radiolabeled octreotide) and incubation 16–24 h at 4°C; and on day 3, addition of secondary antibody with normal rabbit serum and incubation 90 min at room temperature. A precipitate was allowed to form in each tube and was subsequently collected by centrifugation and isolated by aspiration. A standard curve was constructed using a sigmoidal competitive binding equation provided by Graph Pad Prism® software (GraphPad Software Inc, San Diego, CA) (35).
Simulation of Steady-State Drug Serum Levels
Pharmacokinetic parameters were calculated using the pharmacokinetic computer software, WinNonlin® Version 5.2 (Pharsight Co., CA, USA). By the method of successive iterations, the best-fitted parameters and predicted curves were calculated by the Scientist® software program (MicroMath Research, St. Louis, MS). The R2 (goodness-of-fit statistic from model application) was obtained from fitting the equations to evaluate the validity of simulation. The model focuses on a successive fractional release method to convey the phases of drug release from a dosage form (i.e., burst release, diffusion-controlled release, and erosion-controlled release for microspheres). A one-compartment model was identified rat serum concentration and the first-order absorption equation to characterize diffusional burst was followed with another first-order absorption equation to characterize the erosion-controlled phase. Predicted data were obtained from fitting the equations to the drug concentration of each rat versus time profiles. The serum level simulations after repeated doses were subsequently programmed using these parameters into the Scientist® software program (MicroMath Research, St. Louis, MS), and the steady-state levels versus time profiles were obtained from simulations. The serum level simulations after repeated doses were obtained from the least squares fit method to the average concentration versus time profiles.
Statistics
The mean values and standard deviations are presented. The data was compared using Student’s t test with the level of significance set at P < 0.05.
RESULTS
Figure 1a features the mean serum concentration versus time profile of octreotide after single I.M. injection of microsphere formulations at the dose of 3 mg per rat. Figure 1b shows the data collected during the first day of the experiment, highlighting the transient peak which appears during the first 6 h after injection. The simulation of octreotide serum levels based on the experimental data is shown in Fig. 2 and the pharmacokinetic parameters are shown in Table I. There is two-phase bimodal absorption profile for the two octreotide acetate-containing PLGA microsphere batches (Group 2 and 3) and Sandostatin LAR®, as illustrated in Fig. 1a and b. However, three different phases were observed in Oakwood formulation 1 (Group 1). All formulations showed an initial elevation of octreotide levels after administration and peaked at 30 min. Although the AUC0–3 of three Oakwood formulations were not significant (p > 0.05), Cmax1 of all Oakwood formulations were higher than that of Sandostatin LAR® (p < 0.05). All formulations show an initial decline after 1 h followed by a gradual decline, and the drug serum levels are close to zero after 6 h, and certainly by 24 h. Then, drug absorption occurs again after 3 days, and there are discernible levels between 7 and 42 days in formulations 2, 3, and Sandostatin LAR®. In formulation 1, the drug levels gradually increase from days 3 to 7, but the serum drug levels drop from 55.7 to 39.6 pg/10 μL at day 14. The re-elevation of drug levels occurs after 14 days, and the drug levels are maintained between 20 and 42 days. The drug serum levels are less than 8 pg/10 μL by 56 days in all formulations. AUC3–70, AUC0–70 and Cmax2 of formulations 2 and 3 were significantly higher than those of Sandostatin LAR® (p < 0.05). The pharmacokinetic profile of Sandostatin LAR® in this study is in accordance with the study by Lancranjan et al., who reported a similar pharmacokinetic profile after I.M. injection in acromegalic patients (37). A rapid increase in octreotide serum concentrations was measured after I.M. injection of Sandostatin LAR® in acromegalic patients, with a peak occurring within 1 h after the administration followed by a progressive decrease to low octreotide levels within 12 h. On days 2 through 7, octreotide serum concentrations were at lower levels, after single doses of Sandostatin LAR®. Thereafter, an increase in serum drug concentrations occurred, and dose-dependent plateau concentrations were observed between days 14 and 42 followed by a gradual decrease from day 42 onwards (37).
Fig. 1.

