

Non-technical summary
Oxidative stress is a hallmark of various cardiovascular disorders that results in cellular dysfunction and death. Reactive oxygen species (ROS)-induced ROS release (RIRR) is a fundamental mechanism which amplifies ROS levels within the cardiomyocyte resulting in cellular oxidative stress. Despite elegant studies describing the phenomenon of RIRR in isolated myocytes, its biophysical properties and functional consequences in intact myocardium remain unclear. Here, we use ROS imaging to extend the concept of RIRR to the level of the intact heart. We establish regenerative superoxide production as the mediator of RIRR-related arrhythmias and reveal their strong dependence on a key mitochondrial channel, known as the inner membrane anion channel (IMAC). We demonstrate the efficacy of suppressing RIRR and related arrhythmias either by pharmacologically blocking IMAC or scavenging ROS using a synthetic superoxide dismutase/catalase mimetic.
Abstract
Abstract
Reactive oxygen species (ROS)-induced ROS release (RIRR) is a fundamental mechanism by which cardiac mitochondria respond to elevated ROS levels by stimulating endogenous ROS production in a regenerative, autocatalytic process that ultimately results in global oxidative stress (OS), cellular dysfunction and death. Despite elegant studies describing the phenomenon of RIRR under artificial conditions such as photo-induced oxidation of discrete regions within cardiomyocytes, the existence, biophysical properties and functional consequences of RIRR in intact myocardium remain unclear. Here, we used a semi-quantitative approach of optical superoxide (O2−) mapping using dihydroethidium (DHE) fluorescence to explore RIRR, its arrhythmic consequences and underlying mechanisms in intact myocardium. Initially, perfusion of rat hearts with 200 μm H2O2 for 40 min (n = 4) elicited two distinct O2− peaks that were readily distinguished by their timing and amplitude. The first peak (P1), which was generated rapidly (within 5–8 min of H2O2 perfusion) was associated with a relatively limited (10 ± 2%) rise in normalized O2− levels relative to baseline. In contrast, the second peak (P2) occurred 19–26 min following onset of H2O2 perfusion and was associated with a significantly greater amplitude compared to P1. Spatio-temporal ROS mapping during P2 revealed active O2− propagation across the myocardium at a velocity of ∼20 μm s−1. Exposure of hearts (n = 18) to a short (10 min) episode of H2O2 perfusion revealed consistent generation of P2 by high (≥200 μm, 8/8) but not lower (≤100 μm, 3/8) H2O2 concentrations (P < 0.03). In these hearts, onset of P2 occurred following, not during, the 10 min OS protocol, consistent with RIRR. Importantly, P2 (+) hearts exhibited a markedly greater (by 3.8-fold, P < 0.001) arrhythmia score compared to P2 (–) hearts. To explore the mechanism underlying RIRR in intact myocardium, hearts were perfused with either cyclosporin A (CsA) or 4′-chlorodiazepam (4′-Cl-DZP) to inhibit the mitochondrial permeability transition pore (mPTP) or the inner membrane anion channel (IMAC), respectively. Surprisingly, perfusion with CsA failed to suppress (P = 0.75, n.s.) or even delay H2O2-induced P2 or the incidence of arrhythmias compared to untreated hearts. In sharp contrast, perfusion with 4′-Cl-DZP markedly blunted O2− levels during P2, and suppressed the incidence of sustained ventricular tachycardia or ventricular fibrillation (VT/VF). Finally, perfusion of hearts with the synthetic superoxide dismutase/catalase mimetic EUK-134 completely abolished the H2O2-mediated RIRR response as well as the incidence of arrhythmias. These findings extend the concept of RIRR to the level of the intact heart, establish regenerative O2− production as the mediator of RIRR-related arrhythmias and reveal their strong dependence on IMAC and not the mPTP in this acute model of OS.
Introduction
Mitochondria synthesize adenine triphosphate (ATP) through a highly regulated process of oxidative phosphorylation driven by electron transport across the electron transport chain (ETC) (Mitchell, 1961; Honda et al. 2005; Lin & Beal, 2006; O'Rourke et al. 2007; Gustafsson & Gottlieb, 2008). As a natural byproduct of metabolism, reactive oxygen species (ROS) are also generated when a small percentage of electrons leak out of the ETC, combine with molecular oxygen, and form superoxide anions (O2−) (Turrens, 2003). In normal hearts, mitochondrial ROS formation is rapidly countered by efficient anti-oxidant defence systems. In contrast, oxidative stress caused by increased ROS production and/or reduced scavenging capacity is a hallmark of various cardiovascular disorders, including ischaemia–reperfusion injury, diabetes and heart failure (Honda et al. 2005; Huss & Kelly, 2005; O'Rourke et al. 2007; Gustafsson & Gottlieb, 2008). Understanding mechanisms by which local mitochondrial ROS formation causes myocardial oxidative stress and ensuing metabolic, contractile and electrical dysfunction is critical for our ability to combat these prevalent cardiovascular disorders.
Seminal studies by Zorov et al. (2000, 2006) and Aon et al. (2003, 2006, 2007) have advanced the notion of ROS-induced ROS release (RIRR) to explain how local ROS injury within a discrete region of a cardiomyocyte can rapidly accumulate across a critical mass of the mitochondrial network to cause cellular oxidative stress. In these studies, RIRR was described as a fundamental mechanism by which cardiac mitochondria respond to elevated ROS levels by stimulating endogenous ROS production in a regenerative, autocatalytic process that ultimately results in cellular dysfunction and death (Zorov et al. 2006).
Distinct modes of RIRR have been postulated based on their dependence on either the mitochondrial permeability transition pore (mPTP) or the inner membrane anion channel (IMAC) (Brady et al. 2006; Yang et al. 2010). Specifically, Zorov et al. (2000, 2006) demonstrated a convincing relationship between the induction of the mitochondrial permeability transition and the destabilization of mitochondrial membrane potential (ΔΨm) upon mitochondrial oxidation. They also found a direct correlation between mPTP activation and myocyte death. These findings, combined with evidence that ΔΨm depolarization was abolished by the mPTP inhibitor, cyclosporin A (CsA), suggested an important role for mPTP-mediated ΔΨm depolarization in RIRR and eventual apoptosis (Zorov et al. 2000). On the other hand, studies by Aon et al. have provided strong evidence in support of IMAC as a mediator of RIRR and associated electrophysiological and metabolic instabilities. In these studies, photo-induced oxidation of a discrete region within the cardiac myocyte unleashed a regenerative process of ETC-derived ROS that was dependent on IMAC activation. Once a threshold level of ROS was exceeded across a critical mass of the mitochondrial network (i.e. mitochondrial criticality), sustained ΔΨm oscillations were initiated (Aon et al. 2006, 2009). These mitochondrial oscillations resulted in cellular electrophysiological oscillations via cyclical activation of the surface K-ATP current. Interestingly, both ΔΨm and electrophysiological oscillations could be readily abolished by antagonists of the mitochondrial benzodiazepine receptor which modulates IMAC activity but not by CsA (Aon et al. 2003, 2006, 2007, 2009; Akar et al. 2005).
