

Abstract
Two common variants within CFH, the Y402H1–4 and the rs1410996 SNPs5,6, explain 17% of age-related macular degeneration (AMD) liability. However, proof for the involvement of CFH, as opposed to a neighboring transcript, and the potential mechanism of susceptibility alleles are lacking. Assuming that rare functional variants might provide mechanistic insights, we used genotype data and high throughput sequencing to discover a rare high-risk CFH haplotype containing an R1210C mutation. This allele has been implicated previously in atypical hemolytic uremic syndrome, and abrogates C-terminal ligand binding7,8. Genotyping R1210C in 2,423 AMD cases and 1,122 controls demonstrated high penetrance (present in 40 cases versus 1 control, p=7.0×10−6) and six year earlier onset of disease (p=2.3×10−6). This result suggests that loss of function alleles at CFH likely drive AMD risk. This finding represents one of the first instances where a common complex disease variant has led to discovery of a rare penetrant mutation.
Age-related macular degeneration is the leading cause of late-onset blindness in the industrialized world9. Common CFH variants are associated with AMD and meningitis susceptibility. Rare CFH mutations cause atypical hemolytic uremic syndrome (aHUS), an episodic illness that causes acute renal failure, and membranoproliferative glomerulonephritis type II (MPGNII), a chronic renal disease10,11. While the possibility that rare CFH variants confer AMD risk has been raised12, no advanced AMD risk conferring rare variant has yet been identified.
To identify rare penetrant variants within the CFH locus, we phased genotypes for 20 common SNPs spanning the CFH/CFHR1-3 region and a common CFHR1-3 deletion (CFHR1-3Δ) in 711 advanced AMD cases and 1041 controls (“Boston-phased” dataset)13,14. Controls included 304 phenotyped controls without evidence of retinal disease and 737 unphenotyped shared controls presumed to not have disease. Almost all these markers are associated with AMD (Supplementary Table 1). We defined 11 common haplotypes (>0.3% frequency), explaining 97.4% of 3354 chromosomes, and calculated association statistics (Figure 1A, Supplementary Table 2). Consistent with prior observations1–4, the most frequent haplotype (H1) is present in 59% of case chromosomes but only in 37% of control chromosomes; this haplotype exclusively contains the Y402H1–4 risk allele (in LD with rs10801555, r2=0.99 in 288 genotyped controls). Two haplotypes, H2 and H3, containing a proxy to the rs1410996 CFH intronic risk SNP5,6, rs10737680, conferred intermediate risk (OR=0.59 [0.49–0.71] relative to H1). Excepting H4, the remaining haplotypes contained none of the two common risk variants, and in aggregate conferred low risk (OR = 0.30 [0.26–0.36] relative to H1).
Figure 1.

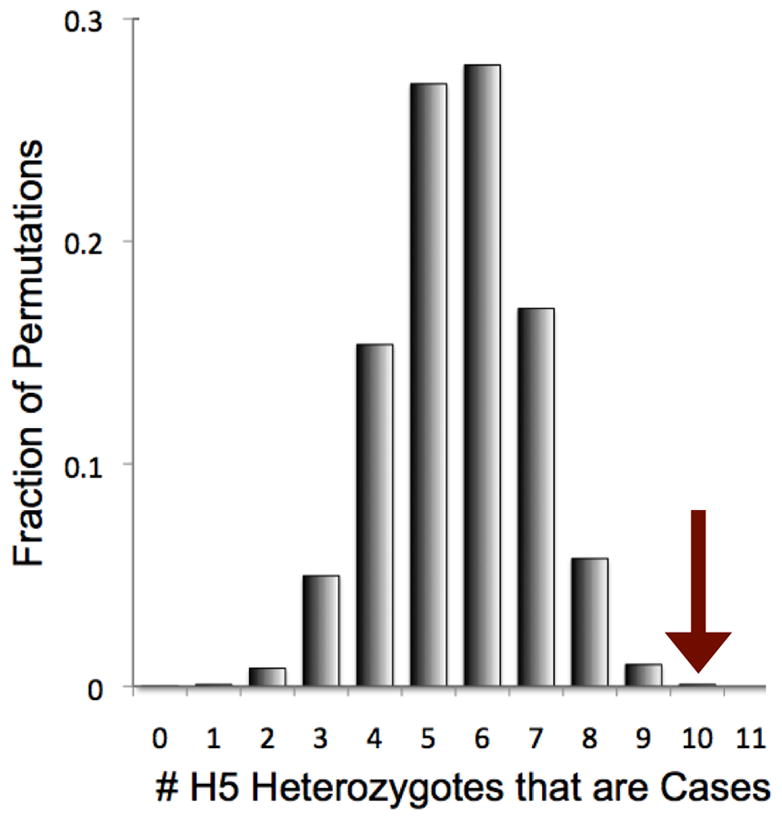
A. Phasing 21 markers in the CFH/CFHR1/CFHR3 region. Here we present association statistics of 11 haplotypes with frequencies >0.3%, resulting from phasing 20 SNP markers and a CFHR1-3 common copy number polymorphism. We specifically note the CFH rs800292 (I62V) SNP; the CFH Y402H proxy, rs10801555; CFH rs1410996 proxy, rs10737680; and the CFHR1-3 deletion. For most SNPs we list the nucleotide, for the CFHR1-3 deletion the empty circle (◎) indicates the region is deleted while the filled circle (◉) indicates the region is not deleted. To the right of each haplotype, we note the observed frequency in cases and controls. To the far right of each haplotype we note the relative ratio of the odds of disease for each haplotye relative to H1. For the H5 haplotype we note that it has a significantly greater allelic odds ratio than H1. The contrast is more striking when compared to the aggregate odds ratio of haplotypes H4-H11 (red dot to the left of the H5 OR). B. Case-control permutations preserving genotype at four common AMD associated markers. Here we present a histogram of the number of H5 heterozygote individuals that are cases for each of the 100,000 permutations. We place an arrow at 10, the observed number of H5 heterozygote individuals that are cases in the actual data (p=0.00081).