a Serum concentration vs. time curves of octreotide after I.M. administrations of three Oakwood formulations and Sandostatin LAR 20 mg at the dose of 3 mg per rat; and b the data collected during the first 6 h after injection. Each point represents mean±S.D. (n = 6 or 7): open circle Oakwood formulation 1; open square Oakwood formulation 2; open upright triangle Oakwood formulation 3; open diamond Sandostatin LAR® 20 mg
Fig. 2.
Simulation of octreotide serum concentration following single I.M. administration of sustained-release microsphrere formulations at the dose of 3 mg per rat, and the data collected during the first 6 h after injection (inset). Each point represents mean±S.D. (n = 6 or 7): a Oakwood formulation 1; b Oakwood formulation 2; c Oakwood formulation 3; and d Sandostatin LAR 20 mg; filled circle experimental data; solid line simulation data
Table I.
Pharmacokinetic Parameters
| Parameters | Group 1a | Group 2a | Group 3a | Sandostatin LAR®b |
|---|---|---|---|---|
| AUC0–3 (day·pg/10 μL) | 14.4 ± 1.57 | 10.6 ± 0.747 | 15.9 ± 1.21 | 14.2 ± 5.23 |
| AUC3–70 (day·pg/10 μL) | 2,000 ± 170 | 2,460 ± 177c | 2,560 ± 275c | 2,020 ± 496 |
| AUC0–70 (day·pg/10 μL) | 2,010 ± 169 | 2,470 ± 177c | 2,570 ± 275c | 2,040 ± 495 |
| Cmax1 (pg/10 μL) | 223 ± 36.4c | 112 ± 17.1c | 253 ± 47.4c | 82.4 ± 12.5 |
| Tmax1 (min) | 30.0 ± 0.0 | 30.0 ± 0.0 | 30.0 ± 0.0 | 30.0 ± 0.0 |
| Cmax2 (pg/10 μL) | 72.4 ± 16.0 | 94.3 ± 8.86c | 93.7 ± 16.4c | 61.3 ± 18.1 |
| Tmax2 (day) | 16.3 ± 7.23c | 24.5 ± 3.83 | 24.5 ± 5.86 | 28.0 ± 10.7 |
| K e 1 (hr−1) | 0.965 ± 0.175 | 0.608 ± 0.137 | 0.993 ± 0.129 | 0.536 ± 0.115 |
| K e 2 (hr−1) | 2.55 × 10−3 ± 1.13 × 10−4 | 2.78 × 10−3 ± 1.23 × 10−4 | 2.74 × 10−3 ± 1.86 × 10−4 | 2.44 × 10−3 ± 5.36 × 10−4 |
| R 2 | 0.904 ± 0.0505 | 0.875 ± 0.0332 | 0.878 ± 0.0404 | 0.912 ± 0.0228 |
a N = 6, Mean±SD
b N = 7, Mean±SD
cSignificantly different from the Sandostatin LAR® (p < 0.05)
Body weight changes of rats were monitored with control group (no drug-treated group). Body weights for the treated rats showed similar patterns for all drug-treated groups through 70 days. Gradual increases in body weight were from ~295 to ~421 g at 70 days in all Oakwood’s batches and the Sandostatin LAR® group. However, the body weight gain of octreotide-treated groups was reduced, in comparison with that of the control group due to the pharmacological effects of the drug (38).