Despite these elegant studies describing the phenomenon of RIRR at a subcellular level, the existence, biophysical properties and functional consequences of RIRR at the tissue-network level and across broad myocardial regions remained unknown. Zhou et al. developed a mathematical, reaction–diffusion model of RIRR that further emphasized the importance of O2− diffusion in mediating mitochondrial dysfunction across 2-dimensional in silico networks of virtual mitochondria (Zhou et al. 2010). In the present study, we experimentally test these model predictions using a semi-quantitative approach of O2− mapping across the heart. We explore RIRR, its arrhythmic consequences and underlying mechanisms in intact myocardium during acute oxidative stress (OS) produced by H2O2 perfusion (Rigoulet et al. 2011), which results in the spontaneous incidence of sustained arrhythmias (Morita et al. 2009; Xie et al. 2009). We further explore the relative importance of IMAC and mPTP on RIRR and associated arrhythmias at the intact heart level. Finally, we test the efficacy of modulating arrhythmia propensity by suppressing RIRR using the synthetic superoxide dismutase/catalase mimetic EUK-134.
Methods
Optical superoxide mapping in ex vivo perfused rat hearts
The authors have read, and the experiments comply with, the policies and regulations of The Journal of Physiology given by Drummond (2009). All procedures involving the handling of animals were approved by the Animal Care and Use Committee of the Mount Sinai School of Medicine and adhered with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. Rats (N = 49) were anaesthetized with ketamine hydrochloride (50 mg kg−1, i.p.). Their hearts were rapidly excised, washed with ice-cold cardioplegic solution, transferred to a Langendorff apparatus, and retrogradely perfused through the aorta with oxygenated (95% O2–5% CO2) Tyrode solution containing (in mm): NaCl 121.7, NaHCO3 25, KCl 4.81, MgSO4 2.74, CaCl2 2.5 and dextrose 5.0, pH = 7.40, temperature = 37 ± 1°C. Perfusion pressure was maintained between 55 and 65 mmHg by adjusting perfusion flow. Hearts were suspended in the buffer-filled, temperature-controlled chamber which was tightly regulated at 37°C throughout the entire protocol, as we have recently reported (Jin et al. 2010b; Lyon et al. 2010). Volume-conducted electrocardiograms were recorded for rhythm analysis using non-contact silver electrodes (Grass Instruments) placed within the chamber. ECG signals were recorded continuously throughout the entire ex vivo perfusion protocol. Left ventricular cavity pressure was also measured using a buffer-filled latex balloon (Harvard apparatus) that was carefully inserted through the mitral valve into the left ventricular cavity. Signals were amplified (ECG100-MP150 Amplifier, Biopac Systems, CA, USA) and displayed in real time using the AcqKnowledge 3.9 software package (Biopac Systems). Hearts were positioned such that the mapping field was centred over a 4 mm × 4 mm region of left ventricular epicardium, midway between apex and base. These preparations remain stable for over 4 h of perfusion (Jin et al. 2010a).
Dihydroethidium (DHE) fluorescence imaging was used to measure relative changes in O2− levels within the intact heart, as reported previously in ischaemia–reperfusion injury (Lu et al. 2006). DHE, which is oxidized by O2− anions to ethidium, has been extensively exploited by multiple groups to monitor changes in myocardial O2− levels in response to oxidative injury (Kevin et al. 2003; Camara et al. 2004; Riess et al. 2004; Lu et al. 2006). Following cannulation, hearts were allowed to stabilize for 10 min at physiological temperature. Hearts were then stained with DHE (10 μm; Molecular Probes Inc.) mixed in a 500 ml volume of Tyrode solution (dye-loading phase) for 20 min. DHE was initially dissolved in DMSO containing 16% Pluronic (weight/volume). The dye-loading phase was followed by a 20–30 min period of dye washout. DHE background fluorescence intensity was measured periodically throughout the dye staining and washout phases using a 6400 pixel CCD-based optical imaging approach that allowed the measurement of normalized O2− levels with 50 μm resolution over a 4 mm × 4 mm region of the epicardial surface. To measure DHE background fluorescence, hearts were excited with filtered light (525 nm) emitted from a 200 W quartz tungsten halogen lamp (Newport Corporation, CT, USA). Emitted fluorescence was filtered (585 nm) and focused onto a CCD camera (Scimeasure, GA, USA). Background fluorescence intensity was measured as the amplitude difference before and after excitation.
During the dye-loading procedure, a marked rise in background DHE fluorescence was verified for multiple pixels in order to confirm adequate dye staining of the heart. During dye washout, the stability of DHE background fluorescence was evaluated from multiple pixels in real time, as this baseline pre-H2O2 level served for normalization purposes during the protocol. In all experiments, the dye washout phase was associated with stable signal intensity, which was required to normalize subsequent changes in DHE fluorescence caused by H2O2 injury. In preliminary experiments, we ensured the stability of background fluorescence for over 2 h of steady-state perfusion. Ex vivo experimental protocols were completed in 90 min (n = 45) or 120 min (n = 4).
High-throughput analysis of optical signals was performed. Peak emitted DHE fluorescence from each of 6400 pixels was measured before and after excitation achieved by a computer-automated filter shutter switch. DHE background fluorescence was baseline corrected by subtracting fluorescence levels before dye staining for each pixel. Background-corrected DHE fluorescence (O2−) during the protocol was then normalized to the value of steady-state DHE fluorescence achieved during the dye washout phase for each of the 6400 individual pixels. Measurements of O2− were taken during diastole, when movement artifact is negligible. Therefore, there was no need to suppress contraction using electromechanical uncoupling agents that are known to affect metabolic processes. Normalized DHE fluorescence (O2− measurements) during H2O2 injury were either averaged or plotted as contour maps using Delta Graph 5.6 (Red Rock Software). These maps served to illustrate the dynamic changes in the spatial distribution of ROS across the heart.
Ex vivo models of RIRR
Perfusion of hearts with H2O2 is a well-established model of acute oxidative stress (OS) that results in triggered activity (Sato et al. 2009) as well as sustained atrial (Morita et al. 2010) and ventricular tachyarrhythmias (Morita et al. 2009). We varied the time and dose of H2O2 perfusion to investigate the presence and functional significance of mitochondrial criticality and RIRR in intact myocardium. The initial proof-of-concept protocol entailed perfusion of hearts with 200 μm H2O2 for 40 min. In this protocol, 100% of hearts exhibit unstable electrical rhythm that culminates in the spontaneous incidence of sustained VT/VF. To further explore the presence of RIRR and its functional significance in intact myocardium, we subjected hearts (n = 18) to short (10 min) episodes of H2O2 perfusion at varying concentrations (50–400 μm). This resulted in diverse outcomes ranging from stable electrical rhythm to sustained VT/VF.