Strikingly, 10 of 11 individuals heterozygote for the rare H5 haplotype had AMD. The unaffected individual was a 49 year-old unphenotyped male, unlikely to have advanced disease given his age. This proportion of H5 heterozygote individuals with disease was unexpected since H5 lacked both the Y402H and rs10737680/rs1410996 risk alleles. We speculated that a rare variant on the H5 haplotype might explain the data. However, prior to pursuing a sequencing experiment, we considered that the haplotype association was the consequence of chance or stratification. Therefore we asked whether (1) H5 associated significantly with disease accounting for known AMD-associated common variants; (2) H5 correlated with earlier age of onset; and (3) the H5 association could be explained by case-control stratification or recent ancestry.
We assessed the statistical significance of the association of H5 with advanced AMD by permuting case-control status across all 1,752 subjects. Since we wanted to conservatively assess whether the association was independent of known risk alleles, we were careful to preserve genotypes for four published AMD-associated common markers: Y402H (rs1080155), rs10737680/rs1410996, CFHR1-3Δ, and rs800292 (I62V). This permutation fixes case-control allele frequencies, association statistics, and odds ratios at those markers, and also constrains association statistics at the other 17 markers due to underlying linkage disequilibrium in region (Supplementary Figure 1). While common marker association statistics are maintained in these permutations, we rarely observed that ten or more affected heterozygote H5 individuals (p=0.00081, Figure 1B). Permuting case-control status without preserving marker genotypes or preserving genotypes at only two common markers (Y402H and rs1410996) yield similar results (p=0.00081 and p<0.00001 respectively). Thus the case-association with this haplotype is beyond what might be expected by chance.
The 10 H5 heterozygote individuals were diagnosed earlier than the other 701 cases in the Boston-phased data set (median age 62.5 years versus 71 years, p=0.0023 by 1-tailed rank-sum test), further suggesting that H5 might influence AMD risk.
To assess the possibility that the H5 association might be the consequence of recent relatedness or population stratification, we selected 79,091 independent SNPs from genome-wide data (Methods). We noted first that there was only modest genome-wide stratification (λgc=1.08)13. We projectied genotype data into principal components14 and observed that H5 heterozygotes do not cluster discretely (Supplementary Figure 2) and that H5 heterozygote individuals were not skewed along any of the top 20 components (p>0.047, Supplementary Table 3). To confirm that H5 was not the consequence of a recent common ancestry, we estimated the proportion of identity by descent to be <3% between H5 heterozygote pairs; at least ~12.5% would be expected for first cousins or more closely related relatives. Taken together, our data suggest that the observed H5 association is unlikely to be artifactual.
To discover the causal mutation we designed a sequencing experiment. We selected 84 samples representing all 11 haplotypes including homozygote individuals when possible; we included all 10 affected individuals with H5. We first applied high-throughput sequencing to a 106.7 kb region containing CFH introns, exons, and promotor. We sequenced a subset of 60 individuals (33 cases and 27 controls), representing each of the 11 haplotypes. These samples included 6 H5 heterozygote individuals. We achieved high coverage with 80% of individuals sequenced at ≥20x for at least 89.3 kb (Supplementary Figure 3). Genotype calls from sequencing were 98.9% accurate in 19 separately genotyped SNPs. We identified a total of 623 variants; to avoid false positive calls, we focused our analyses on 356 variants seen at least twice (Supplementary Table 4, Supplementary Table 5). We were confident that H5 specific CFH alleles, both coding and non-coding, were likely to be among these since two or more H5 heterozygotes achieved >20x coverage in 97.8% of the targetted region. We found only six non-synonymous alleles: (1) rs800292 (I62V); (2) rs1061170 (Y402H); (3) rs1065489 (D936E); (4) rs35274867 (N1050Y); (5) a rare glutamine to histidine variant (Q950H); and (6) a rare exon 22 variant that alters arginine to cysteine, R1210C (Supplementary Figure 4A). Of all observed variants, only R1210C variant was seen exclusively in all of the 6 sequenced H5 heterozygote individuals. To confirm that R1210C was specific for H5, we used capillary electrophoresis to sequence exon 22 in all 84 samples (Supplementary Figure 4B). Only the ten affected individuals with the H5 haplotype had the R1210C mutation (Figure 2); we concluded that R1210C was specific for H5. We found no other exon 22 variants in any of these 84 individuals.