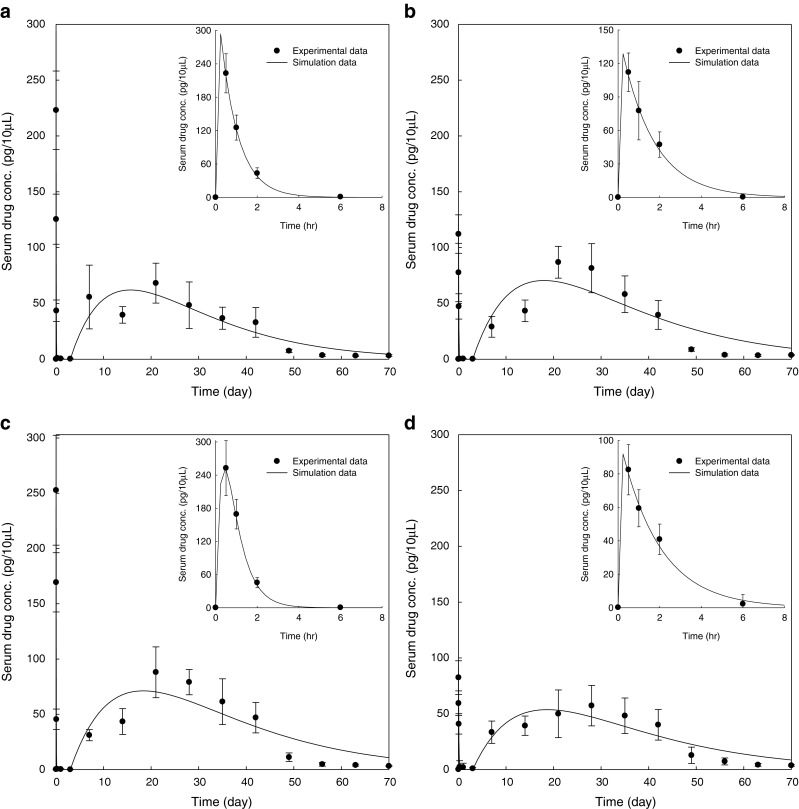
Figure 3 displays the comparison of octreotide serum concentration between formulations, and Fig. 4 shows the simulation of octreotide serum concentration following repeated I.M. injections of sustained-release microsphere formulations every 28 days or every 42 days at the dose of 3 mg per rat. The drug serum level of Oakwood formulation 1 is similar to that of the Sandostatin LAR® (Fig. 3a). Using pharmacokinetic computer simulations, it was estimated that the steady-state octreotide serum drug levels would be predicted to fall in the ranges of 40–100 pg/10 μL and 20–80 pg/10 μL following repeat dosing of the Oakwood formulation 1 and Sandostatin LAR® every 28 days and every 42 days, respectively (Fig. 4a and d). The drug serum levels of Oakwood formulations 2 and 3 are comparable (Fig. 3b), and the steady-state levels of Oakwood formulations 2 and 3 are estimated in the range of 50–130 pg/10 μL (every 28 days) or 30–100 pg/10 μL (every 42 days) after repeated doses (Fig. 4b and c).
Fig. 3.

Comparison of predicted octreotide serum concentration following single I.M. injections of sustained-release microsphrere formulations at the dose of 3 mg per rat. Each point represents mean±S.D. (n = 6 or 7): a Oakwood formulation 1 and Sandostatin LAR® 20 mg; b Oakwood formulation 2 and 3; filled circle predicted data for formulation 1; open circle predicted data for Sandostatin LAR®; filled square predicted data for formulation 2; open square predicted data for formulation 3; solid line predicted curve for formulation 1; dashed line predicted curve for Sandostatin LAR®; dash–dot–dash line predicted curve for formulation 2; dash–dot–dot–dash line predicted curve for formulation 3
Fig. 4.