Arrhythmia propensity
Arrhythmia propensity was scored according to the guidelines described by Curtis and Walker (Walker et al. 1988) using the Lambeth Convention, as we have recently reported (Jin et al. 2010b). Briefly, the following criteria were used to generate an arrhythmia score (AS) for each heart subjected to OS by H2O2 perfusion: 0: <50 ventricular premature beats; 1: 50–499 ventricular premature beats; 2: >500 ventricular premature beats and/or one episode of spontaneously reverting VT/VF; 3: more than one episode of spontaneously reverting VT/VF (< 1 min total combined duration); 4: 1–2 min of total combined VT/VF; 5: >2 min of VT/VF. AS values reflected arrhythmia propensity and severity throughout the entire ex vivo protocol. Time of onset of sustained VT/VF was also documented and compared across groups.
Mitochondrial channel basis of RIRR
To determine the mitochondrial transport mechanism underlying RIRR in intact myocardium, hearts were treated with either 4′-chlorodiazepam (4′Cl-DZP, 64 μm, n = 4) or cyclosporine A (CsA, 0.1 μm,n = 4). In both cases, drug perfusion was initiated 10 min prior to the H2O2 insult and maintained throughout the protocol. In previous studies, we and others demonstrated the efficacy of modulating mitochondrial function and arrhythmias using these inhibitors at these concentrations (Akar et al. 2005; Brown et al. 2008). Therefore, these experiments were designed to investigate the differential efficacy of mPTP versus IMAC blockade on OS-mediated RIRR and associated arrhythmias in the intact heart.
Results
DHE loading of perfused rat hearts for 20 min resulted in a robust and sustained increase in background fluorescence intensity. Following dye washout, background fluorescence remained stable for at least 2 h. As such, H2O2-mediated changes in O2− levels across the heart could be quantitatively measured as a background-subtracted and normalized value relative to the steady-state, pre-H2O2 levels on a pixel-by-pixel basis, as has been performed by Lu et al. (2006).
Optical O2− imaging reveals distinct ROS peaks during H2O2 perfusion
As an initial ‘proof-of-concept’ protocol, acute OS was produced by perfusion of hearts with 200 μm H2O2 for 40 min (Fig. 1). Shown are average, normalized O2− responses of individual hearts (n = 4). This protocol always resulted in the generation of two distinct O2− peaks, which we will herein refer to as P1 and P2 (Fig. 1A). Clearly, the maximum amplitude of P2 (1.5 ± 0.15) was markedly greater than that of P1 (1.1 ± 0.02). Figure 1B shows representative contour maps of normalized DHE fluorescence recorded during the onset of P2. These spatio-temporal maps demonstrate the progressive rise in O2− levels across the heart. As expected, RIRR in this model of acute OS occurred in conjunction with a significant depolarization of ΔΨm indicating mitochondrial dysfunction at the tissue level (Fig. 1C).
Figure 1. Normalized O2− mapping in the intact perfused heart reveals two distinct ROS peaks in response to acute OS.
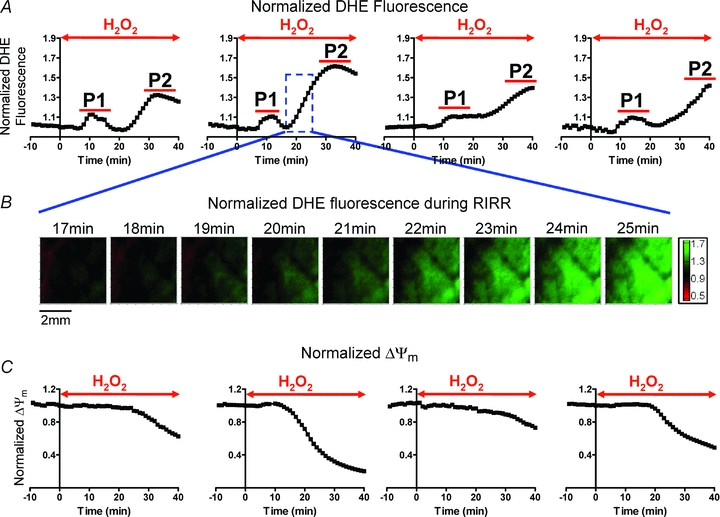
Initial proof-of-concept protocol of OS entailed perfusion of hearts with 200 μm H2O2 for 40 min. This protocol was chosen because it results in sustained ventricular fibrillation (VF) in all hearts. A, average normalized optical O2− levels of individual hearts in response to this protocol of OS. Time of H2O2 perfusion for each heart is indicated by the red arrow. H2O2 perfusion resulted in two distinct O2− peaks (P1 and P2) in all hearts. The maximum amplitude of the delayed peak (P2) was significantly greater than P1. B, contour maps illustrating the dynamic changes in the spatial distribution of O2−. Green represents regions of highest superoxide levels. Representative maps during onset of P2 perfusion demonstrate wave-like propagation of O2− increase, consistent with RIRR. C, normalized ΔΨm levels of 4 Tetra methyl rhodamine methylester (TMRM)-stained hearts in response to this protocol of OS, showing ΔΨm depolarization.
Biophysical properties of RIRR in intact myocardium
Although the presence of P2 caused by 40 min H2O2 perfusion was indeed consistent with RIRR, the onset of P2 always occurred during, not following, this long protocol of acute OS. As such, one could not distinguish between exogenous stress produced by H2O2 perfusion and endogenous autocatalytic ROS generation triggered by the initial insult, which defines RIRR. To address this issue, we systematically investigated the dose dependence of the average myocardial O2− response to OS by varying the concentration (50–400 μm) of H2O2 perfusion (Fig. 2). Specifically, OS was produced by very short (10 min) episodes of H2O2 perfusion to determine if this initial stressor can give rise to sustained ROS bursts that follow the insult in a manner that is more analogous with RIRR observed in isolated myocytes. While high concentrations (>200 μm) of H2O2 for 10 min always produced two O2− peaks (P1 and P2), only 1/4 of 50 μm and 2/4 of 100 μm H2O2 perfused hearts exhibited a secondary peak (P2). The onset of P2 always occurred well after the H2O2 insult, suggesting its generation by endogenous myocardial O2− production and not by direct H2O2 perfusion. The dose dependence of the individual O2− peaks (P1 and P2) is shown in Fig. 3. Clearly, the increase in P2 caused by varying H2O2 concentration was markedly greater than that of P1 (Fig. 3A). Average comparisons between the amplitudes of the two peaks produced by different H2O2 concentrations revealed significantly greater P2 at high (≥200 μm) but not lower H2O2 protocols (Fig. 3B). The dependence of P2 on prior H2O2 exposure of the heart is therefore consistent with RIRR.
Figure 2. Dose dependence of ROS-induced ROS release in intact myocardium.