Figure 2. Sequencing the H5 haplotype identifies an R1210C mutation.

We applied capillary electrophoresis sequencing to 84 individuals representing the common CFH haplotypes depicted in Figure 1. Each circle represents an individual. The position on the grid indicates the two haplotypes for the individual at the CFH locus; individuals along the diagonal are homozygous for a haplotype. Individuals that do not have the R1210C mutation are depicted with an empty circle (◎). Individuals that are heterozygous for the R1210C mutation are indicated with a filled red circle (◉). All 10 individuals with the R1210C mutation are heterozygous for the H5 haplotype, strongly suggesting that the mutation is on that haplotype, and accounts for the increased risk associated with that haplotype.
To confirm the R1210C association and to asses its presence on other haplotypic backgrounds, we genotyped 707 out of the 711 cases and 303 out of 304 examined controls from the Boston-phased data set (Table 1). We observed 13 R1210C heterozygote individuals. In addition to the 10 H5 heterozygote individuals, we identified three more affected individuals with R1210C, two of whom were H1 homozygote and one that was an H1/H11 heterozygote. We did not find the R1210C mutation on any controls. This suggested that in addition to being present consistently on the H5 haplotype, R1210C was also rarely present on H1. One possibility is that the R1210C mutation initially occurred on the common H1 background, and that the H5 haplotype exists because of a historical recombination with H6. These data suggested that R1210C explained the H5 haplotype association to disease and was associated with advanced AMD (p=0.0094 by 1-tailed Fisher’s exact test, Table 1).
Table 1. Genotyping results.
This table presents the genotyping results of individuals from three case-control collections, and also from siblings of individuals in the Boston collections with the R1210C mutation. The first row describes the results of individuals from the Boston-phased data set, used to generate the haplotypes illustrated in Figure 1. The next two rows describe data from two replication collections. The third row presents aggregate statistics of the replication collections. The fourth row presents aggregate statistics from all of the case-control data sets, including both the replication data and the Boston-phased data set. The fifth row presents the results of genotyping siblings of affected individuals with the R1210C mutation. The final row presents statistical significance data of all of the above data sets. For each data set, we present counts of affected and unaffected individuals with and without the R1210C mutation. We also list one-tailed p-values for each row, with and without stratifying on Y402H genotype.
| R1210C Heterozygotes | R1210 Homozygotes | One-tailed p-value | ||||
|---|---|---|---|---|---|---|
| Advanced AMD | Unaffected | Advanced AMD | Unaffected | Not conditional on Y402H | Conditional on Y402H | |
| 1. Boston-Phased | 13 | 0 | 694 | 303 | 0.0094 | 0.0030 |
| 2. Boston Replication | 10 | 1 | 1195 | 656 | 0.058 | 0.0086 |
| 3. Baltimore Replication | 10 | 0 | 492 | 160 | 0.062 | 0.016 |
| All Replication (2+3) | 0.0052 | 0.00021 | ||||
| All Case-Control (1+2+3) | 8.0E-05 | 9.1E-07 | ||||
| 4. First Degree Relatives (Boston) | 7 | 0 | 2 | 2 | 0.033 | |
| Meta-Analysis (1+2+3+4) | 7.0E-06 | 1.3E-07 | ||||
To replicate the R1210C association, we genotyped an independent set of 1,707 cases and 817 controls from Boston and Baltimore sample sets (Table 1). We assessed significance with exact statistics since the R1210C heterozygotes were so rare (Methods). In this independent case-control replication we observed a statistically significant association (20 in cases vs 1 in controls, one-tailed p=0.0052). Combining our case-control replication together with the Boston-phased discovery data set we observed that R1210C heterozygotes were seen in 33/2414 cases (1.4%) compared to 1/1120 controls (<0.1%), suggesting that this mutation confers a high-degree of risk (one-tailed combined p=8.0×10−5) and acts as an almost fully penetrant allele. Since Y402H is an influential risk factor at the population level1–4, we also assessed significance by stratifying for the Y402H genotype (Methods). We observed that stratifying on Y402H only increased significance of the R1210C association (one tailed p=9.1×10−7). We concluded that R1210C confers risk independently of the common Y402H risk allele.
To further replicate the R1210C association in independent first-degree relatives, we identified 11 siblings of 6 cases with R1210C from the Boston-phased and Boston replication. These siblings were either unambiguously affected with advanced AMD (n=9), or had absolutely no clinical evidence of disease (n=2) (Table 1, Supplementary Table 6). We observed that 7 of 9 affected individuals were R1210C heterozygotes and 0 of 2 controls were R1210C heterozygotes (p=0.033, one-tailed binomial). In total, aggregating all of the results from our family and case-control data together, we observed a highly significant association (p=7.0×10−6, p=1.3×10−7 stratifying for Y402H, Table 1). We estimate that this mutation accounts for ~14% increase in disease prevalence among siblings of affected probands.