Computer simulation of octreotide serum concentration following repeated I.M. injections of sustained-release microsphrere formulations every 28 days or 42 days at the dose of 3 mg per rat: a Oakwood formulation 1; b Oakwood formulation 2; c Oakwood formulation 3; d Sandostatin LAR® 20 mg; solid line simulation data for every 28 days; broken line simulation data for every 42 days
DISCUSSION
Overall, the in vivo release profile of octreotide from microspheres presented two phases, initial burst release phase due to the localization of the peptide near the surface or within pores or channels near the surface, followed by continuous release phase corresponding to the peptide entrapped in polymer matrix (35). After I.M. administration of microspheres, the initial burst released drug was rapidly absorbed (Tmax1, 30 min; Table I), and was almost eliminated within 6 h (Fig. 1b). As shown in Fig. 1b, the rate of drug elimination at first phase is directly proportional to the serum drug concentration (first-order elimination), and these elimination profiles are consistent with a previous report by Lemaire et al. (39). One major obstacle of injectable microspheres is the high initial burst of drug release (typically 10–80%) during the first day (40). In this study, the initial transient absorption completed within several hours, and the ratios of initial burst AUC0–3 to the total AUC0–70 were less than 0.75% in all formulations (Table I). No more than 1% of the drug was absorbed during the initial burst period, and over 99% of the drug absorption took place during the last period. Although the burst release phase was included in the modeling, the parameter Ka1 (burst-phase absorption constant in phase-1) and the parameter Ke1(elimination constant in phase-1) could not be accurately determined owing to the lack of sufficient data points in the first 6 h after administration. In fact, the first sample time point performed after drug administration at 30 min was the maximal value for the burst phase of release. As shown in Table I, the elimination rates of octreotide were rapid at the first phase, but the drug elimination rates were retarded at the second phase because the drug elimination rate was governed by the drug absorption rate, which is the rate-determining step. In fact, the absorption rates of the second phase were almost same as the elimination rates in all formulations, and these results suggest that the drug release rates were precisely controlled by all microsphere formulations at the second phase.
Octreotide from all formulations have a very slow (close to zero) absorption period for three days after the initial burst period, and then the drug absorption occurs again after 3 days (Fig. 2). The second continuous absorption phase was obtained in Oakwood formulation 2, 3 and the Sandostatin LAR®, and the main mechanism of this phase has been suggested as polymer erosion and bulk degradation (35). However, the second and the third phase were observed in Oakwood formulation 1. The triphasic drug release mechanism from microspheres has been reported in many papers (41–45). For triphasic release, the drug initially diffuses from the surface or close to the surface of microspheres. After this initial phase, drug released during the secondary stage of slower more sustained release probably diffuses out of the more porous polymer matrix as a result of polymer hydration (matrix release). This was followed by phase 3 release of drug which appeared to correspond to bulk degradation of the polymer devices (41–46). This different absorption pattern may be ascribed to the lower molecular weight of PLGA in formulation 1 compared to other two formulations (Oakwood formulations 2 and 3) (42,43). Erosion-controlled release of a luteinizing hormone analogue from a PLGA polymer was shown to exhibit biphasic kinetics over 2 months when injected intramuscularly to rhesus monkeys (47), and the triphasic pattern was reported in the in vivo absorption study of octreotide pamoate (41).
To describe the pharmacokinetic profiles of the drug, we used the simplest model, a 1-compartmental model with first-order input because a complex model can only explain the specific data set. The biphasic model was used to explain the octreotide pharmacokinetic profiles, and biphasic modeling can be commonly applied to all data set. Predicted data were obtained from fitting the equations to the drug concentration of each rat versus time profiles, therefore, the simulations seem to underestimate the observed values as in Fig. 2b, c and d. As shown in Table I, more than 0.87 of R2 (goodness-of-fit statistic from model application) values were obtained from all data sets.
Because of its potent inhibitory effect on hormone secretion the use of octreotide in clinical disorders related to excessive hormone secretion such as acromegaly has been proposed. Therefore, the use of octreotide can decrease the body weight in normal subjects and the body weights significantly decreased (p < 0.05) by about 13.5–17.7% in all octreotide-treated rats after 70 days of administration. From these results, it is expected that the pharmacological effect of the drug was maintained during the drug release period.
Despite the small difference in absorption pattern, the drug serum levels of Oakwood formulation 1 and the Sandostatin LAR® between 7 and 42 days are comparable (Fig. 3a). The drug serum level of Oakwood formulation 2 between 7 and 42 days is similar to that of Oakwood formulation 3 (Fig. 3b) although the initial burst concentration of Oakwood formulation 3 was two times higher than that of Oakwood formulation 2.