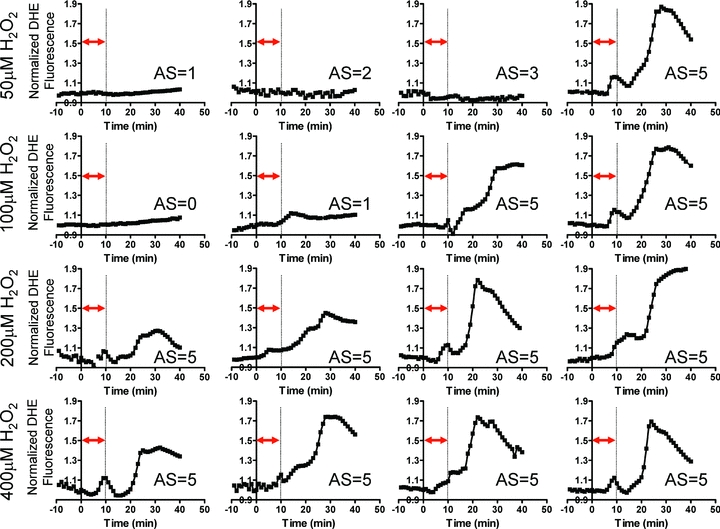
Average O2− responses of individual hearts subjected to a short (10 min) episode of OS by perfusion with 50 μm (n = 4), 100 μm (n = 4), 200 μm (n = 4) and 400 μm (n = 4) H2O2 followed by reperfusion with normal Tyrode solution. The timing of H2O2 perfusion is indicated by the double arrow for each heart. The delayed second peak (P2) is clearly evident in 1/4 (50 μm), 2/4 (100 μm), 4/4 (200 μm), and 4/4 (400 μm) H2O2 protocols. The arrhythmia score (AS) for each heart is also indicated. All hearts exhibiting P2 had an AS of 5, reflecting incidence of sustained VT/VF.
Figure 3. Dose dependence of O2− release in intact myocardium.
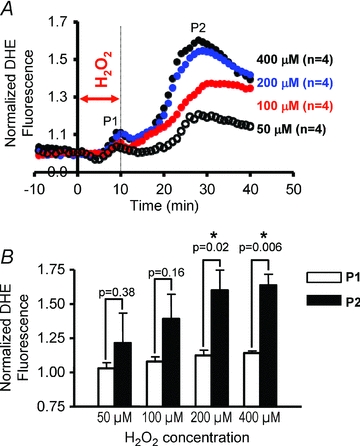
A, average O2− responses (n = 4 each group) caused by 10 min H2O2 perfusion at 50, 100, 200 and 400 μm. Higher concentrations of H2O2 produced markedly greater amplitude of P2 compared to P1. B, average P1 and P2 peak O2− amplitudes generated by various H2O2 concentrations. P2 is significantly greater than P1 at high but not low H2O2 doses.
Functional electrical consequences of RIRR in intact myocardium
To gain further insights into the functional implications of RIRR in intact myocardium, we compared differences in arrhythmia propensity, quantified as an arrhythmia score (AS), between hearts that exhibited a second ROS peak (P2) versus those that did not. Figure 4A shows representative O2− responses of P2 (–) and P2 (+) hearts. Also shown are optical O2− contour maps that serve to illustrate the sequential changes in O2− levels across representative P2 (–) and P2 (+) hearts.
Figure 4. RIRR-mediated arrhythmias in intact myocardium.
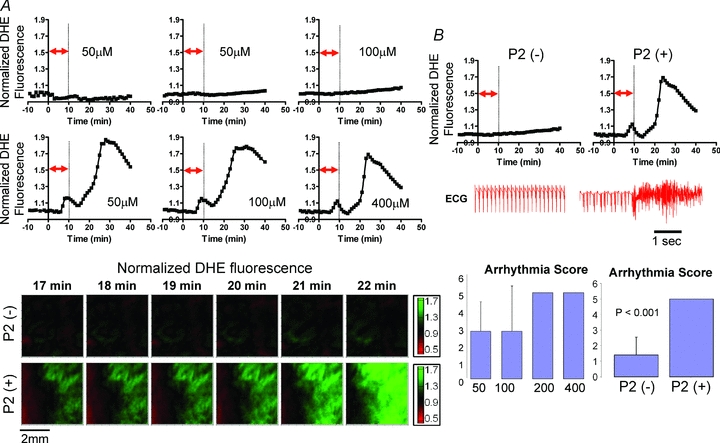
A, representative O2− responses to OS in P2 (–) and P2 (+) hearts. Contour maps of O2− indicating absence (top) and presence (bottom) of increased O2− levels in hearts perfused with different concentrations of H2O2. B, volume-conducted ECG traces recorded from P2 (+) but not P2 (–) hearts exhibit incidence of sustained arrhythmias. Arrhythmia severity was quantified as an arrhythmia score in hearts challenged by increasing concentrations of H2O2 for 10 min (bottom, left). P2 (–) hearts exhibited a significantly lower AS value compared to P2 (+) hearts (bottom, right).
While P2 (–) hearts exhibited relatively stable electrical rhythm (volume-conducted ECG) throughout the entire protocol, P2 (+) hearts were always associated with the genesis of sustained arrhythmias (Fig. 4B). Arrhythmia scores were compared as a function of either H2O2 dose or presence/absence of P2. Interestingly, all hearts that were perfused with high (200 and 400 μm) concentrations of H2O2 for 10 min exhibited AS values of 5 (Fig. 2), reflecting the incidence of sustained VT/VF. In contrast, hearts perfused with lower H2O2 concentrations exhibited variable AS values, ranging from 0 to 5. Finally, P2 (+) hearts were associated with significantly (P < 0.001) greater AS values compared to P2 (–) hearts, irrespective of H2O2 dose (Fig. 4B), further implicating RIRR in arrhythmia severity.
Role of mPTP versus IMAC in RIRR and associated arrhythmias
To determine the underlying mechanism of RIRR and its arrhythmic consequences in intact myocardium, we investigated the dependence of P2 on mPTP versus IMAC activation. Specifically, we compared changes in normalized O2− levels during P2 in control (Fig. 5; blue) and 0.1 μm CsA (red)-treated hearts (n = 4 each). As expected, mPTP blockade with CsA did not alter O2− levels during baseline, H2O2-free perfusion. Surprisingly, however, mPTP blockade with CsA also failed to reduce (P = 0.45 for P1, P = 0.75 for P2) or even delay the H2O2-mediated ROS peaks, indicating that mPTP activation did not underlie RIRR in this model of OS. Moreover, CsA treatment did not suppress the incidence of arrhythmias which were observed in all control (n = 4) and CsA-treated (n = 4) hearts subjected to an identical OS protocol. Paradoxically, CsA-treated hearts exhibited a strong trend towards an earlier rise in O2− levels during P2 (P = 0.06, Fig. 5A and D) and arrhythmia onset (Fig. 5B), indicating a higher sensitivity to OS-mediated electrical dysfunction.
Figure 5. Role of mPTP in RIRR-mediated arrhythmias in intact myocardium.