Since R1210C apparently has a strong genetic effect, we tested whether the mutation also drives an early age of disease onset. We observed that unrelated individuals with the R1210C mutation had significantly earlier disease onset (median of 65 versus 71 years, p=2.3×10−6 by rank-sum test, Figure 3A). Of the 55 individuals with an age of onset <55 years, 5 had the R1210C mutation (8.3%); no individual with onset after 75 years had the R1210C mutation. The retinal phenotype for individual patients with R1210C typically included numerous small, medium, and large drusen (Figure 3B).
Figure 3. The phenotype of the R1210C mutation.


A. Here we plot a histogram of the age of onset for 23 individuals with the R1210C mutation and also for 1,887 individuals without the R1210C mutation from Boston-phased and Boston-replication with available data. Age of onset is defined as the age when the patient starts showing signs of AMD. While the median age of diagnosis for affected individuals with the R1210C mutation is six years less than those without (65 versus 71 years), the mean age is 8.6 years less (61.9 versus 70.5 years). B. Fundus photograph of the right eye of a subject with the R1210C mutation showing central geographic atrophy (advanced dry age-related macular degeneration) surrounded by numerous large and very large drusen in the posterior pole and along the vascular arcades. Drusen were present nasal and temporal to the macula and in all four quadrants out to the mid-peripheral retina.
Importantly, in our sequencing experiment we found only one rare intronic allele that might potentially contribute to the phenotype; this was a SNP (at 194905360 (HG18)) in 5 of the 6 H5 heterozygotes (Supplementary Table 4). However, we consider this allele unlikely to be causal. First, this variant has no evidence of mammalian conservation or regulatory function. Second, applying capillary electrophoresis sequencing to 17 R1210C heterozygote individuals revealed six who individuals lacking the intronic allele, indicating again that the R1210C allele is on multiple haplotypes and that the intronic change is more recent.
The R1210C mutation had been described as an incompletely penetrant allele that causes familial forms of aHUS15–19, and has also been associated with primary glomerulonephritis20. This allele has been observed worldwide in multiple aHUS families and in 1–4% of individuals in aHUS cohorts16. The R1210C mutation has been shown to compromise C-terminal CFH function; mutant CFH protein has normal cofactor activity but exhibits defective binding to C3d, C3b, heparin, and endothelial cells7,8,21. Moreover, mutant protein product in plasma produces a high-molecular-weight factor H protein, resulting from a covalent interaction with human serum albumin15.
We assessed renal function by estimating the glomerular filtration rate from serum creatinine measurements in 17 unrelated R1210C heterozygotes with advanced AMD. None had evidence of clinically significant renal dysfunction (creatinine>1.7 mg/dl, eGFR<30 mL/min), though we noted subclinical renal dysfunction (median eGFR of 62 mL/min). Since AMD patients in general have subclinical renal dysfunction22, we compared renal function to 17 individuals matched on disease severity, age, and gender but without R1210C. We observed no significant difference (one-tailed p=0.54, Supplementary Figure 5).
A previous study proposed that rare CFH variants might be compound heterozygous alleles acting in trans with common alleles such as Y402H or I62V12. We found no evidence of this for the R1210C mutation. For the 13 Boston-phased cases with the R1210C (with complete phase information), we observed no excess beyond chance of Y402H risk alleles in trans [61% (=8/13) versus 59% frequency in all cases] or I62V risk [85% (=11/13) versus 87% frequency] in trans.
Our data suggest that compromised CFH function contributes to AMD pathogenesis and facilitates the transition from association to causality that is ubiquitously necessary to advance studies of complex disorders. We do not exclude possible contributions of neighboring CFH-like loci. Nonetheless, abrogated C-terminal activity of this protein represents potentially the first mechanistic clue about pathogenesis, and could inform attempts to understand the action of other cis and trans susceptibility alleles. This highlights the value of finding rare alleles with experimentally tractable biological effect in complex traits.
Our data link two clinically unrelated diseases, AMD and aHUS, with common underlying pathology. We note that AMD has common variants in multiple complement pathway genes (CFI23, CFH1–6, CFB24, and C34,25), and that rare predisposing mutations to aHUS have been observed in these same genes26. Future studies sequencing these genes in large numbers of patients might also reveal rare, high penetrant alleles that could contribute to understanding AMD mechanism.
METHODS
Study Sample Descriptions
Case Definitions
All individuals, except MIGEN shared controls, were evaluated by a board certified ophthalmologist. For all individuals we either (1) clinically evaluated with visual acuity measurements, dilated slit lamp biomicroscopy, and stereoscopic color fundus photography or (2) reviewed full ophthalmologic medical records. Case patients had either geographic atrophy (advanced dry AMD) or neovascular disease (wet AMD) (Clinical Age-Related Maculopathy Grading System (CARMS) stages 4 and 5)27. Controls were without macular degeneration, and without early or intermediate disease, categorized as stage 1. All Boston controls and most Baltimore samples (>80%) were ≥60 years old. Individuals with early or intermediate disease were excluded from this study.