Two different dosing intervals, every 28 days and every 42 days, were utilized for multiple dosing simulations because Sandostatin LAR® is designed to be injected once every 28 days, and the drug serum levels were discernible between days 7 and 42 in all formulations. Figure 4 shows the time to reach the steady-state drug–serum concentrations and the drug–serum concentration range between the maximum drug concentration and the minimum drug concentration. Vertical lines mean the serum–drug concentration arising from initial burst release of the drug from microspheres after each administration. Following multiple doses of the long-acting depot dosage form given every 28 days or every 42 days, steady-state octreotide serum concentrations were achieved after the third injection (Fig. 4). The higher steady-state drug serum levels were obtained from every 28 days administration compared to every 42 days dosing. One of the major technical challenges in injectable microspheres, is a lag-time period, which is a very slow (close to zero) absorption period after the initial burst period because during this lag time, the patient may not be effectively treated due to the lack of sufficient drug absorption (40). The lag time of all Oakwood formulations and the Sandostatin LAR® was about 3 days, and the drug absorption was almost zero in this period (Fig. 3). However, if repeat dosing were given every 28 days or every 42 days, the serum drug levels can be maintained higher than 40 or 20 pg/10 μL after second dosing (Fig. 4).
CONCLUSIONS
In vivo sustained-absorption of the polypeptide therapeutic drug, octreotide acetate, was successfully achieved for at least 6 weeks following intramuscular depot injection of polymeric microspheres composed of the biodegradable and biocompatible heteropolymer, PLGA. In vivo absorption profile of octreotide from PLGA microspheres was bimodal with an initial rapid absorption followed by extended absorption between 3–56 days. Using pharmacokinetic computer simulations, the steady-state drug levels were predicted to be in the range of 40–130 pg/10 μL (every 28 days) or 20–100 pg/10 μL (every 42 days) depending on the dosing intervals.
ACKNOWLEDGMENT
This research was supported by Oakwood Laboratories. Senior laboratory technicians, Ms. Wei Qiu and Mr. Charles Ritchie, are gratefully acknowledged for their technical assistance.
REFERENCES
- 1.Jiang W, Gupta RK, Deshpande MC, Schwendeman SP. Biodegradable poly(lactic-co-glycolic acid) microparticles for injectable delivery of vaccine antigens. Adv Drug Deliv Rev. 2005;57(3):391–410. doi: 10.1016/j.addr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 2.Mansour HM, Sohn M, Al-Ghananeem A, Deluca PP. Materials for pharmaceutical dosage forms: molecular pharmaceutics and controlled release drug delivery aspects. Int J Mol Sci. 2010;11(9):3298–3322. doi: 10.3390/ijms11093298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ignatius AA, Claes LE. In vitro biocompatibility of bioresorbable polymers: poly(l, dl-lactide) and poly(l-lactide-co-glycolide) Biomaterials. 1996;17(8):831–839. doi: 10.1016/0142-9612(96)81421-9. [DOI] [PubMed] [Google Scholar]
- 4.Shive MS, Anderson JM. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Deliv Rev. 1997;28(1):5–24. doi: 10.1016/S0169-409X(97)00048-3. [DOI] [PubMed] [Google Scholar]
- 5.Kang J, Lambert O, Ausborn M, Schwendeman SP. Stability of proteins encapsulated in injectable and biodegradable poly(lactide-co-glycolide)-glucose millicylinders. Int J Pharm. 2008;357(1–2):235–243. doi: 10.1016/j.ijpharm.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Faisant N, Siepmann J, Benoit JP. PLGA-based microparticles: elucidation of mechanisms and a new, simple mathematical model quantifying drug release. Eur J Pharm Sci. 2002;15(4):355–366. doi: 10.1016/S0928-0987(02)00023-4. [DOI] [PubMed] [Google Scholar]
