

Non-technical summary
The movement of receptors to and from the surface of nerve cells determines whether nerves can detect and respond to stimulants in the extracellular fluid. It is well established that stimulated receptors move from the surface of nerves to intracellular compartments, but the processes that control movement of internalized receptors back to the surface are poorly understood. We show that an intracellular enzyme, endothelin-converting enzyme-1, controls the translocation of receptors back to the surface of neurones of the intestinal tract that control the normal process of digestion. This process is likely to regulate digestion, and abnormalities in the mechanism could cause digestive diseases.
Abstract
Abstract
Neuropeptide signalling at the plasma membrane is terminated by neuropeptide degradation by cell-surface peptidases, and by β-arrestin-dependent receptor desensitization and endocytosis. However, receptors continue to signal from endosomes by β-arrestin-dependent processes, and endosomal sorting mediates recycling and resensitization of plasma membrane signalling. The mechanisms that control signalling and trafficking of receptors in endosomes are poorly defined. We report a major role for endothelin-converting enzyme-1 (ECE-1) in controlling substance P (SP) and the neurokinin 1 receptor (NK1R) in endosomes of myenteric neurones. ECE-1 mRNA and protein were expressed by myenteric neurones of rat and mouse intestine. SP (10 nm, 10 min) induced interaction of NK1R and β-arrestin at the plasma membrane, and the SP–NK1R–β-arrestin signalosome complex trafficked by a dynamin-mediated mechanism to ECE-1-containing early endosomes, where ECE-1 can degrade SP. After 120 min, NK1R recycled from endosomes to the plasma membrane. ECE-1 inhibitors (SM-19712, PD-069185) and the vacuolar H+ATPase inhibitor bafilomycin A1, which prevent endosomal SP degradation, suppressed NK1R recycling by >50%. Preincubation of neurones with SP (10 nm, 5 min) desensitized Ca2+ transients to a second SP challenge after 10 min, and SP signals resensitized after 60 min. SM-19712 inhibited NK1R resensitization by >90%. ECE-1 inhibitors also caused sustained SP-induced activation of extracellular signal-regulated kinases, consistent with stabilization of the SP–NK1R–β-arrestin signalosome. By degrading SP and destabilizing endosomal signalosomes, ECE-1 has a dual role in controlling endocytic signalling and trafficking of the NK1R: promoting resensitization of G protein-mediated plasma membrane signalling, and terminating β-arrestin-mediated endosomal signalling.
Introduction
Peptidergic neurotransmission requires the presence of receptors at the plasma membrane, where they can interact with neuropeptides in the extracellular fluid. The mechanisms that regulate signalling and trafficking of seven transmembrane domain (7TMD) neuropeptide receptors at the plasma membrane have been thoroughly investigated (reviewed in Gainetdinov et al. 2004). Mechanisms that terminate plasma membrane signalling include neuropeptide degradation by cell-surface peptidases such as neprilysin, which inactivates substance P (SP) and bradykinin and attenuates their proinflammatory actions (Okamoto et al. 1994; Lu et al. 1997; Sturiale et al. 1999), and receptor phosphorylation by G protein-coupled receptor kinases. Phosphorylation increases the affinity of 7TMD receptors for β-arrestins, which translocate from the cytosol to interact with agonist-occupied receptors at the plasma membrane. β-Arrestins uncouple receptors from heterotrimeric G proteins and thereby desensitize G protein-dependent signalling. β-Arrestins also couple receptors to the clathrin-mediated endocytic machinery, which depletes receptors from the cell surface and consequently diminishes cellular responsiveness to extracellular agonists. In addition, β-arrestins are adaptor molecules that interact with diverse signalling proteins. By recruiting receptors and signalling partners such as mitogen-activated protein kinases (MAPKs) to multi-protein signalling complexes (signalosomes), β-arrestins can transduce a second wave of G protein-independent signalling from the endosomal network (Murphy et al. 2009). Thus, β-arrestins have a dual role in regulating 7TMD receptors, terminating G protein-mediated signalling from plasma membranes, and initiating β-arrestin-mediated signalling from endosomal membranes. However, β-arrestin-mediated endosomal signals are distinct from G protein-mediated plasma membrane signals, in terms of mechanism, subcellular location, duration and outcome.
Compared to these mechanisms that regulate neuropeptide receptor signalling and trafficking at the plasma membrane, little is known about the processes that control neuropeptide receptor functions in endosomes. In particular, the processes that terminate β-arrestin-mediated signalling of 7TMD receptors in endosomes of neurones are unexplored, but are likely to involve destabilization of signalosomes, which induces dissociation of the neuropeptide–receptor–β-arrestin complex. This dissociation allows the receptor, freed from β-arrestins, to recycle, which is necessary for resensitization of G protein-mediated signalling at the plasma membrane (Oakley et al. 1999).
We recently identified a proteolytic mechanism for controlling signalling and trafficking of neuropeptide receptors in endosomes that is analogous to well-defined mechanisms that control receptors at the plasma membrane (Padilla et al. 2007; Roosterman et al. 2007; Cottrell et al. 2009). Endothelin-converting enzyme-1 (ECE-1) is a neprilysin-related metallo-endopeptidase that is present at the plasma membrane and in endosomes. In the acidic endosomal environment, ECE-1 degrades neuropeptides and thereby destabilizes peptide–receptor–β-arrestin–MAPK signalosomes, allowing receptors to recycle and re-engage G protein-dependent signalling at the plasma membrane, and terminating β-arrestin-mediated MAPK signalling from endosomes. This mechanism regulates neuropeptides that are ECE-1 substrates at acidic endosomal pH and controls receptors that form stable interactions with β-arrestins in endosomes, including SP and the neurokinin 1 receptor (NK1R), and calcitonin gene-related peptide and the calcitonin receptor-like receptor. However, this mechanism has been studied thus far only in cell lines. Nothing is known about the role of ECE-1 in controlling endosomal signalling and trafficking of neuropeptide receptors in neurones.
We examined whether ECE-1 regulates endosomal signalling and trafficking of the NK1R in myenteric neurones of the intestine. The NK1R is expressed by several classes of myenteric neurones and mediates ascending contraction of the peristaltic reflex (Sternini et al. 1995; Grider, 2003). Although the contractile effects of SP in the intestine desensitize and resensitize (Gaddum, 1953), and SP-induces endocytosis and recycling of the NK1R in myenteric neurones (Grady et al. 1996b; McConalogue et al. 1998), the mechanisms underlying these regulatory processes are not defined. We report that ECE-1, which is widely expressed in enteric neurones, promotes recycling and resensitization of plasma membrane G protein signalling, and terminates endosomal β-arrestin-mediated signalling of the NK1R.
Methods
Ethical approval
The Institutional Animal Care and Use Committee of the University of California, San Francisco approved all procedures on experimental animals.
Animals
C57/BL6 mice (6–8 weeks, 20–25 g, male and female) and Sprague–Dawley rats (6 weeks, 200–250 g, male) were from Charles River Laboratories (Hollister, CA, USA). Animals were maintained under temperature- (22 ± 4°C) and light- (12 h light–dark cycle) controlled conditions with free access to food and water. Animals were anaesthetized with sodium pentobarbital (200 mg kg−1i.p.) and killed by bilateral thoracotomy before tissue collection.
Reagents
SP was from Bachem (Torrance, CA, USA). The ECE-1 inhibitor PD-069185 (Ahn et al. 1998) was a gift from Pfizer Pharmaceuticals (Pfizer, UK). The NK1R antagonist RP-67580 (Garret et al. 1992) was from Tocris Bioscience (Ellisville, MO, USA). Alexa Fluor 594 protein labelling kit, Fura 2-AM and Prolong Gold containing DAPI were from Invitrogen (Carlsbad, CA, USA). SP was labelled with Alexa Fluor 594 (Alexa-SP) and purified as described (Bunnett et al. 1995). Duolink Proximity Ligation Assay (PLA) was from Olink Biosciences (Uppsala, Sweden). Other reagents were from Sigma-Aldrich (St Louis, MO, USA).