Boston
Subjects were recruited through ongoing AMD study protocols5,28–31; sample ascertainment and genotyping has been previously described5,23,25. All samples were unrelated self-described white individuals of European descent. A subset of these samples were previously genotyped using the Affymetrix SNP 6.0 GeneChip32. We combined those samples passing strict quality-control genome-wide with 100% successful SNP and CNV genotype calls at the CFH locus (n=711 cases, 304 controls) with shared control samples from the MIGEN33 study passing the same strict quality control criteria (n=737) to constitute the Boston-phased data set34. Remaining unrelated individuals (n=1,205 cases, 657 controls) comprised the Boston Replication sample.
Baltimore
Unrelated subjects recruited at Johns Hopkins Hospital in Baltimore, MD as previously described32,35–37, that were self-described white individuals of European descent constituted the Baltimore Replication collection.
Siblings
For individuals with advanced AMD who had the R1210C mutation of interest, we identified siblings within the Boston cohort. We included only extremely discordant siblings with advanced AMD or no evidence of disease to avoid ambiguous diagnoses. Sibling samples are independent of the original Boston-phased and Boston Replication collections.
High throughput sequencing
We use long-range PCR tp sequence the whole 107 kb region around CFH.
We designed long-range PCR primers to be 28–35 nucleotides long to assure high melting temperature/specificity, without G stretches (4+ Gs), without secondary structure, and without multiple annealing locations genome-wide (Supplementary Table 7A). We used the high-fidelity Takara LA DNA polymerase for PCR amplification. Primers for each amplicon was QC tested on control DNA. We selected primer pairs that that successfully generated PCR products and in aggregate covered the whole region of interest. We used the PreCR™ (New England Biolabs) repair approach in order to remediate samples that performed poorly during the PCR amplification step.
We cleaned amplicons using Ampure Beads, quantified using Picogreen, and then pooled in equimolar amounts. We constructed libraries with Nextera™ (Epicentre Biotechnologies). During the subsequent enrichment step unique index sequences are added for each DNA sample. Ten index libraries were pooled together in a single lane for sequencing. We sequenced pooled index libraries on Illumina IIGX genome analyzer To generate 50 nucleotides paired end read.
We used Burrows-Wheeler Aligner (BWA)38 to align the short reads to the human genome build GRCh37. Duplicate reads were marked and removed using Picard (http://picard.sourceforge.net) to avoid recurrent errors in DNA molecules. We performed quality score recalibration and local realignment around indels using the Genome Analysis Tool Kit (GATK)39. We called SNP genotypes with the Unified Genotyper module in GATK based on three metrics a) mapping quality score b) coverage and c) strand bias (see http://www.broadinstitute.org/software/syzygy/). Low coverage samples (<10x depth for >20kb) and samples with >15% genotype error with 19 SNPs genotyped on the Affymetrix array were removed. For this study we required that the minimum mapping quality score be at least 10, coverage be at least 20 and the strand bias be at most 0 (where a positive strand bias suggests that variant reads are emerging predominantly from only one of the two strands).
Confirmation with electrophoresis capillary sequencing
We confirmed genotype for sequenced samples with capillary electrophoresis sequencing using the Applied Biosystems™ 3730xl platform (see Supplementary Table 7B for primers). We analyzed traces using Phred/Phrap/Consed40,41 and mutations were identified with Polyphred42.
Genotyping R1210C
Once we identified the R1210C mutation, we genotyped it in all available samples – this includes all samples described above with the exception of MIGEN shared controls. We genotyped Boston samples were genotyped at the Clinical and Translation Research Center Core Lab of Tufts Clinical and Translation Science Institute with a custom TaqMan approach (see Supplementary Table 8). We genotyped Baltimore samples at the Duke University Center for Human Disease Modeling using a custom made TaqMan genotyping assay for CFH R1210C by Applied Biosystems and with the ABI 7900 Real-Time PCR system.
Statistical Analyses
Defining Haplotypes
To insure accurate phasing, the individuals in this data set had 0% missing genotype for CFH markers. For all markers we constructed haplotypes using the Boston-phased data set across the CFH locus with Beagle 43 and PLINK44. We selected haplotypes with frequencies >0.3%, and calculated case and control frequencies. For each haplotype we calculated odds ratios and 95% confidence intervals relative to the most frequent haplotype.
Assessing haplotypic association accounting for common SNP associations
To quantify the probability that the number of H5 heterozygote individuals might be affected by chance, we permuted the case-control status across all 1,752 individuals in the Boston-phased data set. But, to control for the effects of common associated risk alleles we preserved genotypes at four markers: Y402H (rs1080155), at rs10737680/rs1410996, at CFHR3/1 deletion, and rs800292 (I62V) for each individual. We selected those markers since they have been most commonly suggested as causal alleles in the published literature. All individuals were grouped by genotypes at each of those four markers, and then case-control status within each group was permuted. We conducted 100,000 permutations. For each permutation we calculated the association statistics and odds ratios for each common marker; using all of the permutations we calculated the 95% confidence range for odds ratios and association statistics for each common marker. To assess the significance of our H5 association we compared the observed number of H5 individuals with disease to the distribution from permuted results. For comparison, we conducted similar analyses where we permuted case-control status preserving genotyped at no markers at all, and preserving genotypes at only two markers (Y402H (rs1080155) and rs10737680/rs1410996).