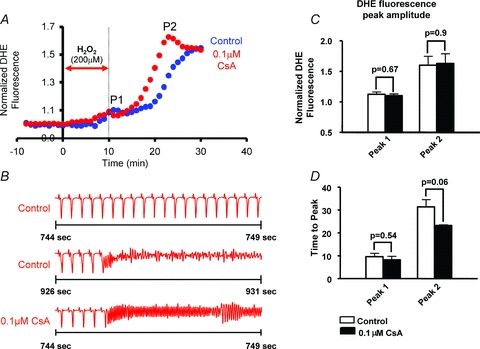
A, average normalized O2− responses of control (blue, n = 4) and 0.1 μm CsA (red, n = 4)-treated hearts to perfusion with 200 μm H2O2 for 10 min. In the treatment group, CsA perfusion was introduced 10 min prior to the H2O2 challenge and maintained throughout the entire protocol. The timing of H2O2 challenge is indicated by the red arrow. B, ECG traces indicating incidence of sustained VT/VF in both control and CsA-treated hearts. Onset of VF occurred earlier in CsA-treated hearts indicating higher sensitivity to OS. C, bar graph comparing maximum O2− amplitudes associated with P1 and P2 peaks in CsA versus control hearts. CsA failed to reduce the amplitude of O2− levels during RIRR. D, bar graphs showing the time at which P1 and P2 occur in CsA-treated versus control hearts. Onset of P2 appears to occur at an earlier time point in CsA-treated hearts, indicating a strong trend (P = 0.06) towards a higher propensity for H2O2-mediated RIRR.
Since mPTP blockade did not suppress RIRR or arrhythmia propensity in this acute model of H2O2-induced OS, we investigated the role of IMAC using the mitochondrial benzodiazepine receptor antagonist, 4′-Cl-DZP. In sharp contrast to CsA, perfusion of hearts with 64 μm 4′-Cl-DZP markedly blunted P2 amplitude (Fig. 6A). Spatio-temporal ROS imaging failed to detect actively propagating waves of O2− in any of the 4′-Cl-DZP-treated hearts. Remarkably, P2 suppression by IMAC blockade coincided with protection of hearts against sustained arrhythmias, as reflected by lower AS values and absence of VT/VF (Fig. 6B).
Figure 6. Role of IMAC in RIRR and arrhythmias in intact myocardium.
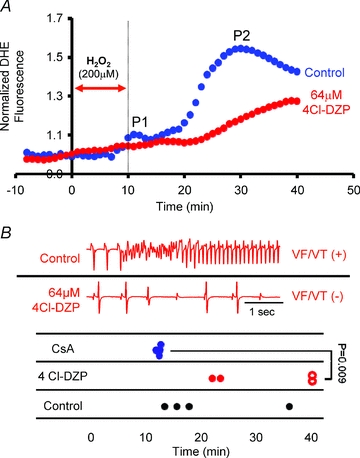
A, average normalized O2− responses of control (blue, n = 4) and 64 μm 4′-Cl-DZP (red, n = 4)-treated hearts to perfusion with 200 μm H2O2 for 10 min. In the treatment group, 4′-Cl-DZP perfusion was introduced 10 min prior to the H2O2 challenge and maintained throughout the entire protocol. The timing of H2O2 challenge is indicated by the red arrow. B, representative volume-conducted ECG traces from control and 4′-Cl-DZP-treated hearts challenged with an identical H2O2 perfusion protocol indicating the presence and absence of sustained VT in control and 4′-Cl-DZP-treated hearts, respectively. Time of onset of VT/VF in control (black), CsA- (blue), and 4′-Cl-DZP- (red) treated hearts is plotted. Open red circles indicate absence of arrhythmias at the end of the experimental protocol in 4′-Cl-DZP hearts. 100% of CsA- and 50% of 4′-Cl-DZP-treated hearts exhibited VT/VF. Time of onset was earlier in CsA-treated hearts.
Prevention of RIRR by EUK-134 suppresses the incidence of arrhythmias
To further implicate RIRR in the mechanism of arrhythmias, we next tested whether perfusion with a synthetic superoxide dismutase/catalase mimetic (EUK-134) altered RIRR and arrhythmia propensity in this model of acute OS. As shown in Fig. 7, perfusion of hearts with 50 μm EUK-134 (red) completely abolished the RIRR response elicited by H2O2 perfusion as neither peak (P1 or P2) was evident compared to untreated hearts (blue). Interestingly, suppression of RIRR by EUK-134 was also associated with complete abrogation of H2O2-mediated arrhythmias (Fig. 7B, P = 0.0048). In addition to its potent protective effects against electrical dysfunction, EUK-134 was also highly effective in preserving the left ventricular cavity pressure compared to untreated hearts (P < 0.01), indicating marked protection against OS-mediated contractile dysfunction (Fig. 7C and D).
Figure 7. Prevention of RIRR and arrhythmias by EUK-134.
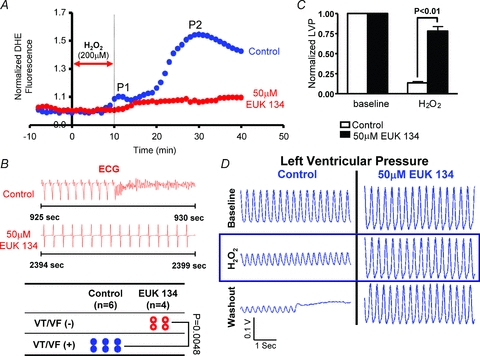
A, average normalized DHE fluorescence of control (blue) and EUK-134 (50 μm, red)-treated hearts to perfusion with 200 μm H2O2 for 10 min. In the treatment group, perfusion with the synthetic superoxide dismutase/catalase mimetic prevented the rise in ROS levels. EUK-134 perfusion was introduced 10 min prior to the H2O2 challenge and maintained throughout the entire protocol. B, prevention of RIRR by EUK-134 was associated with full protection against H2O2-mediated arrhythmias. C, average normalized left ventricular cavity pressure measurements following H2O2 challenge as compared to baseline indicate significant preservation of contractility in EUK-134-treated hearts compared to controls. D, representative traces of left ventricular cavity pressure recordings measured in control and EUK-134-treated hearts indicating preservation of contractility in EUK-134-treated but not control hearts following challenge with the RIRR protocol.
Discussion
In the present study, we extended the concept of ROS-induced ROS release (RIRR) from a subcellular phenomenon to one occurring at the organ level, in which functional electrical instability and arrhythmias can be directly examined. This allowed us to further investigate the biophysical features of RIRR at the tissue-network level and assess mitochondrial channel mechanisms. The main findings of the present report are as follows. (1) Acute OS by H2O2 perfusion elicits two O2− peaks which can be distinguished by their relative timing, amplitude, and recovery properties. (2) The presence of OS-mediated RIRR at the tissue-network level is highly associated with the genesis of sustained arrhythmias. (3) Acute OS-mediated RIRR and associated arrhythmias in otherwise ‘normal’ myocardium is mediated by IMAC and not the mPTP. (4) Suppression of RIRR by EUK-134 prevents the incidence of OS-mediated arrhythmias.