- 7.Perugini P, Genta I, Conti B, Modena T, Pavanetto F. Long-term Release of Clodronate from Biodegradable Microspheres. AAPS PharmSciTech. 2001;2(3):article 10. [DOI] [PMC free article] [PubMed]
- 8.Hamishehkar H, Emami J, Najafabadi AR, Gilani K, Minaiyan M, Mahdavi H, et al. The effect of formulation variables on the characteristics of insulin-loaded poly(lactic-co-glycolic acid) microspheres prepared by a single phase oil in oil solvent evaporation method. Colloids Surf B Biointerfaces. 2009;74(1):340–349. doi: 10.1016/j.colsurfb.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 9.Eratalay A, Coskun-Ari FF, Oner F, Ozcengiz E. In vitro and in vivo evaluations of PLGA microsphere vaccine formulations containing pDNA coexpressing Hepatitis B surface antigen and Interleukin-2. J Microencapsul. 2009. [DOI] [PubMed]
- 10.Ren XH, Zhang YL, Luo HY, Li HY, Liu SC, Zhang MJ, et al. PLGA microsphere-mediated growth hormone release hormone expression induces intergenerational growth. Anim Biotechnol. 2009;20(3):124–132. doi: 10.1080/10495390902945787. [DOI] [PubMed] [Google Scholar]
- 11.Chung HJ, Kim HK, Yoon JJ, Park TG. Heparin immobilized porous PLGA microspheres for angiogenic growth factor delivery. Pharm Res. 2006;23(8):1835–1841. doi: 10.1007/s11095-006-9039-9. [DOI] [PubMed] [Google Scholar]
- 12.Brandhonneur N, Loizel C, Chevanne F, Wakeley P, Jestin A, Le Potier MF, et al. Mucosal or systemic administration of rE2 glycoprotein antigen loaded PLGA microspheres. Int J Pharm. 2009;373(1–2):16–23. doi: 10.1016/j.ijpharm.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 13.van der Walle CF, Sharma G, Ravi Kumar M. Current approaches to stabilising and analysing proteins during microencapsulation in PLGA. Expert Opin Drug Deliv. 2009;6(2):177–186. doi: 10.1517/17425240802680169. [DOI] [PubMed] [Google Scholar]
- 14.Jiang W, Schwendeman SP. Stabilization of tetanus toxoid encapsulated in PLGA microspheres. Mol Pharm. 2008;5(5):808–817. doi: 10.1021/mp800027f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geng Y, Yuan W, Wu F, Chen J, He M, Jin T. Formulating erythropoietin-loaded sustained-release PLGA microspheres without protein aggregation. J Control Release. 2008;130(3):259–265. doi: 10.1016/j.jconrel.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 16.Gebrekidan S, Woo BH, DeLuca PP. Formulation and in vitro transfection efficiency of poly (d,l-lactide-co-glycolide) microspheres containing plasmid DNA for Gene delivery AAPS PharmSciTech. 2000;1(4):article 28. [DOI] [PMC free article] [PubMed]
- 17.Abouelfadel Z, Crawford ED. Leuprorelin depot injection: patient considerations in the management of prostatic cancer. Therapeut Clin Risk Manag. 2008;4(2):513–526. doi: 10.2147/tcrm.s6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kundi SA, Wazir S, Afzal S, Afzal N, Qayum I, Forsling ML. Release of vasopressin during suppression of oestrous cycle in rat by zoladex and hypovolemic challenge. J Ayub Med Coll Abbottabad. 2005;17(4):63–66. [PubMed] [Google Scholar]
- 19.Horvath JE, Bajo AM, Schally AV, Kovacs M, Herbert F, Groot K. Effects of long-term treatment with the luteinizing hormone-releasing hormone (LHRH) agonist Decapeptyl and the LHRH antagonist Cetrorelix on the levels of pituitary LHRH receptors and their mRNA expression in rats. Proc Natl Acad Sci USA. 2002;99(23):15048–15053. doi: 10.1073/pnas.232579499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berges R, Bello U. Effect of a new leuprorelin formulation on testosterone levels in patients with advanced prostate cancer. Curr Med Res Opin. 2006;22(4):649–655. doi: 10.1185/030079906X96425. [DOI] [PubMed] [Google Scholar]