Assessing common ancestry and stratification with genome-wide SNPs
To assess relatedness between samples and population stratification, we used the genome-wide SNP data passing stringent quality control from the Affymetrix array on the Boston-phase samples for all 1,752 samples. We required that SNPs met the following criteria (1) Hardy-Weinberg with p> 0.001, (2) missing genotypes < 2%, and (3) minor allele frequency > 5%. We removed SNPs in regions with known association to AMD, the CFH locus (chr1, 194–196 MB in HG18 coordinates) and the ARMS2 locus (chr10, 123–125 MB), along with SNPs in the highly stratified the Major Histocompatability Complex (chr 6, 25–35 MB) and the chromosome 8 inversion region (chr 8, 7–13 MB). Pruning SNPs in linkage disequilibrium (r2<0.2) to insure independence resulted in 79,091 SNPs. To assess whether any of the pairs of H5 individuals had a recent common ancestor, we used these SNPs with Plink44 to estimate identity by descent between pairwise H5 heterozygotes. To assess whether the haplotype association was the consequence of stratification, we applied Eigenstrat to generate principal components for these individuals14. Then we separately plotted cases, controls, and H5 heterozygotes along the top two principal components. To assess whether H5 individuals were skewed along any of the individual top 20 components, we sampled 11 individuals randomly (to match the number of H5 heterozygotes) 10,000 times. For each component we noted the median value for the H5 heterzygotes and compared it to the medians of the randomly sampled individuals. The p-value was the percentage of sampling instances with more extreme than observed median values in either direction.
Selecting samples for sequencing
We aimed to select 90 samples based on available DNA stores for sequencing. We selected only those samples that could be phased into the common identified haplotypes within the CFH locus. Where it was possible, we selected 6 homozygotes for each haplotype randomly from available samples with an equal number cases and controls. When sufficient numbers of homozygotes were not available, up to 16 heterozygotes were selected randomly with an equal number of cases and controls. We selected samples to insure that each common haplotype was represented.
Statistical test for association of R1210C
For single strata case-control sample collections we used a 2×2 Fisher’s exact test to calculate a one-tailed p-value. Assuming a single strata that there are a total of N individuals, of whom ncase are cases and nR1210C are heterozygotes for the R1210C mutation, we can calculate the one-tailed significance of observing nR1210C,cases individuals who have the R1210C mutation and also have advanced AMD as follows:
where hypergeometric is the hyper-geometric probability distribution assuming that there are nR1210C draws from a total of N samples, of which x of a total of ncase are drawn. We used this approach to calculate the significance values reported in Table 1 for Boston-phased, Boston-replication, and Duke-replication not stratified on Y402H genotype.
If multiple strata are present, for example if we are stratifying genotype counts on Y402H genotypes or combining multiple case-control collections together, we expand the above strategy to calculate an exact p-value. Assume that we observe a total of nR1210C,case R1210C heterozygotes who are affected across all strata then, for each strata j, we can calculate significance as follows:
Here, for each strata j we have separate numbers of total individuals (Nj), separate numbers of individuals who are cases (nj,case), and individuals with the R1210C genotype (nj,R1210C). So, we calculate the hyper-geometric probability for each individual strata for all the possible counts that would result in an equal or greater than nR1210C,case total number of heterozygtes associated with advanced AMD, and total these probabilities together to obtain a p-value.
For first-degree siblings we calculated statistical significance using the binomial distribution. Given that siblings were selected from affected probands that had the R1210C mutation, the probability that each additional relative would have the mutation is 0.5. We assign each sibling a score, si which is 1 if the sibling has both advanced AMD and R1210C or if the sibling neither advanced AMD or R1210C and 0 if the sibling has advanced AMD without R1210C or R1210C without advanced AMD. We obtain an aggregate score by summing over all independent siblings:
Under the null hypothesis, the aggregate score should be distributed according to a binomial distribution. So if there are a total of nsiblings we can calculate psibling, the one-tailed significance:
where the function binomial represents the binomial distribution for x successful draws out of nsiblings each with a 0.5 probability.
In order to calculate an aggregate meta-analysis we define a score, saggregate which is the total of nR1210C,case across all strata and s. We can calculate the probability of obtaining the score s or a more significant score to determine the exact one-tailed p-value:
Statistical analysis of age of onset
For 1910 out of 1912 unrelated individuals affected with advanced AMD from Boston-phased and Boston-replication samples we had information on age of disease onset. Age of onset is defined as the age at which the individual was first diagnosed with AMD, which might predate the age at which patients fulfilled the case-definition of advanced AMD. We used a non-parametric Wilcoxon rank-sum test to test if affected individuals with the R1210C mutation had earlier age of onset than those without this mutation.