Antibodies
Rabbit anti-NK1R (94168) antibody has been described (Grady et al. 1996a). Mouse anti-β-arrestin1/2 (610551) and mouse anti-early endosomal antigen 1 (EEA1, 610457) antibodies were from BD Biosciences (San Jose, CA, USA). Goat anti-human ECE-1 (AF1784) antibody was from R&D Systems, Inc. (Minneapolis, MN, USA). Rabbit antibody to the N terminus of human ECE-1 that recognizes ECE-1b/d isoforms (Padilla et al. 2007) was from Invitrogen. Rabbit anti-ERK1/2 (9102) and mouse anti-phospho (p)-ERK1/2 (9106) antibodies were from Cell Signalling Technology (Danvers, MA, USA). Chicken anti-PGP9.5 (72910) was from Abcam (Cambridge, UK). Donkey anti-chicken, -rabbit, -mouse and -goat IgG conjugated to rhodamine red X, fluorescein isothiocyanate or cyanine 5 were from Jackson Immunoresearch (West Grove, PA, USA).
ECE-1 and NK1R RT-PCR
The muscularis externa and myenteric plexus (ME-MP) of mouse ileum was obtained by sharp dissection. Total RNA was extracted, treated with DNase I and reverse transcribed to amplify ECE-1 isoforms and NK1R. The following primers were used to detect mouse ECE-1 isoforms: ECE-1a forward: 5′-CCCTGGTCTCATGGTCTCGCT-3′; ECE-1b forward: 5′-GGTGCAGCATGCGGACCGTGT-3′; ECE-1c1 forward: 5′-GAGCCTTAGCGGGAGGTGCAT-3′; ECE-1c2 forward: 5′-GCAGCCTGCCCACCAGGGTAA-3′; ECE-1d forward: 5′-GCAATGGAGACGCTGAGGGAGT-3′; ECE-1 reverse: 5′-CGTAGCTGAAGAAGTCCTGGCA-3′. The following primers were used to detect the mouse NK1R: forward: 5′-GTGCAACCTACCTGGCAAAT-3′; reverse: 5′-CTACTTCCTGCCTCTGCTGGT-3′. Products were separated by electrophoresis and sequenced.
ECE-1 Western blotting
Membranes were prepared from ME-MP of the mouse ileum and from HEK cells as described previously (Cottrell et al. 2009). Proteins (15 μg) were separated by SDS-PAGE and ECE-1 was detected by Western blotting (Cottrell et al. 2009).
NK1R trafficking in organotypic preparations of myenteric plexus
Terminal ileum or distal colon from rats and mice were placed in ice-cold Hank's balanced salt solution (HBSS) containing nicardipine (1 μm) and tetrodotoxin (100 nm). Segments were opened, pinned mucosa-side down onto silicone elastomer-lined dishes, and incubated in physiological salt solution (PSS, mm: NaCl 118; KCl 4.8; NaHCO3 25; NaH2PO4 1.0; MgSO4 1.2; d-glucose 11.1; CaCl2 2.5). Tissues were incubated with SP (10 nm, 10 min, 37°C) or vehicle (H2O), washed and recovered in SP-free PSS for 0–120 min at 37°C. To examine endocytosis of fluorescent SP, tissues were incubated with Alexa-SP (100 nm, 60 min, 4°C), washed and recovered in SP-free PSS for 0–120 min at 37°C. To induce release of endogenous SP from nerve fibres, preparations were depolarized with KCl (50 mm, 5 min, 37°C), washed and recovered in PSS for 0–60 min, all in the presence of peptidase inhibitors (thiorphan (100 nm), phosphoramidon, captopril and bestatin (all 10 μm)) (Grady et al. 1996b). Preparations were fixed in 4% paraformaldehyde in 0.1 m PBS (pH 7.4, overnight, 4°C), and whole-mounts were prepared of the longitudinal muscle and myenteric plexus (Poole et al. 2002).
NK1R trafficking in cultured myenteric neurones
Myenteric neurones were isolated and cultured from the small intestine of rats and mice as described previously (Grady et al. 1996b; McConalogue et al. 1998). Intestinal segments were placed in ice-cold HBSS containing nicardipine (1 μm) and antibiotic/antimycotic solution (penicillin G, 100 U ml−1; streptomycin sulphate, 10 mg ml−1; amphotericin B, 25 mg ml−1). Segments were opened, pinned flat and the ME-MP was dissected. Tissue was digested with collagenase type IA (1 mg ml−1) and DNase IV (0.1 mg ml−1, 150 kU) in Dulbecco's modified Eagle's medium (DMEM) (1 h, 37°C) with agitation. Tissue was then digested with trypsin (0.25% solution with EDTA) (15 min, 37°C). Digestion was stopped by adding 20% fetal bovine serum (FBS) in DMEM containing soybean trypsin inhibitor (10 mg ml−1) followed by centrifugation. A single cell suspension was achieved by sequential trituration. Neurones were plated onto poly-l-lysine/laminin-coated glass coverslips, and were cultured in DMEM supplemented with normal horse serum (NHS) (5%), FBS (5%), l-glutamine (2 mm), antibiotic/antimycotic and N1 neuronal supplement (1:100). Cytosine arabinoside (1 μm) was included in the culture media from day 2 onwards to inhibit proliferation of non-neuronal cells. Neurones were studied after 5–7 days. Cultured neurones were incubated in DMEM (0.1% BSA without phenol red, 5% CO2–95% O2) and were stimulated with SP (10 nm, 10 min, 37°C) or vehicle (H2O). Cultures were washed and recovered in SP-free DMEM for 0–120 min. Neurones were fixed in 4% paraformaldehyde (20 min, 4°C) and processed for immunofluorescence or for examination of NK1R and β-arrestin1/2 or total ERK1/2 and pERK1/2 interactions by proximity ligation assay (PLA).
Immunofluorescence
Whole-mounts and cultures were incubated in blocking buffer (10% NHS, 0.1% Triton X-100, 0.1 m PBS) for 30 min at room temperature, and then incubated with the following primary antibodies in blocking buffer: NK1R (1:500), β-arrestin1/2 (1:100), ECE-1 (1:100), EEA1 (1:200) and PGP9.5 (1:100) (all 48 h, 4°C). Slides were washed and incubated with secondary antibodies in PBS (1:200, 2 h, room temperature). Slides were mounted in Prolong and examined by confocal microscopy.
Proximity ligation assay
Interactions between NK1R and β-arrestin1/2 or total ERK1/2 and pERK1/2 were examined using in situ PLA (Soderberg et al. 2008) with anti-mouse PLA probe plus, anti-rabbit PLA probe minus and detection kit 563, according to the manufacturer's instructions. Neurones were permeabilized with 0.1% Triton X-100 in PBS for 4 min, blocked with 1% normal goat serum in PBS (blocking buffer) for 1 h at room temperature, and incubated with pairs of primary antibodies: NK1R (1:500) plus β-arrestin1/2 (1:100), or total ERK1/2 (1:100) plus pERK1/2 (1:200) (overnight, 4°C). Neurones were washed in PBS and then incubated with PLA probes in a humidified chamber for 2 h at 37°C. Neurones were incubated with hybridization mixture for 15 min, washed with Tris-buffered saline containing 0.05% Tween 20 (TBS-T), and incubated with ligation mixture for 15 min. Neurones were washed with TBS-T, incubated with amplification mixture for 90 min, washed with TBS-T and incubated with detection probes for 60 min. Finally, neurones were washed with 2× saline-sodium citrate buffer (SSC), 1× SSC, 0.2× SSC, 0.02× SSC and 70% ethanol. Coverslips were air-dried and mounted in Prolong. Images of PLA signals were collected using the 543 nm laser and five optical sections were taken (63× objective) at intervals of 0.32 μm. Collection settings were optimized for stimulated preparations and all subsequent images were taken using these settings. The number of PLA signals per neurone (‘blobs’) was counted using the BlobFinder software using the cell-average analysis mode (Allalou & Wahlby, 2009) (http://www.cb.uu.se/~amin/BlobFinder/).