Mitochondrial criticality and RIRR
The biophysical concept of mitochondrial criticality was first advanced by Aon and colleagues who described emergent behaviour of cardiac mitochondria once a critical level of ROS was accumulated across the mitochondrial network. Specifically, they found that a ‘global’ transition in the cellular energy state (i.e. ΔΨm depolarization) occurred once ROS levels reached a certain threshold. This ‘tipping point’ was followed by sustained oscillations in key metabolic and electrophysiological properties. Since, in these experiments, mitochondrial criticality was achieved by a limited insult to a discrete region of the cardiac myocyte (involving approximately 50 mitochondria only), RIRR constituted an important feed-forward mechanism that systematically amplified ROS levels across the entire myocyte, eventually causing OS and associated metabolic and cellular electrophysiological oscillations. Whether a similar phenomenon underlies ROS elevation across broad myocardial regions that can account for global left ventricular dysfunction and arrhythmias remained unknown. In the present study, we found that a short episode of OS produced by high but not low concentrations of H2O2 always elicits two distinct superoxide peaks that can be readily distinguished by their relative timing, maximum amplitude, and persistence after the triggering insult. As such, these findings are indeed consistent with cellular studies of RIRR. Notably, since the large secondary peak (P2) was always initiated following, not during, the initial insult, it most likely reflects the capacity of the myocardium to form a regenerative RIRR process that can outlast the initial perturbation.
A major advantage of our approach was the ability to directly examine the functional significance of RIRR in terms of electrical instability. Remarkably, we found that electrical rhythm in all hearts (17/17) that exhibited P2 (and therefore RIRR) also exhibited spontaneous onset of sustained VT/VF. In contrast, hearts that did not exhibit P2 were protected against sustained arrhythmias (Figs 4B and 7). As such, these findings extend previous in vitro studies in which RIRR was shown to precede cyclical oscillations of the action potential duration and myocyte excitability by directly implicating RIRR in arrhythmia propensity and severity.
Moreover, the propagating waves of O2− elevation, which could be observed during P2, are reminiscent of the waves of ΔΨm collapse that we had identified in a recent study (Lyon et al. 2010). Indeed, these data support the notion that actively propagating waves of ΔΨm collapse across the intact myocardium are either mediated or accompanied by an organized, regenerative wave of O2− elevation. Similarly, these findings are consistent with theoretical models that predict widespread mitochondrial entrainment to oscillatory dynamics under conditions of oxidative stress, and adduce the scalability of these phenomena to the whole heart. Indeed, our findings of mitochondrial dysfunction and RIRR produced by exogenous H2O2 perfusion are consistent with previous findings of Slodzinski et al. and Brown et al. who demonstrated that oxidative stress produced by glutathione oxidation or simulated ischaemia–reperfusion in the intact heart acts as a potent trigger for mitochondrial depolarization, ROS (Slodzinski et al. 2008) and arrhythmias (Brown et al. 2010).
Finally, our findings of two distinct peaks with different amplitudes at the tissue level may reflect different numbers of cells with depolarized mitochondrial networks and increased ROS levels. In fact the genesis of P2 may represent a critical mass of de-energized cells beyond which arrhythmias are readily generated.
Mitochondrial ion channels and arrhythmogenesis
Discrepancies regarding the culprit mitochondrial transport mechanism that underlies RIRR have been reported (Aon et al. 2003; Zorov et al. 2006). While some studies link RIRR-mediated ΔΨm depolarization and eventual apoptosis to the mPTP, other studies support the importance of IMAC as the mechanism for ROS amplification during acute OS. Recently, computational models have successfully recapitulated key features of RIRR including ΔΨm depolarization and wave propagation (Yang et al. 2010). Interestingly, two distinct modes of RIRR were also observed in silico that appeared to be distinguished by their dependence on IMAC versus mPTP, further fuelling the uncertainty regarding the mechanism underlying RIRR-mediated electrical dysfunction in the intact heart. A major finding of the present report is the demonstration of the efficacy of IMAC but not mPTP blockade in suppressing H2O2-mediated RIRR at the intact tissue level (Figs 5 and 6). In fact, these findings are completely consistent with those of Zhou et al. (2010) who used reaction–diffusion models of one- and two-dimensional networks of virtual mitochondria to highlight the importance IMAC-mediated O2− release in the mechanism of RIRR. Our findings are also consistent with those of Yang et al. who found that the relatively ‘fast’ (approx. 20 μm s−1) waves of ΔΨm collapse which we observed experimentally during ischaemia (Lyon et al. 2010), also appear to be dependent on IMAC activation in their elegant computational model of RIRR (Yang et al. 2010).
We and others have previously found that IMAC blockade is more effective in suppressing the incidence of reperfusion-mediated arrhythmias than CsA blockade (Akar et al. 2005; Brown et al. 2008, 2010). This efficacy, however, was not directly linked to mitochondrially derived OS because of technical challenges in measuring spatio-temporal gradients in ROS levels within the intact heart. Instead, the link between arrhythmias observed at the tissue level and RIRR observed at the myocyte level remained indirect and largely speculative. Here, we demonstrate that prevention of RIRR by IMAC blockade is indeed protective against OS-mediated arrhythmias. Finally, the finding that IMAC blockade protects against H2O2-mediated VF extends our earlier finding regarding its efficacy in suppressing ischaemia–reperfusion arrhythmias, further highlighting a central role of OS in these common, clinically relevant rhythm disorders.
Conclusions
Using optical superoxide mapping in intact myocardium, we have extended the concept of RIRR from a subcellular phenomenon to one occurring at the organ level, and demonstrated its functional significance in terms of arrhythmia propensity. In states of oxidative challenge, accelerated superoxide production overwhelms antioxidant defence systems resulting in OS. As superoxide levels increase, IMAC open probability also increases in a positive feed-forward process that accumulates ROS across the intact myocardium (Zhou et al. 2010), triggering the onset of sustained ventricular arrhythmias. Indeed, our current report highlights IMAC-mediated O2− accumulation as the driving force for RIRR and associated arrhythmias in intact myocardium.
Limitations
The experimental approach described in this manuscript assumes a regionally homogeneous distribution of O2− under baseline conditions of normal perfusion. Similar assumptions have been made by other investigators using similar techniques in the past (Salama et al. 1987; Lu et al. 2006). Moreover, our experimental design allowed for measurements of relative (not absolute) changes in ROS during the course of an experiment.
Non-superoxide sources may contribute to DHE fluorescence signals. Therefore, in general, one should interpret DHE fluorescence measurements with caution. However, DHE has been successfully used to accurately assess changes in superoxide levels in the heart by multiple groups worldwide (Kevin et al. 2003; Riess et al. 2004; Lu et al. 2006; Shimoni et al. 2008; Lu et al. 2009; Viola & Hool, 2010). Numerous studies have documented that increased DHE fluorescence resulting from enhanced ROS (particularly O2−) levels in the heart is indeed reversible when ROS levels are restored to baseline (Kevin et al. 2003; Camara et al. 2004; Riess et al. 2004; Lu et al. 2006; Aldakkak et al. 2008, 2011). However, the reversibility of DHE fluorescence remains a matter of debate that requires further clarification (Zielonka & Kalyanaraman, 2010).
Furthermore, our findings that the increase in normalized DHE fluorescence in this model of RIRR is completely abolished by IMAC blockade and superoxide scavengers strongly suggests that superoxide is indeed the main contributor to DHE fluorescence in our experiments.