- 21.Sophocleous AM, Zhang Y, Schwendeman SP. A new class of inhibitors of peptide sorption and acylation in PLGA. J Control Release. 2009;137(3):179–184. doi: 10.1016/j.jconrel.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kemp SF, Fielder PJ, Attie KM, Blethen SL, Reiter EO, Ford KM, et al. Pharmacokinetic and pharmacodynamic characteristics of a long-acting growth hormone (GH) preparation (nutropin depot) in GH-deficient children. J Clin Endocrinol Metab. 2004;89(7):3234–3240. doi: 10.1210/jc.2003-030825. [DOI] [PubMed] [Google Scholar]
- 23.Cook DM, Biller BM, Vance ML, Hoffman AR, Phillips LS, Ford KM, et al. The pharmacokinetic and pharmacodynamic characteristics of a long-acting growth hormone (GH) preparation (nutropin depot) in GH-deficient adults. J Clin Endocrinol Metab. 2002;87(10):4508–4514. doi: 10.1210/jc.2002-020480. [DOI] [PubMed] [Google Scholar]
- 24.Gefvert O, Eriksson B, Persson P, Helldin L, Bjorner A, Mannaert E, et al. Pharmacokinetics and D2 receptor occupancy of long-acting injectable risperidone (Risperdal Consta) in patients with schizophrenia. Int J Neuropsychopharmacol. 2005;8(1):27–36. doi: 10.1017/S1461145704004924. [DOI] [PubMed] [Google Scholar]
- 25.Irani J, Mottet NA, Salomon L, Oba R. Randomized, double blind study comparing efficacy and tolerance of venlafaxine vs medroxyprogesterone acetate vs cyproterone for hot flushes in men under leuprorelin 11.25 mg (Enantone) for prostate cancer. J Urol. 2009;181(4):231–231. doi: 10.1016/S0022-5347(09)60657-1. [DOI] [Google Scholar]
- 26.Grischke EM, Fehm T, Solomayer E, Hillger H, Wallwiener D. Effect of a 3 month depot injection of leuprorelin acetate (trenantone (R)) on pre-menopausal breast cancer—results of a post-marketing surveillance study. Geburtshilfe Frauenheilkd. 2008;68(10):1008–1012. doi: 10.1055/s-2008-1038944. [DOI] [Google Scholar]
- 27.Rhee YS, Park CW, DeLuca PP, Mansour HM. Sustained-release injectable drug delivery systems. Pharm Tech. 2010;11:16–22. [Google Scholar]
- 28.Anthony L, Freda PU. From somatostatin to octreotide LAR: evolution of a somatostatin analogue. Curr Med Res Opin. 2009;25(12):2989–2999. doi: 10.1185/03007990903328959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Susini C, Buscail L. Rationale for the use of somatostatin analogs as antitumor agents. Ann Oncol. 2006;17(12):1733–1742. doi: 10.1093/annonc/mdl105. [DOI] [PubMed] [Google Scholar]
- 30.Kendall-Taylor P, Miller M, Gebbie J, Turner S, al-Maskari M. Long-acting octreotide LAR compared with lanreotide SR in the treatment of acromegaly. Pituitary. 2000;3(2):61–65. doi: 10.1023/A:1009997506216. [DOI] [PubMed] [Google Scholar]
- 31.Parames F, Freitas I, Fragata J, Trigo C, Pinto MF. Octreotide–additional conservative therapy for postoperative chylothorax in congenital heart disease. Rev Port Cardiol. 2009;28(7–8):799–807. [PubMed] [Google Scholar]
- 32.Lieb JG, 2nd, Shuster JJ, Theriaque D, Curington C, Cintron M, Toskes PP. A pilot study of Octreotide LAR vs. octreotide tid for pain and quality of life in chronic pancreatitis. JOP. 2009;10(5):518–522. [PubMed] [Google Scholar]
- 33.Zuidema J, Kadir F, Titulaer HAC, Oussoren C. Release and absorption rates of intramuscularly and subcutaneously injected pharmaceuticals (II) Int J Pharm. 1994;105(3):189–207. doi: 10.1016/0378-5173(94)90103-1. [DOI] [Google Scholar]