Confocal microscopy
Preparations were observed using a Zeiss Axiovert microscope with a Zeiss 510 Meta confocal system, with Plan Apochromat ×100 (NA 1.4), Fluar Plan Apochromat ×63 (NA 1.4) and ×40 Plan-Neofluar (NA 1.3) objectives. The subcellular distribution of NK1R was analysed from captured images using Image J (http://rsbweb.nih.gov/ij/) as described previously (Poole et al. 2007). Single optical sections of the neuronal soma, including the nucleus, were analysed. The threshold of individual images was defined as the fluorescence pixel intensity of the nucleus, which takes advantage of the absence of nuclear NK1R-IR. Positive staining (i.e. NK1R-IR above background) was set to 255 and negative staining was set to 0. A region of interest was manually drawn around the soma of NK1R-IR neurones and the total positive pixels of the soma were measured. A second region of interest was drawn immediately beneath the plasma membrane (readily detected by autofluorescence) and the intracellular NK1R-IR positive pixels were measured. Any positive nuclear staining (e.g. non-specific high-intensity spots) was subtracted from both values. The membrane and intracellular NK1R pixels were then expressed as the relative percentages of the total cellular pixels (100%).
Ratiometric Ca2+ imaging of cultured myenteric neurones
[Ca2+]i was measured in cultured myenteric neurones by ratiometric imaging of Fura 2-AM as described previously (McConalogue et al. 1998). Cultured neurones were incubated in HBSS (containing 0.1% BSA and 20 mm Hepes, pH 7.4) and loaded with Fura 2-AM (5 μm, 45 min, 37°C). Coverslips were washed and mounted on a temperature-controlled open chamber at 37°C. Fluorescence of individual neurones was measured at 340 nm and 380 nm excitation, and 510 nm emission. Test substances were directly added to the chamber (50 μl injection). To assess desensitization and resensitization of the NK1R, neurones were stimulated with SP (10 nm, 5 min), washed, recovered in SP-free medium for 10, 30 or 60 min, and then re-challenged with SP (10 nm). Responses of the same neurones to both SP challenges were examined. Finally, neurones were stimulated with carbachol (1 μm) and KCl (50 mm). Neurone identity was confirmed based on size, morphology and responsiveness to KCl. The magnitude of the change in emission ratio at 340/380 nm excitation, which is proportional to [Ca2+]i, and the proportion of the total number of KCl-responsive neurones that responded to SP challenge were determined. The extent of resensitization was calculated as the percentage of all KCl-responsive neurones that responded to the second SP challenge compared to the first SP challenge (normalized to 100%).
Drug treatments
Organotypic whole-mounts or cultured neurones were preincubated with the following drugs: ECE-1 inhibitors SM-19712 (Matsumura et al. 2000) (10 μm) or PD-069185 (Ahn et al. 1998) (30 μm) (60 min); H+ATPase inhibitor bafilomycin A1 (1 μm, 30 min); NK1R antagonist RP-67580 (Garret et al. 1992) (100 nm, 5 min); dynamin GTPase inhibitor Dynasore (Macia et al. 2006) (80 μm, 10 min); or vehicle (control). Inhibitors were included throughout the experiments. As a positive control for the ERK1/2.pERK1/2 PLA, neurones were stimulated with phorbol 12,13-dibutyrate (PDBu, 100 nm, 10 min).
Statistical analysis
Data are presented as mean ± SEM of n≥ 5 experiments or animals. Results were compared by Student's t test (2 comparisons) or by ANOVA and Student–Newman–Keuls test (multiple comparisons). P < 0.05 was considered to be significantly different at the 95% confidence level.
Results
ECE-1 is expressed in myenteric neurones
We examined ECE-1 expression in ME-MP by RT-PCR and Western blotting. Transcripts corresponding to ECE-1a (293 bp), ECE-1b (390 bp), ECE-1c1 (387 bp), ECE-1c2 (473 bp) and NK1R (557 bp), but not ECE-1d, were amplified from ME-MP of mouse ileum and identified by sequencing (Fig. 1A). ECE-1a, ECE-1b, ECE-1c and ECE-1d mRNA have been similarly identified in ME-MP of the rat ileum (Cottrell et al. 2009). ECE-1 immunoreactivity (IR) was detected by Western blotting of mouse ileum ME-MP with an apparent molecular mass of ∼118 kDa, similar to that detected in HEK cells, which naturally express ECE-1 (Padilla et al. 2007) (Fig. 1B). Smaller bands are likely to represent ECE-1 degradation products, although this possibility remains to be investigated. We localized ECE-1-IR in myenteric plexus whole-mounts of rat ileum by immunofluorescence using an antibody to ECE-1b and d isoforms (Roosterman et al. 2007). This antibody was unsuitable for detection of ECE-1 in mice. ECE-1-IR was localized to endosomes but not to the plasma membrane of myenteric neurones that were identified by PGP9.5-IR (Fig. 1C), consistent with the predominant endosomal localization of ECE-1b/d isoforms (Padilla et al. 2007; Roosterman et al. 2007). ECE-1 was also localized to interstitial cells of Cajal, but was not detected in the longitudinal or circular muscle cells (not shown). Pre-adsorption of the antibody with ECE-1 abolished staining, as we have previously reported (Cattaruzza et al. 2009; Cottrell et al. 2009), indicating specific detection of endosomal ECE-1 in enteric neurones.
Figure 1. Expression and localization of ECE-1.
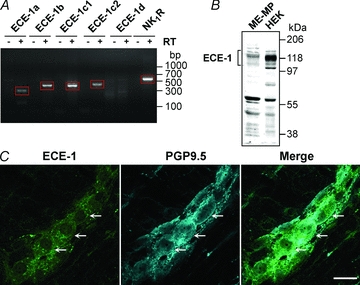
A, RT-PCR amplification of mRNA from isolated ME-MP of the mouse ileum encoding ECE-1a (293 bp), ECE-1b (390 bp), ECE-1c1 (387 bp), ECE-1c2 (473 bp) and NK1R (557 bp). ECE-1d was not detected. RT, reverse transcriptase. B, Western blotting for ECE-1 in ME-MP of mouse ileum and in HEK cells (which naturally express ECE-1). C, co-localization of ECE-1-IR and PGP9.5-IR in whole-mount of rat myenteric plexus, showing vesicular localization of ECE-1-IR in neurones. Arrows denote ECE-1-expressing neurones. Scale, 20 μm.
SP induces NK1R and β-arrestin interactions at the plasma membrane and in endosomes
Despite the established importance of β-arrestins in regulating signalling and trafficking of GPCRs (Gainetdinov et al. 2004), including the NK1R expressed in cell lines (McConalogue et al. 1999; DeFea et al. 2000), little is known about their ability to regulate endogenous receptors in neurones. We examined SP-induced trafficking of NK1R and β-arrestins in organotypic preparations of myenteric plexus from rat ileum, since β-arrestin antibodies were raised in mice and were unsuitable for use in that species. In unstimulated myenteric neurones, NK1R-IR was at the plasma membrane and β-arrestin-IR was cytosolic (Fig. 2A). SP (10 nm, 10 min, wash, 10 min recovery, 37°C) induced the redistribution of the NK1R-IR and β-arrestin-IR to the same endosomes in close proximity to the plasma membrane (Fig. 2A). To examine SP-induced interactions between NK1R and β-arrestins, we used an in situ PLA, which localizes protein–protein interactions at the cellular level (Soderberg et al. 2008). If two proteins are in close proximity (<40 nm), short DNA strands attached to secondary antibodies are ligated, and the DNA can be amplified by rolling circle amplification and detected using fluorescent oligonucleotides. NK1R–β-arrestin interactions were examined in rat myenteric neurones in culture, since high levels of non-specific PLA signals were detected in whole-mounts. SP (10 nm) induced a >4-fold increase in the number of NK1R–β-arrestin interactions within 10 min, and interactions persisted for at least 30 min (Fig. 2B and C). Omission of either of the primary antibodies prevented PLA signals, indicating specificity. These results indicate that SP stimulates NK1R–β-arrestin interactions in endosomes of myenteric neurones.
Figure 2. SP-induced trafficking and interaction of NK1R and β-arrestins in myenteric neurones.