Assessing renal function
For 17 unrelated individuals from Boston-phased with advanced AMD and the R1210C mutation, we determined the creatinine concentration using the DetectX Serum Creatinine Detection Kit (Cat# KB02-H1, Arbor Assays, Ann Arbor, Michigan) on stored serum. We calculated eGFR using the MDRD formula45. We then selected 17 other individuals from Boston-phased, also with advanced AMD, but without the R1210C mutation. We matched individuals for gender, age at the time of serum collection, and stage of AMD for each eye at the time of serum collection. We measured serum creatinine on stored serum for these individuals, and calculated eGFR. We compared eGFR of R1210C individuals to matched controls with a paired non-parametric Wilcoxan signed rank test.
Supplementary Material
Acknowledgments
We appreciate the contribution of an anonymous donor to the research of J.M.S., without whom this genome-wide association study would not have been possible. We thank the participants and numerous ophthalmologists throughout the country who participated in this study, the Age-Related Eye Disease Study Research Group. This research was supported in part by grants RO1-EY11309 (JMS), K08AR055688-01A1 (Raychaudhuri), and U01 MH085520-01 (Ripke) from the NIH; Massachusetts Lions Eye Research Fund, Inc.; the Foundation for Fighting Blindness; the Macular Vision Research Foundation; Research to Prevent Blindness Challenge Grant to the New England Eye Center, Department of Ophthalmology, Tufts University School of Medicine; and the Macular Degeneration Research Fund of the Ophthalmic Epidemiology and Genetics Service, New England Eye Center, Tufts Medical Center, Tufts University School of Medicine. NK is a Distinguished Brumley Professor. We thank the Myocardial Infarction Genetics Consortium (MIGen) study for the use of their genotype data as control data in our study. The MIGen study was funded by grants from the NIH-NLBI (R01HL087676) and the NIH-NCRR. We acknowledge informal and helpful discussions from our colleagues Benjamin Neale, Robert Plenge, David Reich, Eli Stahl, Barbara Stranger, and Benjamin Voight.
Footnotes
CONTRIBUTIONS
Raychaudhuri, JMS, NK, and MJD conceptualized this study, wrote the initial manuscript, and interpreted all results. Raychaudhuri oversaw the statistical analyses and coordinated collaborative experimental efforts. JMS and MJD oversaw genome-wide genotyping of Boston-phased. Ripke analyzed the genome-wide Boston-phased genotype data to assess population stratification and recent ancestry. JMS, KC, RR, and YY organized the Boston clinical cohort. BC, PC, and DZ organized the Baltimore clinical cohort. PT and NK genotyped Baltimore samples. AT genotyped Boston samples. OI, SG, SV, and KM sequenced the CFH gene.
References
- 1.Hageman GS, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102:7227–32. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haines JL, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–21. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 3.Klein RJ, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–9. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Edwards AO, et al. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–4. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 5.Maller J, et al. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat Genet. 2006;38:1055–9. doi: 10.1038/ng1873. [DOI] [PubMed] [Google Scholar]
- 6.Li M, et al. CFH haplotypes without the Y402H coding variant show strong association with susceptibility to age-related macular degeneration. Nat Genet. 2006;38:1049–54. doi: 10.1038/ng1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jozsi M, et al. Factor H and atypical hemolytic uremic syndrome: mutations in the C-terminus cause structural changes and defective recognition functions. J Am Soc Nephrol. 2006;17:170–7. doi: 10.1681/ASN.2005080868. [DOI] [PubMed] [Google Scholar]
- 8.Manuelian T, et al. Mutations in factor H reduce binding affinity to C3b and heparin and surface attachment to endothelial cells in hemolytic uremic syndrome. J Clin Invest. 2003;111:1181–90. doi: 10.1172/JCI16651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bressler NM. Age-related macular degeneration is the leading cause of blindness. JAMA. 2004;291:1900–1. doi: 10.1001/jama.291.15.1900. [DOI] [PubMed] [Google Scholar]
- 10.Davila S, et al. Genome-wide association study identifies variants in the CFH region associated with host susceptibility to meningococcal disease. Nat Genet. 2010;42:772–6. doi: 10.1038/ng.640. [DOI] [PubMed] [Google Scholar]
- 11.Jozsi M, Zipfel PF. Factor H family proteins and human diseases. Trends Immunol. 2008;29:380–7. doi: 10.1016/j.it.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 12.Boon CJ, et al. Basal laminar drusen caused by compound heterozygous variants in the CFH gene. Am J Hum Genet. 2008;82:516–23. doi: 10.1016/j.ajhg.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 14.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 15.Sanchez-Corral P, et al. Structural and functional characterization of factor H mutations associated with atypical hemolytic uremic syndrome. Am J Hum Genet. 2002;71:1285–95. doi: 10.1086/344515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez-Barricarte R, et al. The complement factor H R1210C mutation is associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2008;19:639–46. doi: 10.1681/ASN.2007080923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caprioli J, et al. The molecular basis of familial hemolytic uremic syndrome: mutation analysis of factor H gene reveals a hot spot in short consensus repeat 20. J Am Soc Nephrol. 2001;12:297–307. doi: 10.1681/ASN.V122297. [DOI] [PubMed] [Google Scholar]