OS results in highly complex electrophysiological changes which ultimately render the heart prone to arrhythmias. In this work, we focused on mitochondrial function and RIRR. In humans and animal models, OS-related arrhythmias are caused by multi-factorial mechanisms including, but not limited to, changes in ion channel function, calcium cycling proteins, and cell-to-cell coupling. Therefore, one should not expect that a single metric of cardiac function will fully predict arrhythmias in this setting. However, our present findings strongly implicate mitochondrial dysfunction in general, and RIRR in particular, as an important mechanism of arrhythmias in acute OS.
Acute OS in humans typically results from ischaemia–reperfusion injury that causes a transmural gradient of blood flow and tissue perfusion. As such, this injury is expected to produce highly complex alterations in local metabolic properties across the ventricular wall. Our measurements in the present study were limited to the epicardial layer (<250 μm in depth; Girouard et al. 1996). Therefore, future investigations of transmural heterogeneities in metabolic properties in response to OS are warranted. These measurements can be achieved by applying the optical technique, which we describe here, to the transmural surface of arterially perfused wedge preparations, as we and others have reported in the past (Akar et al. 2002; Akar & Rosenbaum, 2003; Glukhov et al. 2010). Finally, in this study we focused on RIRR caused by acute OS in otherwise normal myocardium. Future studies are required to determine the role of RIRR in OS-mediated arrhythmias associated with chronic remodelling, including hypertrophy, heart failure and diabetes.
Acknowledgments
This work was supported by grants from the National Institutes of Health (HL097108, F.G.A.; HL091923, F.G.A.), the American Heart Association (0830126N, F.G.A.), and the Irma T. Hirschl and Monique Weill Caullier Trusts (F.G.A.). The authors have no conflicts of interest to disclose.
Glossary
Abbreviations
- AS
arrhythmia score
- 4′-Cl-DZP
4′-chlorodiazepam
- CsA
cyclosporin A
- DHE
dihydroethidium
- ETC
electron transport chain
- IMAC
inner membrane anion channel
- mPTP
mitochondrial permeability transition pore
- OS
oxidative stress
- RIRR
ROS-induced ROS release
- ROS
reactive oxygen species
- VT/VF
ventricular tachycardia or ventricular fibrillation
Author contributions
Experiments were performed in the Cardiovascular Research Institute of Mount Sinai School of Medicine in New York City. F.G.A. conceived and designed the experiments. N.B., C.X. and J.K. performed the experiments and analysed the data. F.G.A., C.X., N.B., and J.K. wrote and critically revised the manuscript. All authors have approved the final version of the manuscript.
References
- Akar FG, Aon MA, Tomaselli GF, O'Rourke B. The mitochondrial origin of postischemic arrhythmias. J Clin Invest. 2005;115:3527–3535. doi: 10.1172/JCI25371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akar FG, Rosenbaum DS. Transmural electrophysiological heterogeneities underlying arrhythmogenesis in heart failure. Circ Res. 2003;93:638–645. doi: 10.1161/01.RES.0000092248.59479.AE. [DOI] [PubMed] [Google Scholar]
- Akar FG, Yan GX, Antzelevitch C, Rosenbaum DS. Unique topographical distribution of M cells underlies reentrant mechanism of torsade de pointes in the long-QT syndrome. Circulation. 2002;105:1247–1253. doi: 10.1161/hc1002.105231. [DOI] [PubMed] [Google Scholar]
- Aldakkak M, Stowe DF, Chen Q, Lesnefsky EJ, Camara AK. Inhibited mitochondrial respiration by amobarbital during cardiac ischaemia improves redox state and reduces matrix Ca2+ overload and ROS release. Cardiovasc Res. 2008;77:406–415. doi: 10.1016/j.cardiores.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Aldakkak M, Stowe DF, Heisner JS, Riess ML, Camara AK. Adding ROS quenchers to cold K+ cardioplegia reduces superoxide emission during 2-hour global cold cardiac ischemia. J Cardiovasc Pharmacol Ther. 2011 doi: 10.1177/1074248410389815. DOI: 10.1177/1074248410389815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aon MA, Cortassa S, Akar FG, Brown DA, Zhou L, O'Rourke B. From mitochondrial dynamics to arrhythmias. Int J Biochem Cell Biol. 2009;41:1940–1948. doi: 10.1016/j.biocel.2009.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aon MA, Cortassa S, Akar FG, O'Rourke B. Mitochondrial criticality: a new concept at the turning point of life or death. Biochim Biophys Acta. 2006;1762:232–240. doi: 10.1016/j.bbadis.2005.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aon MA, Cortassa S, Maack C, O'Rourke B. Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. J Biol Chem. 2007;282:21889–21900. doi: 10.1074/jbc.M702841200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aon MA, Cortassa S, Marban E, O'Rourke B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J Biol Chem. 2003;278:44735–44744. doi: 10.1074/jbc.M302673200. [DOI] [PubMed] [Google Scholar]
- Brady NR, Hamacher-Brady A, Westerhoff HV, Gottlieb RA. A wave of reactive oxygen species (ROS)-induced ROS release in a sea of excitable mitochondria. Antioxid Redox Signal. 2006;8:1651–1665. doi: 10.1089/ars.2006.8.1651. [DOI] [PubMed] [Google Scholar]
- Brown DA, Aon MA, Akar FG, Liu T, Sorarrain N, O'Rourke B. Effects of 4′-chlorodiazepam on cellular excitation-contraction coupling and ischaemia-reperfusion injury in rabbit heart. Cardiovasc Res. 2008;79:141–149. doi: 10.1093/cvr/cvn053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, Aon MA, Frasier CR, Sloan RC, Maloney AH, Anderson EJ, O'Rourke B. Cardiac arrhythmias induced by glutathione oxidation can be inhibited by preventing mitochondrial depolarization. J Mol Cell Cardiol. 2010;48:673–679. doi: 10.1016/j.yjmcc.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camara AK, Riess ML, Kevin LG, Novalija E, Stowe DF. Hypothermia augments reactive oxygen species detected in the guinea pig isolated perfused heart. Am J Physiol Heart Circ Physiol. 2004;286:H1289–H1299. doi: 10.1152/ajpheart.00811.2003. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girouard SD, Laurita KR, Rosenbaum DS. Unique properties of cardiac action potentials recorded with voltage-sensitive dyes. J Cardiovasc Electrophysiol. 1996;7:1024–1038. doi: 10.1111/j.1540-8167.1996.tb00478.x. [DOI] [PubMed] [Google Scholar]