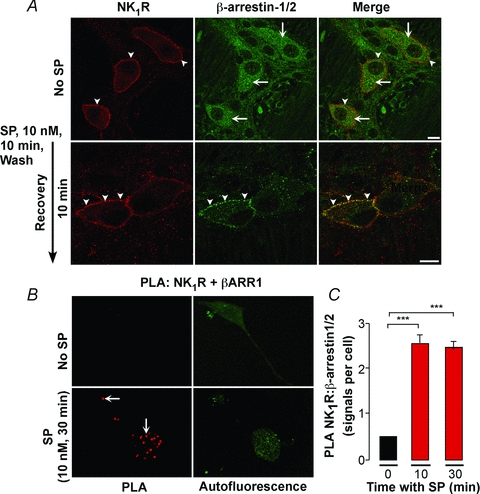
A, organotypic preparations of rat ileum were unstimulated or incubated with SP (10 nm, 10 min), washed and recovered for 10 min. NK1R and β-arrestin1/2 were detected in whole-mount preparations by immunofluorescence. In unstimulated neurones, NK1R-IR was at the plasma membrane (arrowheads) and β-arrestin-IR was cytosolic (arrows). SP induced translocation of NK1R-IR and β-arrestin-IR to the same endosomes (arrowheads). Scale, 10 μm. B and C, PLA for NK1R and β-arrestin in rat myenteric neurones in culture. PLA signals are in red, and green is autofluorescence showing the localization of the neurones. In the absence of SP, PLA signals were minimal. SP (10 nm, 10 or 30 min) induced PLA signals (arrows) that denote NK1R/β-arrestin interactions. ***P < 0.001. n > 3 experiments (i.e. neuronal cultures from 1 rat per preparation).
Dynamin mediates SP-induced endocytosis of the NK1R in myenteric neurones
SP stimulates NK1R endocytosis in neurones by unknown mechanisms (Mantyh et al. 1995; Grady et al. 1996b; McConalogue et al. 1998). We used Dynasore, a selective inhibitor of dynamin GTPase (Macia et al. 2006), to determine the role of dynamin in SP-induced endocytosis of the NK1R in myenteric neurones. Organotypic preparations of mouse colon were incubated with Dynasore or vehicle (control), and were stimulated with SP (10 nm, 10 min, 37°C), washed and recovered for 30 min. The NK1R was localized in whole-mounts of myenteric plexus by immunofluorescence. Dynasore alone did not alter the subcellular localization of NK1R-IR, which remained at the plasma membrane of the soma and neurites of myenteric neurones (Fig. 3A). SP stimulated NK1R endocytosis in the soma and in neurites, and Dynasore abolished endocytosis. Quantitative analysis of total cellular NK1R-IR pixels indicated that 13.3 ± 3.8% were in endosomes of unstimulated neurones, 37.2 ± 2.1% were in endosomes of SP-stimulated neurones, and 15.1 ± 1.1% were in endosomes of Dynasore-treated SP-stimulated neurones (Fig. 3B; P < 0.001, Dynasore compared to vehicle). Thus, dynamin mediates SP-induced endocytosis of the NK1R in myenteric neurones.
Figure 3. Dynamin-mediated NK1R endocytosis in myenteric neurones.
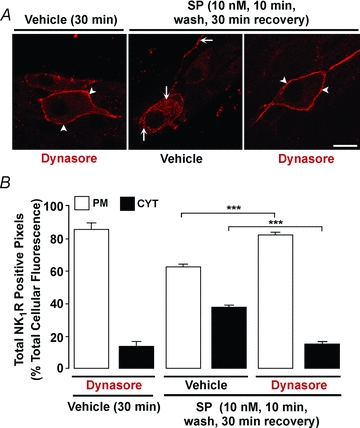
Organotypic preparations of mouse ileum were incubated with Dynasore or vehicle, and were unstimulated or stimulated with SP (10 nm, 10 min, wash, 30 min recovery). NK1R was localized in whole-mounts of myenteric plexus by immunofluorescence (A), and the pixel intensity at the plasma membrane (PM) and in the cytosol (CYT) was determined (B). Dynasore alone for 30 min did not affect the distribution of NK1R-IR, which remained at the plasma membrane (arrowheads). In vehicle-treated neurones, SP induced NK1R-IR translocation to endosomes (arrows). Dynasore abolished SP-induced endocytosis of the NK1R-IR, causing receptor retention at the plasma membrane (arrowheads). Scale, 10 μm. ***P < 0.001. 12–91 neurones per group, n = 3 mice.
ECE-1 mediates SP-induced recycling of the NK1R in myenteric neurones
When heterologously expressed, all ECE-1 isoforms localize to early endosomes, although ECE-1a and ECE-1c are also present at the plasma membrane (Padilla et al. 2007; Roosterman et al. 2007). SP and the NK1R traffic to early endosomes containing ECE-1 in cells that heterologously express NK1R and ECE-1 isoforms. To determine whether SP traffics with the NK1R to ECE-1-containing endosomes of myenteric neurones, organotypic preparations of the rat ileum were incubated with Alexa-SP (100 nm, 60 min, 4°C), washed and recovered in SP-free medium for 10 or 30 min at 37°C. NK1R, ECE-1 and EEA-1 were localized in whole-mounts of myenteric plexus by immunofluorescence. At 4°C, Alexa-SP was confined to the plasma membrane (not shown). After incubation at 37°C, Alexa-SP was detected in vesicles throughout the soma of myenteric neurones that also contained EEA1-IR (Fig. 4A), NK1R-IR and ECE-1-IR (Fig. 4B).
Figure 4. Translocation of SP to early endosomes containing ECE-1 in myenteric neurones.
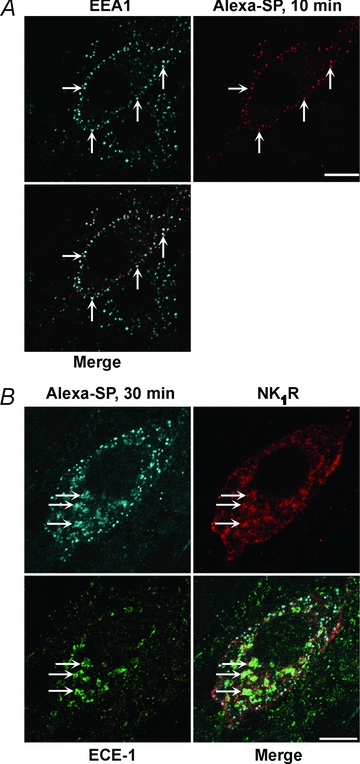
Organotypic preparations of rat ileum were incubated with Alexa-SP (100 nm, 60 min, 4°C), washed and recovered in SP-free medium for 10 min (A) or 30 min (B) at 37°C. Alexa-SP was simultaneously localized in whole-mounts of myenteric plexus with EEA1 (A) and with NK1R and ECE-1 (B), which were detected by immunofluorescence. Arrows show colocalization of Alexa-SP with EEA1-IR, NK1R-IR and ECE-1-IR. Scale, 10 μm.
To examine the post-endocytic sorting of NK1R and ECE-1 in myenteric neurones, organotypic preparations of rat ileum were incubated with SP (10 nm, 10 min, 37°C), washed and recovered in SP-free medium for 30–120 min. NK1R and ECE-1 were localized in whole-mounts of myenteric plexus by immunofluorescence. In unstimulated neurones, NK1R-IR was confined to the plasma membrane of the soma and neurites, and ECE-1-IR was detected in endosomes (Fig. 5). At 30 min recovery, NK1R-IR and ECE-1-IR colocalized in endosomes, and after 60 and 120 min recovery, NK1R-IR was recycled to the plasma membrane. Quantitative analysis indicated that 77.7 ± 1.8% of total cellular NK1R-IR pixels were at the plasma membrane of untreated neurones, 49.8 ± 1.4% were at the plasma membrane after 30 min recovery, and 75.9 ± 1.4% were at the plasma membrane after 120 min recovery (Fig. 6A; P < 0.005, 30 min recovery compared to unstimulated neurones). Thus, SP and the NK1R initially traffic to early endosomes of myenteric neurones that also contain ECE-1. The NK1R then recycles to the plasma membrane and ECE-1 remains in endosomes.
Figure 5. SP-induced endocytosis and recycling of the NK1R in myenteric neurones.