- 18.Caprioli J, et al. Complement factor H mutations and gene polymorphisms in haemolytic uraemic syndrome: the C-257T, the A2089G and the G2881T polymorphisms are strongly associated with the disease. Hum Mol Genet. 2003;12:3385–95. doi: 10.1093/hmg/ddg363. [DOI] [PubMed] [Google Scholar]
- 19.Neumann HP, et al. Haemolytic uraemic syndrome and mutations of the factor H gene: a registry-based study of German speaking countries. J Med Genet. 2003;40:676–81. doi: 10.1136/jmg.40.9.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Servais A, et al. Primary glomerulonephritis with isolated C3 deposits: a new entity which shares common genetic risk factors with haemolytic uraemic syndrome. J Med Genet. 2007;44:193–9. doi: 10.1136/jmg.2006.045328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferreira VP, et al. The binding of factor H to a complex of physiological polyanions and C3b on cells is impaired in atypical hemolytic uremic syndrome. J Immunol. 2009;182:7009–18. doi: 10.4049/jimmunol.0804031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weiner DE, Tighiouart H, Reynolds R, Seddon JM. Kidney function, albuminuria and age-related macular degeneration in NHANES III. Nephrol Dial Transplant. 2011 doi: 10.1093/ndt/gfr022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fagerness JA, et al. Variation near complement factor I is associated with risk of advanced AMD. Eur J Hum Genet. 2009;17:100–4. doi: 10.1038/ejhg.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gold B, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–62. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maller JB, et al. Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nat Genet. 2007;39:1200–1. doi: 10.1038/ng2131. [DOI] [PubMed] [Google Scholar]
- 26.Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:1676–87. doi: 10.1056/NEJMra0902814. [DOI] [PubMed] [Google Scholar]
- 27.Seddon JM, Sharma S, Adelman RA. Evaluation of the clinical age-related maculopathy staging system. Ophthalmology. 2006;113:260–6. doi: 10.1016/j.ophtha.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 28.Seddon JM, Santangelo SL, Book K, Chong S, Cote J. A genomewide scan for age-related macular degeneration provides evidence for linkage to several chromosomal regions. Am J Hum Genet. 2003;73:780–90. doi: 10.1086/378505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seddon JM, Cote J, Davis N, Rosner B. Progression of age-related macular degeneration: association with body mass index, waist circumference, and waist-hip ratio. Arch Ophthalmol. 2003;121:785–92. doi: 10.1001/archopht.121.6.785. [DOI] [PubMed] [Google Scholar]
- 30.Seddon JM, Cote J, Page WF, Aggen SH, Neale MC. The US twin study of age-related macular degeneration: relative roles of genetic and environmental influences. Arch Ophthalmol. 2005;123:321–7. doi: 10.1001/archopht.123.3.321. [DOI] [PubMed] [Google Scholar]
- 31.Seddon JM, et al. Dietary fat and risk for advanced age-related macular degeneration. Arch Ophthalmol. 2001;119:1191–9. doi: 10.1001/archopht.119.8.1191. [DOI] [PubMed] [Google Scholar]
- 32.Neale BM, et al. Genome-wide association study of advanced age-related maular degeneration identifies a role of the hepatic lipase gene (LIPC) Proc Natl Acad Sci U S A. 2010;107:7395–400. doi: 10.1073/pnas.0912019107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kathiresan S, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41:334–41. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raychaudhuri S, et al. Associations of CFHR1–CFHR3 deletion and a CFH SNP to age-related macular degeneration are not independent. Nat Genet. 2010;42:553–555. doi: 10.1038/ng0710-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang Z, et al. Toll-like receptor 3 and geographic atrophy in age-related macular degeneration. N Engl J Med. 2008;359:1456–63. doi: 10.1056/NEJMoa0802437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Z, et al. Genetic and functional dissection of HTRA1 and LOC387715 in age-related macular degeneration. PLoS Genet. 2010;6:e1000836. doi: 10.1371/journal.pgen.1000836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen W, et al. Genetic variants near TIMP3 and high-density lipoprotein-associated loci influence susceptibility to age-related macular degeneration. Proc Natl Acad Sci U S A. 2010;107:7401–6. doi: 10.1073/pnas.0912702107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–85. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- 41.Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- 42.Stephens M, Sloan JS, Robertson PD, Scheet P, Nickerson DA. Automating sequence-based detection and genotyping of SNPs from diploid samples. Nat Genet. 2006;38:375–81. doi: 10.1038/ng1746. [DOI] [PubMed] [Google Scholar]
- 43.Browning BL, Browning SR. A unified approach to genotype imputation and haplotype phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84:210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levey AS, et al. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999;130:461–70. doi: 10.7326/0003-4819-130-6-199903160-00002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.