- Glukhov AV, Fedorov VV, Lou Q, Ravikumar VK, Kalish PW, Schuessler RB, Moazami N, Efimov IR. Transmural dispersion of repolarization in failing and nonfailing human ventricle. Circ Res. 2010;106:981–991. doi: 10.1161/CIRCRESAHA.109.204891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson AB, Gottlieb RA. Heart mitochondria: gates of life and death. Cardiovascular research. 2008;77:334–343. doi: 10.1093/cvr/cvm005. [DOI] [PubMed] [Google Scholar]
- Honda HM, Korge P, Weiss JN. Mitochondria and ischemia/reperfusion injury. Ann N Y Acad Sci. 2005;1047:248–258. doi: 10.1196/annals.1341.022. [DOI] [PubMed] [Google Scholar]
- Huss JM, Kelly DP. Mitochondrial energy metabolism in heart failure: a question of balance. J Clin Invest. 2005;115:547–555. doi: 10.1172/JCI200524405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Chemaly ER, Lee A, Kho C, Hadri L, Hajjar RJ, Akar FG. Mechanoelectrical remodeling and arrhythmias during progression of hypertrophy. FASEB J. 2010a;24:451–463. doi: 10.1096/fj.09-136622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Nass RD, Joudrey PJ, Lyon AR, Chemaly ER, Rapti K, Akar FG. Altered spatiotemporal dynamics of the mitochondrial membrane potential in the hypertrophied heart. Biophys J. 2010b;98:2063–2071. doi: 10.1016/j.bpj.2010.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kevin LG, Camara AK, Riess ML, Novalija E, Stowe DF. Ischemic preconditioning alters real-time measure of O2 radicals in intact hearts with ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2003;284:H566–H574. doi: 10.1152/ajpheart.00711.2002. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Lu LS, Liu YB, Sun CW, Lin LC, Su MJ, Wu CC. Optical mapping of myocardial reactive oxygen species production throughout the reperfusion of global ischemia. J Biomed Opt. 2006;11:021012. doi: 10.1117/1.2186321. [DOI] [PubMed] [Google Scholar]
- Lu YM, Han F, Shioda N, Moriguchi S, Shirasaki Y, Qin ZH, Fukunaga K. Phenylephrine-induced cardiomyocyte injury is triggered by superoxide generation through uncoupled endothelial nitric-oxide synthase and ameliorated by 3-[2-[4-(3-chloro-2-methylphenyl)-1-piperazinyl]ethyl]-5,6-dimethoxyindazole (DY-9836), a novel calmodulin antagonist. Mol Pharmacol. 2009;75:101–112. doi: 10.1124/mol.108.050716. [DOI] [PubMed] [Google Scholar]
- Lyon AR, Joudrey PJ, Jin D, Nass RD, Aon MA, O'Rourke B, Akar FG. Optical imaging of mitochondrial function uncovers actively propagating waves of mitochondrial membrane potential collapse across intact heart. J Mol Cell Cardiol. 2010;49:565–575. doi: 10.1016/j.yjmcc.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961;191:144–148. doi: 10.1038/191144a0. [DOI] [PubMed] [Google Scholar]
- Morita N, Lee JH, Xie Y, Sovari A, Qu Z, Weiss JN, Karagueuzian HS. Suppression of re-entrant and multifocal ventricular fibrillation by the late sodium current blocker ranolazine. J Am Coll Cardiol. 2010;57:366–375. doi: 10.1016/j.jacc.2010.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita N, Sovari AA, Xie Y, Fishbein MC, Mandel WJ, Garfinkel A, Lin SF, Chen PS, Xie LH, Chen F, Qu Z, Weiss JN, Karagueuzian HS. Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am J Physiol Heart Circ Physiol. 2009;297:H1594–H1605. doi: 10.1152/ajpheart.00579.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke B, Cortassa S, Akar F, Aon M. Mitochondrial ion channels in cardiac function and dysfunction. Novartis Found Symp. 2007;287:140–151. doi: 10.1002/9780470725207.ch10. discussion 152–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riess ML, Camara AK, Kevin LG, An J, Stowe DF. Reduced reactive O2 species formation and preserved mitochondrial NADH and [Ca2+] levels during short-term 17 degrees C ischemia in intact hearts. Cardiovasc Res. 2004;61:580–590. doi: 10.1016/j.cardiores.2003.09.016. [DOI] [PubMed] [Google Scholar]
- Rigoulet M, Yoboue ED, Devin A. Mitochondrial ROS generation and its regulation: mechanisms involved in H2O2 signaling. Antioxid Redox Signal. 2011;14:459–468. doi: 10.1089/ars.2010.3363. [DOI] [PubMed] [Google Scholar]
- Salama G, Lombardi R, Elson J. Maps of optical action potentials and NADH fluorescence in intact working hearts. Am J Physiol Heart Circ Physiol. 1987;252:H384–H394. doi: 10.1152/ajpheart.1987.252.2.H384. [DOI] [PubMed] [Google Scholar]
- Sato D, Xie LH, Sovari AA, Tran DX, Morita N, Xie F, Karagueuzian H, Garfinkel A, Weiss JN, Qu Z. Synchronization of chaotic early afterdepolarizations in the genesis of cardiac arrhythmias. Proc Natl Acad Sci U S A. 2009;106:2983–2988. doi: 10.1073/pnas.0809148106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoni Y, Chen K, Emmett T, Kargacin G. Aldosterone and the autocrine modulation of potassium currents and oxidative stress in the diabetic rat heart. Br J Pharmacol. 2008;154:675–687. doi: 10.1038/bjp.2008.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slodzinski MK, Aon MA, O'Rourke B. Glutathione oxidation as a trigger of mitochondrial depolarization and oscillation in intact hearts. J Mol Cell Cardiol. 2008;45:650–660. doi: 10.1016/j.yjmcc.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viola HM, Hool LC. Qo site of mitochondrial complex III is the source of increased superoxide after transient exposure to hydrogen peroxide. J Mol Cell Cardiol. 2010;49:875–885. doi: 10.1016/j.yjmcc.2010.07.015. [DOI] [PubMed] [Google Scholar]
- Walker MJ, Curtis MJ, Hearse DJ, Campbell RW, Janse MJ, Yellon DM, Cobbe SM, Coker SJ, Harness JB, Harron DW, et al. The Lambeth Conventions: guidelines for the study of arrhythmias in ischaemia infarction, and reperfusion. Cardiovasc Res. 1988;22:447–455. doi: 10.1093/cvr/22.7.447. [DOI] [PubMed] [Google Scholar]
- Xie LH, Chen F, Karagueuzian HS, Weiss JN. Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ Res. 2009;104:79–86. doi: 10.1161/CIRCRESAHA.108.183475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Korge P, Weiss JN, Qu Z. Mitochondrial oscillations and waves in cardiac myocytes: insights from computational models. Biophys J. 2010;98:1428–1438. doi: 10.1016/j.bpj.2009.12.4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Aon MA, Almas T, Cortassa S, Winslow RL, O'Rourke B. A reaction-diffusion model of ROS-induced ROS release in a mitochondrial network. PLoS Comput Biol. 2010;6:e1000657. doi: 10.1371/journal.pcbi.1000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielonka J, Kalyanaraman B. Hydroethidine- and MitoSOX-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: another inconvenient truth. Free Radic Biol Med. 2010;48:983–1001. doi: 10.1016/j.freeradbiomed.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta. 2006;1757:509–517. doi: 10.1016/j.bbabio.2006.04.029. [DOI] [PubMed] [Google Scholar]