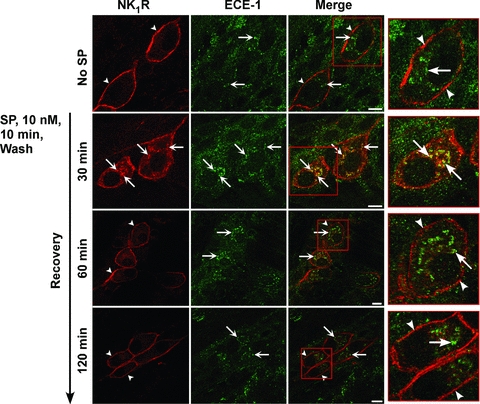
Organotypic preparations of rat ileum were unstimulated or stimulated with SP (10 nm, 10 min), washed, and recovered for 30–120 min. NK1R and ECE-1 were localized in whole-mounts of myenteric plexus by immunofluorescence. In unstimulated neurones, NK1R-IR was at the plasma membrane (arrowheads) and ECE-1-IR was in endosomes (arrows). At 30 min recovery, NK1R-IR colocalized with ECE-1-IR in endosomes (arrows). At 60 and 120 min recovery, NK1R-IR had recycled to the plasma membrane (arrowheads). Scale, 10 μm.
Figure 6. Quantitative analysis of ECE-1 regulation of NK1R recycling in myenteric neurones.
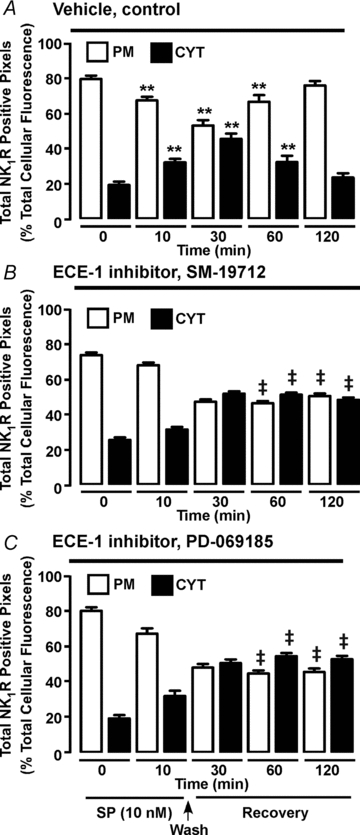
Organotypic preparations of rat ileum were incubated with vehicle (A) or the ECE-1 inhibitors SM-19712 (B) or PD-069185 (C). Neurones were unstimulated or stimulated with SP (10 nm, 10 min), washed and recovered for 30–120 min. NK1R was localized in whole-mounts of myenteric plexus by immunofluorescence and pixel intensity at the plasma membrane (PM) and in the cytosol (CYT) was determined. In vehicle-treated preparations before SP stimulation (0 min), NK1R-IR was predominantly at the plasma membrane. SP reduced plasma membrane intensity and increased endosomal intensity at 10 and 30 min, indicating endocytosis. At 60–120 min recovery, plasma membrane intensity was increased and endosomal intensity was decreased, indicating recycling. SM-19712 and PD-069185 did not affect NK1R-IR endocytosis, but caused NK1R-IR retention in endosomes at 60 and 120 min, indicating inhibition of recycling. **P < 0.005 to 0 min vehicle control. ‡P < 0.005 to vehicle control. 25–88 neurones per group from n = 6 rats.
By degrading SP in acidified endosomes of model cell lines, ECE-1 destabilizes the SP–NK1R–β-arrestin complex and promotes NK1R recycling (Roosterman et al. 2007; Cottrell et al. 2009). To evaluate whether ECE-1 similarly controls post-endocytic trafficking of the NK1R in myenteric neurones, organotypic preparations of rat ileum were treated with the ECE-1 selective inhibitors SM-19712 (Umekawa et al. 2000) or PD-069185 (Ahn et al. 1998). These are highly selective ECE-1 inhibitors that do not suppress the activity of other proteases that degrade SP, such as neprilysin. Inhibition of ECE-1 did not affect SP-induced endocytosis of NK1R-IR, which translocated from the plasma membrane to early endosomes that also contained ECE-1-IR (Fig. 6A and B). However, in marked contrast to neurones with active ECE-1, in which NK1R recycled after 60 and 120 min recovery (Fig. 5, Fig. 6A), in neurones treated with either ECE-1 inhibitor, NK1R-IR was retained in endosomes containing ECE-1 at both time points (Fig. 7A and B). Quantitative analysis of NK1R-IR localization at the plasma membrane and endosomes confirmed these observations. In vehicle-treated control preparations, the proportion of NK1R-IR pixels that were detected in endosomes was 21.7 ± 1.8% before SP treatment, 49.0 ± 1.4% after 30 min recovery post-SP, and 23.3 ± 1.4% at 120 min (Fig. 6A; P < 0.005, 30 min recovery compared to unstimulated neurones), indicative of NK1R endocytosis and recycling. In SM-19712-treated neurones, these proportions were 25.6 ± 1.4% before treatment, 47.2 ± 1.3% after 30 min recovery, and 48.3 ± 1.4% at 120 min recovery (Fig. 6B; P < 0.005, 120 min recovery, SM-19712 compared to vehicle), and in PD-069185-treated neurones, the proportions were 19.4 ± 2.0% before treatment, 50.8 ± 2.1% after 30 min recovery post-SP, and 53.0 ± 1.9% at 120 min (Fig. 6C; P < 0.005, 120 min recovery, PD-069185 compared to vehicle). Thus, SM-19712 caused a 2-fold (48.2/23.3%) and PD-069185 caused a 2.3-fold (52.9/23.3%) increase in levels of NK1R-IR in endosomes at 120 min, when compared to controls.
Figure 7. Effects of ECE-1 inhibition on recycling of the NK1R.
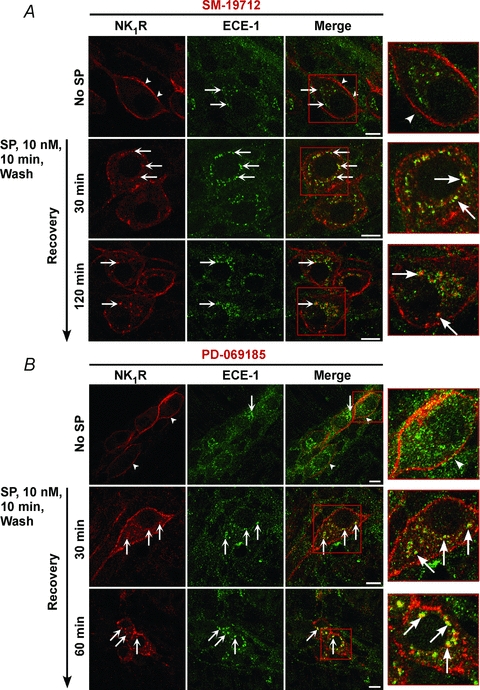
Organotypic preparations of rat ileum were incubated with the ECE-1 inhibitors SM-19712 (A) or PD-069185 (B), and were unstimulated or stimulated with SP (10 nm, 10 min), washed and recovered for 30–120 min. NK1R and ECE-1 were localized in whole-mounts of myenteric plexus by immunofluorescence. In unstimulated neurones, NK1R-IR was at the plasma membrane (arrowheads) and ECE-1-IR was in endosomes (arrows). At 30 min recovery, NK1R-IR colocalized with ECE-1-IR in endosomes (arrows). At 60 and 120 min recovery, NK1R-IR was prominently detected in endosomes (arrows). Scale, 10 μm.
We similarly determined whether active ECE-1 is required for SP-induced endocytosis and recycling of the NK1R in myenteric neurones of the mouse colon. Organotypic preparations from mouse colon were incubated with SP (10 nm, 10 min, 37°C), washed and recovered in SP-free medium for 30–120 min. In neurones with active ECE-1, SP induced trafficking of NK1R-IR to endosomes after 30 min recovery, and by 60 min NK1R-IR was completely recycled (Fig. 8A and B). SM-19712 did not affect NK1R endocytosis, but caused retention of NK1R-IR in endosomes at 60 min recovery (Fig. 8A and C). The proportion of NK1R-IR pixels in endosomes at 60 min recovery was 14.5 ± 1.2% in vehicle-treated neurones and 63.6 ± 2.7% in SM-19712-treated neurones (P < 0.005, 60 min recovery, SM-19712 compared to vehicle). Thus, SM-19712 caused a 4.4-fold (63.3/14.47%) increase in levels of NK1R in endosomes at 60 min, when compared to controls. Bafilomycin A1, an inhibitor of vacuolar H+ATPase, prevents endosomal acidification and thereby suppresses ECE-1-mediated degradation of SP in acidified endosomes (Roosterman et al. 2007). Bafilomycin A1 also caused marked retention of NK1R-IR in endosomes at 60 min recovery (Fig. 8A).