- 34.Zuidema J, Pieters FAJM, Duchateau GSMJE. Release and absorption rate aspects of intramuscularly injected pharmaceuticals. Int J Pharm. 1988;47(1–3):1–12. doi: 10.1016/0378-5173(88)90209-8. [DOI] [Google Scholar]
- 35.Murty SB, Wei Q, Thanoo BC, DeLuca PP. In vivo release kinetics of octreotide acetate from experimental polymeric microsphere formulations using oil/water and oil/oil processes. AAPS PharmSciTech. 2004;5(3):e49. doi: 10.1208/pt050349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Logue S, Miller M, Hancock T, McCush F, Velagaleti P, de Chasteigner S. A radioimmunoassay method for the quantitation of octreotide in rat plasma. AAPS PharmSci. 2001;3(S1):549. [Google Scholar]
- 37.Lancranjan I, Bruns C, Grass P, Jaquet P, Jervell J, Kendall-Taylor P, et al. Sandostatin LAR®: pharmacokinetics, pharmacodynamics, efficacy, and tolerability in acromegalic patients. Metabolism. 1995;44(Supplement 1):18–26. doi: 10.1016/0026-0495(95)90306-2. [DOI] [Google Scholar]
- 38.Flyvbjerg A, Jorgensen KD, Marshall SM, Orskov H. Inhibitory effect of octreotide on growth hormone-induced Igf-I generation and organ growth in hypophysectomized rats. Am J Physiol. 1991;260(4):E568–E574. doi: 10.1152/ajpendo.1991.260.4.E568. [DOI] [PubMed] [Google Scholar]
- 39.Lemaire M, Azria M, Dannecker R, Marbach P, Schweitzer A, Maurer G. Disposition of sandostatin, a new synthetic somatostatin analogue, in rats. Drug Metab Dispos. 1989;17(6):699–703. [PubMed] [Google Scholar]
- 40.Wang J, Wang BM, Schwendeman SP. Characterization of the initial burst release of a model peptide from poly(d,l-lactide-co-glycolide) microspheres. J Control Release. 2002;82(2–3):289–307. doi: 10.1016/S0168-3659(02)00137-2. [DOI] [PubMed] [Google Scholar]
- 41.Comets E, Mentre F, Nimmerfall F, Kawai R, Mueller I, Marbach P, et al. Nonparametric analysis of the absorption profile of octreotide in rabbits from long-acting release formulation OncoLAR. J Control Release. 1999;59(2):197–205. doi: 10.1016/S0168-3659(98)00194-1. [DOI] [PubMed] [Google Scholar]
- 42.Cleland JL. Solvent evaporation processes for the production of controlled release biodegradable microsphere formulations for therapeutics and vaccines. Biotechnol Prog. 1998;14(1):102–107. doi: 10.1021/bp970128t. [DOI] [PubMed] [Google Scholar]
- 43.Lewis KJ, Irwin WJ, Akhtar S. Development of a sustained-release biodegradable polymer delivery system for site-specific delivery of oligonucleotides: characterization of P(LA-GA) copolymer microspheres in vitro. J Drug Target. 1998;5(4):291–302. doi: 10.3109/10611869808995882. [DOI] [PubMed] [Google Scholar]
- 44.Bodmer D, Kissel T, Traechslin E. Factors influencing the release of peptides and proteins from biodegradable parenteral depot systems. J Control Release. 1992;21(1–3):129–137. doi: 10.1016/0168-3659(92)90014-I. [DOI] [Google Scholar]
- 45.Chen X, Ooi CP, Lye WS, Lim TH. Sustained release of ganciclovir from poly(lactide-co-glycolide) microspheres. J Microencapsul. 2005;22(6):621–631. doi: 10.1080/02652040500162782. [DOI] [PubMed] [Google Scholar]
- 46.Wang HT, Palmer H, Linhardt RJ, Flanagan DR, Schmitt E. Degradation of poly(ester) microspheres. Biomaterials. 1990;11(9):679–685. doi: 10.1016/0142-9612(90)90026-M. [DOI] [PubMed] [Google Scholar]
- 47.Sanders LM, Kent JS, McRae GI, Vickery BH, Tice TR, Lewis DH. Controlled release of a luteinizing hormone-releasing hormone analogue from poly(d,l-lactide-co-glycolide) microspheres. J Pharm Sci. 1984;73(9):1294–1297. doi: 10.1002/jps.2600730927. [DOI] [PubMed] [Google Scholar]