Figure 8. ECE-1-mediated recycling of the NK1R in myenteric neurones.
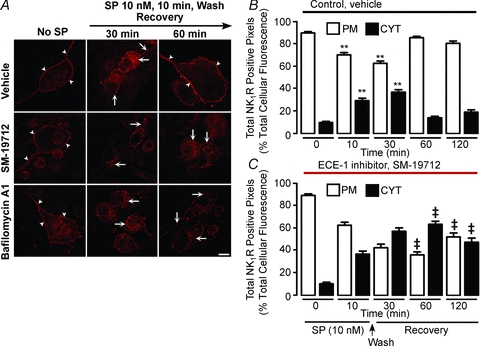
Organotypic preparations of mouse colon were unstimulated or stimulated with SP (10 nm, 10 min), washed and recovered for 30–120 min. NK1R was localized in whole-mounts of myenteric plexus by immunofluorescence (A) and pixel intensity at the plasma membrane (PM) and in the cytosol (CYT) was determined (B and C). In unstimulated neurones, NK1R-IR was at the plasma membrane (arrowheads). At 30 min recovery, NK1R-IR was detected in endosomes (arrows), and after 60 min recovery, NK1R-IR had recycled to the plasma membrane (arrowheads). SM-19712 did not affect endocytosis, but caused retention of the NK1R-IR in endosomes at 60 and 120 min (arrows), indicating inhibition of recycling. Bafilomycin A1 also caused retention of the NK1R in endosomes at 60 min recovery (arrows). Scale, 10 μm. **P < 0.005 to 0 min. ‡P < 0.005 to vehicle control. 25–84 neurones per group from n = 3 mice.
To evaluate whether ECE-1 regulates NK1R recycling after stimulation of neurones with physiologically relevant concentrations of SP, organotypic preparations of mouse colon were incubated with KCl (50 mm, 5 min, 37°C), which stimulates the release of endogenous SP (Grady et al. 1996b), or were unstimulated. Preparations were then washed, recovered in SP-free medium, and the NK1R was localized in whole-mounts of myenteric plexus. In unstimulated neurones, NK1R-IR was confined to the plasma membrane (89.8 ± 1.1% of total NK1R-IR pixels) (Fig. 9A and B). KCl induced trafficking of NK1R to endosomes at 30 min (37.2 ± 2.1% of total NK1R-IR pixels) and depletion from the plasma membrane. Preincubation of tissue with the NK1R antagonist RP-67580 abolished KCl-induced endocytosis of the NK1R (Fig. 9A), which is thus attributable to release of SP and activation of the NK1R. After 60 min recovery, the NK1R was detected at the plasma membrane (85.5 ± 1.2% of total NK1R-IR pixels), indicating recycling. SM-19712 did not affect NK1R endocytosis at 30 min, but caused retention of NK1R-IR in endosomes at 60 min. The proportion of NK1R-IR pixels in endosomes at 60 min recovery was 14.4 ± 1.2% in vehicle-treated neurones and 47.7%± 3.6% in SM-19712-treated neurones (Fig. 9A and B; P < 0.005, 60 min recovery, SM-19712 compared to vehicle).
Figure 9. ECE-1-mediated recycling of the NK1R in response to endogenous SP in myenteric neurones.
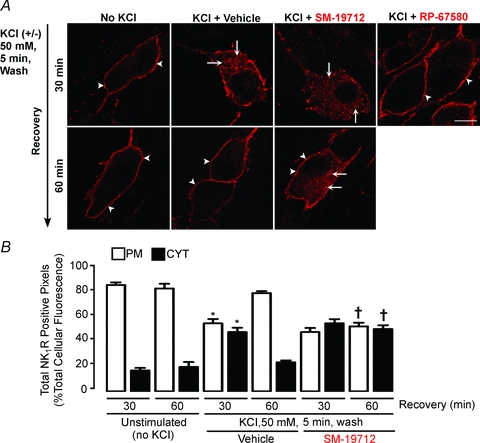
Organotypic preparations of mouse colon were unstimulated or stimulated with KCl (50 mm, 5 min), washed and recovered for 30 or 60 min. NK1R was localized in whole-mounts of myenteric plexus by immunofluorescence (A) and pixel intensity at the plasma membrane (PM) and within the cytosol (CYT) was determined (B). In unstimulated neurones, NK1R-IR remained at the plasma membrane (arrowheads). KCl stimulated trafficking to endosomes at 30 min (arrows), and recycling to the plasma membrane at 60 min (arrowheads). The ECE-1 inhibitor SM-19712 did not affect endocytosis at 30 min, but caused NK1R retention in endosomes (arrows), and thus suppressed recycling at 60 min. The NK1R antagonist RP-67580 prevented KCl-induced endocytosis at 30 min. Scale, 10 μm. *P < 0.05 compared to no KCl at 30 min; †P < 0.05 compared to KCl at 60 min. 12–39 neurones per group from n = 3 mice.
These results indicate that endocytic sorting of the NK1R to the plasma membrane requires ECE-1 activity in acidified endosomes, where ECE-1 can degrade SP to promote signalosome disassembly.
ECE-1 promotes NK1R resensitization in myenteric neurones
Although ECE-1-induced recycling of the NK1R is a major mechanism of resensitization of responses to SP in cell lines (Roosterman et al. 2007), the role of ECE-1 in NK1R resensitization in neurones is unknown. To assess desensitization and resensitization, myenteric neurones cultured from the mouse intestine were repeatedly challenged with SP (10 nm) and [Ca2+]i was measured by ratiometric imaging. Both the magnitude of the increase in [Ca2+]i and the proportion of the KCl-responsive neurones that also responded to SP were determined. SP (10 nm, 5 min) caused a rapid and transient increase in [Ca2+]i in 54 ± 6.4% of myenteric neurones (Fig. 10A). When the same neurones were re-challenged with SP (10 nm) 10 min after the initial receptor stimulation, SP signals were undetectable (Fig. 10A and C), indicating complete desensitization of the NK1R. These neurones maintained responsiveness to carbachol. Responses were partially recovered when the interval between SP challenges was 30 min (not shown). Both the magnitude of the response and the proportion of neurones with a detectable response were almost fully recovered when the interval between SP challenges was 60 min, indicating a high degree of resensitization (Fig. 10A and C). This resensitization coincided with recycling of NK1R-IR to the plasma membrane of cultured myenteric neurones (not shown). The ECE-1 inhibitor SM-19712 did not affect the initial increase in [Ca2+]i in response to SP, which was detected in 60.2 ± 6.1% of myenteric neurones, nor the subsequent desensitization of this response to a second SP challenge at 10 min (Fig. 10B and C). However, SM-19712 strongly inhibited resensitization after 60 min (% resensitized neurones compared to initial SP response (100%), vehicle control, 74 ± 20%; SM-19712, 10.8 ± 3.8%; P < 0.05). SM-19712 also inhibited recycling of NK1R-IR in cultured myenteric neurones (not shown). These data are consistent with a major role of ECE-1 in regulating resensitization of NK1R-mediated Ca2+ signalling in myenteric neurones. The NK1R antagonist RP-67580 (Garret et al. 1992) markedly inhibited responses to SP, which were detectable in only 7.9 ± 3.6% of cultured neurones (P < 0.005 compared to vehicle), indicating that SP responses are attributable to activation of the NK1R (Fig. 10D and E).
Figure 10. ECE-1-mediated resensitization of SP-induced Ca2+ transients in myenteric neurones.
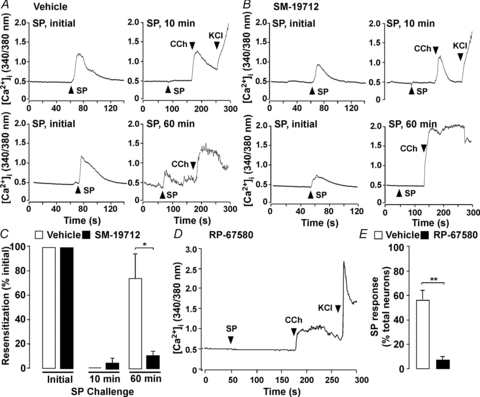
A–C, mouse myenteric neurones in culture were challenged with SP (initial, 10 nm, 5 min), washed, and the same neurones were re-challenged (10 nm SP) after 10 or 60 min recovery. After the final SP challenge, neurones were exposed to carbachol (CCh, 1 μm) and then KCl (50 mm). A and B, Ca2+ transients from the same neurones in each experiment (upper panel: 10 min re-challenge; lower panel: 60 min re-challenge) treated with vehicle (A) or SM-19712 (B). C, resensitization of neurones treated with vehicle or SM-19712. The proportion of neurones responsive to initial SP were recorded and are represented as 100% when compared to the proportion of the same neurones responsive to SP re-challenge. *P < 0.05 to vehicle, 46–102 neurones per group, n > 4 experiments (i.e. neuronal cultures from 3–5 mice per preparation). D and E, mouse myenteric neurones in culture were incubated with vehicle or the NK1R antagonist RP-67580, which suppressed detectable changes in [Ca2+]i. **P < 0.005 compared to vehicle, 149–176 neurones per group, n > 4 mice.
To examine whether SP selectively desensitizes the NK1R and not other receptors, we examined the responses of neurones to carbachol. Carbachol (1 μm) increased [Ca2+]i in myenteric neurones (Fig. 11A and B). The magnitude of the response to carbachol was unaffected by preincubation of neurones with SP (10 nm, 5 min), both in neurones that responded to SP and thus expressed the NK1R, and in non-SP responsive neurones that lacked detectable NK1R. In contrast, preincubation with SP caused marked desensitization of the NK1R at the same time point (Fig. 10). The response to carbachol was unaffected by SM-19712 (10 μm, continuous) compared to vehicle (control, Fig. 11A and B). Thus, preincubation with SP selectively desensitizes the NK1R but does not heterologously desensitize muscarinic receptors in myenteric neurones. ECE-1 does not regulate the responses of neurones to carbachol.
Figure 11. Responses of myenteric neurones to carbachol.
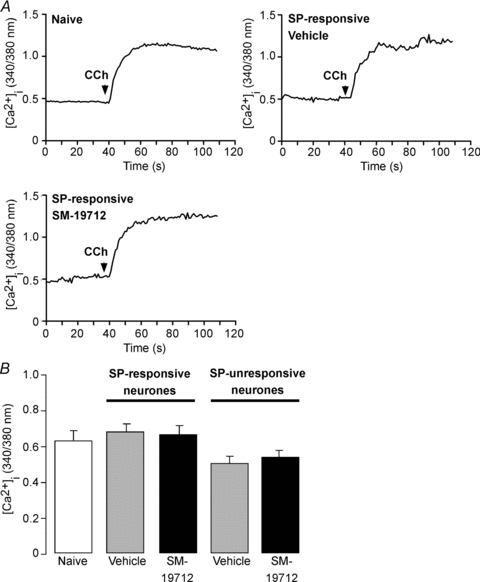
Mouse myenteric neurones in culture were challenged with carbachol (CCh, 1 μm), and Ca2+ transients were measured. A, Ca2+ transients to carbachol in naïve neurones (i.e. no prior stimulation) and in SP-responsive neurones preincubated with SP (10 nM, 5 min). SP-responsive neurones preincubated with SM-19712 (10 μm, throughout experiment) or vehicle (control). B, mean responses to carbachol in naïve neurones, in SP-responsive neurones preincubated with SP (10 nM, 5 min), and in non-SP responsive neurones lacking detectable NK1R. Both SP-responsive and non-responsive neurones preincubated with SM-19712 or vehicle control. 26–43 neurones per group, n = 3 experiments (i.e. neuronal cultures from 3–5 mice per preparation). Note that the response to carbachol was unaffected by preincubation with SP or SM-19712.
ECE-1 attenuates NK1R-mediated ERK1/2 activation in myenteric neurones
β-Arrestins assemble a SP–NK1R–β-arrestin–Src signalosome in endosomes that mediates G protein-independent activation of ERK1/2 (DeFea et al. 2000). The mechanisms that terminate β-arrestin-mediated MAPK signalling in neuronal endosomes are unknown. We examined whether ECE-1, by degrading SP and destabilizing the signalosome, could attenuate MAPK signalling. To enable sensitive detection of ERK1/2 activation at the cellular level, we used a PLA with primary antibodies to ERK1/2 and pERK1/2. Rat myenteric neurones in culture were stimulated with PDBu (100 nm) or SP (10 nm) and pERK1/2:ERK1/2 PLA signals were quantified. In unstimulated neurones there were few signals by PLA. PDBu (positive control) stimulated a 4-fold increase in PLA signals within the cytosol and nucleus within 10 min, indicating formation of a pERK1/2 and ERK1/2 complex and ERK1/2 activation (Fig. 12A and B). SP also stimulated a 4-fold increase in PLA signals within 10–30 min that declined after 120 min (Fig. 12A and B). The ECE-1 inhibitor SM-19712 did not affect the magnitude of the early phases of SP-induced ERK1/2 activation, but at 120 min, the response was completely maintained and >2-fold higher than in vehicle-treated preparation (Fig. 12A and B). Omission of primary antibodies abolished PLA signals (not shown). These results are consistent with SM-19712 causing endosomal retention of NK1R and suggest that endosomal ECE-1, by destabilizing the β-arrestin signalosome, attenuates SP-induced ERK1/2 activation.
Figure 12. ECE-1 attenuation of SP-induced ERK1/2 signals in myenteric neurones.
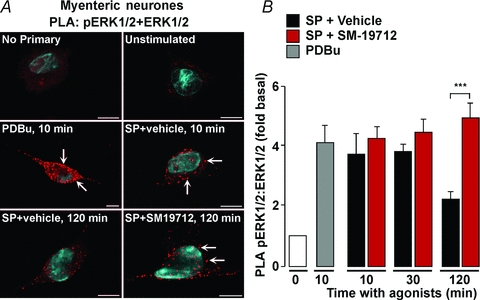
Rat myenteric neurones in culture were challenged with PDBu (100 nm) or SP (10 nm), and neurones were processed for PLA for ERK1/2 and pERK1/2. PDBu and SP strongly induced PLA signals (in red) within 10–30 min, indicative of activation of pERK1/2 (arrows). SP-induced PLA signals were attenuated after 120 min. The ECE-1 inhibitor SM-19712 did not affect the initial increase in PLA signals, but caused sustained signals at 120 min, indicative of prolonged activation of pERK1/2. The blue stain in A (DAPI) denotes the nucleus. ***P < 0.001. n > 3 experiments (i.e. neuronal cultures from 1 rat per preparation).
Discussion
In contrast to the extensively studied mechanisms that control G protein signalling and trafficking of neuropeptide receptors at the plasma membrane, very little is known about the control of receptor signalling and trafficking in endosomes. Cell-surface peptidases that degrade neuropeptides in the extracellular fluid, and β-arrestins, which uncouple receptors from G proteins and couple receptors to the endocytic machinery, rapidly attenuate plasma membrane signalling. Our results suggest that an analogous proteolytic mechanism regulates the association of the NK1R with β-arrestins in endosomes, and thereby controls the post-endocytic trafficking and signalling of the NK1R in enteric neurones. We observed that neurones of the myenteric plexus of the intestine that express the NK1R also express ECE-1 isoforms within endosomes. SP and the NK1R traffic to endosomes containing ECE-1, where we have previously reported that ECE-1 can degrade neuropeptides, including SP, in the acidic endosomal environment (Padilla et al. 2007; Roosterman et al. 2007, 2008). Inhibition of ECE-1 caused retention of the NK1R in endosomes, preventing recycling back to the plasma membrane and inhibiting resensitization of plasma membrane signalling. ECE-1 inhibition also resulted in markedly sustained activation of ERK1/2, presumably by a β-arrestin-mediated, endosomal mechanism. These observations reveal a new proteolytic mechanism that controls endosomal signalling and trafficking of the NK1R in myenteric neurones.
ECE-1 is expressed in the enteric nervous system
We amplified mRNA that encodes multiple isoforms of ECE-1 (ECE-1a, ECE-1b, ECE-1c1 and ECE-1c2) from ME-MP of the mouse intestine by RT-PCR, and confirmed ECE-1 expression and localization to endosomes using an antibody that detects ECE-1b/d isoforms. When expressed in cell lines, ECE-1b/d isoforms are most prominently localized to early endosomes and ECE-1a/c isoforms are most prominently present at the plasma membrane (Padilla et al. 2007; Roosterman et al. 2007). However, all isoforms are found in endosomes and constitutively internalize and possibly recycle via the endosomal network. These results support other reports of ECE-1 expression in myenteric neurones of rats (Cottrell et al. 2009), and are consistent with the detection of ECE-1 substrates (big-endothelin) and products (endothelin-1) in enteric neurones of humans (Escrig et al. 1992). ECE-1-IR has also been detected in endothelial, epithelial, smooth muscle and neuroendocrine cells of the intestine, and in neurones of sympathetic ganglia, indicating widespread expression (Hsu & Huang, 2003). Although we did not attempt to classify the subtypes of myenteric neurones that express ECE-1, ECE-1-IR was detected in neurones expressing the NK1R, as well as other cell types that express this receptor, including interstitial cells of Cajal. Further studies will be necessary to identify those classes of enteric neurones that express ECE-1 by neurochemical coding analyses.
ECE-1 promotes recycling and resensitization of NK1R signalling at the plasma membrane of myenteric neurones
Although several studies have reported that SP stimulates endocytosis and recycling of the NK1R in enteric neurones (Grady et al. 1996b; McConalogue et al. 1998; Southwell et al. 1998; Mann et al. 1999), the mechanism and function of this trafficking have not been defined. We observed that brief exposure of neurones to SP resulted in translocation of β-arrestins to the plasma membrane and then to endosomes, where β-arrestins colocalized with the NK1R. By using a PLA that is sensitive in detecting protein–protein interactions at the subcellular level, we observed interaction of the NK1R and β-arrestins in SP-stimulated neurones. These results support the well-defined role of β-arrestins in mediating desensitization and endocytosis of the NK1R in cell lines (McConalogue et al. 1999; DeFea et al. 2000). Moreover, NK1R–β-arrestin interactions in myenteric neurones coincided with desensitization of SP-mediated Ca2+ signalling, which is probably mediated by β-arrestins. The dynamin GTPase inhibitor Dynasore (Macia et al. 2006) abolished SP-stimulated endocytosis of the NK1R in myenteric neurones, consistent with clathrin- and dynamin-mediated endocytosis (Grady et al. 1995, 1996b; Schmidlin et al. 2001).
In myenteric neurones with active ECE-1, the internalized NK1R recycled from early endosomes to the plasma membrane, and recycling coincided with resensitization of G protein-mediated NK1R signalling at the plasma membrane, assessed by measurement of SP-stimulated Ca2+ transients. The inhibition of endosomal ECE-1 by several approaches blocked NK1R recycling and resensitization, indicating a major role for ECE-1 in these processes. Two distinct ECE-1 inhibitors, SM-19712 (Umekawa et al. 2000) and PD-069185 (Ahn et al. 1998), inhibited these events. Bafilomycin A1, a vacuolar H+ATPase inhibitor that prevents endosomal acidification and suppresses ECE-1 degradation of SP in endosomes, which only occurs at acidic endosomal pH (Roosterman et al. 2007), also inhibited recycling. A limitation of this approach is the reliance on pharmacological tools to block ECE-1 activity, since we could not efficiently transfect myenteric neurones with ECE-1 cDNA or siRNA. However, we have previously reported that whereas overexpression of ECE-1 accelerates receptor recycling and resensitization, ECE-1 knockdown has the opposite effects (Padilla et al. 2007; Roosterman et al. 2007). In the present study we observed that ECE-1 inhibition prevented NK1R recycling after release of endogenous SP by K+ depolarization, suggesting that this mechanism is physiologically relevant.
We did not directly determine the mechanism by which ECE-1 promotes recycling and resensitization of the NK1R in myenteric neurones, due to difficulty in examining neuropeptide degradation in myenteric neurones. In cell lines, ECE-1 makes a major contribution to the degradation of internalized neuropeptides, including SP, calcitonin gene-related peptide and somatostatin-14, whereas ECE-1 inhibitors and bafilomycin A1 prevent this degradation (Padilla et al. 2007; Roosterman et al. 2007, 2008). Moreover, ECE-1 degrades these neuropeptides only at endosomal pH 5.5 and not at extracellular pH 7.4. Thus, a likely explanation of the effects of ECE-1 inhibition on NK1R recycling and resensitization is suppressed degradation of SP in endosomes, and consequent stabilization of the endosomal SP–NK1R–β-arrestin signalosome, which traps the NK1R in endosomes. Dissociation of β-arrestins and receptor recycling is necessary for resensitization of other 7TMD receptors (Oakley et al. 1999), including the NK1R (Schmidlin et al. 2001; Roosterman et al. 2004).
We observed that the NK1R is only partially internalized at a time when SP-induced Ca2+ signalling is fully desensitized (10 min). Thus, endocytosis does not mediate desensitization, which is likely to depend on NK1R phosphorylation by G protein-coupled receptor kinases and interaction with β-arrestins. Although our results suggest that ECE-1-dependent NK1R recycling is a major mechanism for resensitization, we cannot exclude the possibility that surface-retained receptors could resensitize without the requirement for internalization. In support of this possibility, we have recently reported a role for protein phosphatase 2A in resensitization of non-internalized NK1R (Murphy et al. 2011). This mechanism proposes that some NK1Rs are phosphorylated at the plasma membrane and desensitized by a second messenger kinase such as protein kinase C. These receptors neither interact with β-arrestins nor internalize. Protein phosphatase 2A translocates to the plasma membrane and dephosphorylates the surface-retained NK1Rs to mediate resensitization. Further studies are required to determine the contribution of protein phosphatase 2A to resensitization of the NK1R in enteric neurones.
ECE-1 terminates endosomal signalling of the NK1R in myenteric neurones
There is a growing realization that 7TMD receptors can continue to signal from endosomes by mechanisms that are distinct from those operating at the plasma membrane, and that β-arrestins play a critical role in these processes (Murphy et al. 2009). Whereas β-arrestins terminate G protein signalling at the plasma membrane, they recruit receptors and signalling partners to signalosomes, and thereby transmit a second wave of G protein independent signals. SP interacts with Src and induces assembly of a SP–NK1R–β-arrestin–Src signalosome that mediates sustained ERK1/2 signalling (DeFea et al. 2000). We observed that SP stimulated ERK1/2 activation in myenteric neurones, and that an ECE-1 inhibitor caused markedly sustained ERK1/2 signalling, probably by stabilizing the signalosome. A limitation of our approach is that we could not directly assess the contribution of β-arrestins to this signalling pathway due to difficulties in expressing β-arrestin siRNA in enteric neurones. However, disruption of β-arrestins in cell lines attenuates SP-stimulated ERK1/2 activation (DeFea et al. 2000), and ECE-1 inhibition with SM-19712 or bafilomycin A1 stabilizes the signalosome and prolonged ERK1/2 signalling in model cell systems (Cottrell et al. 2009).
Cell-surface membrane metallo-endopeptidases, such as neprilysin, that degrade neuropeptides in the extracellular fluid, protect against sustained neuropeptide signalling. Neprilysin deletion or inhibition results in plasma extravasation and exacerbated inflammation due to the impaired degradation and amplified signalling of the proinflammatory neuropeptides SP and bradykinin (Lu et al. 1997; Sturiale et al. 1999). ECE-1 may similarly protect against uncontrolled signalling of receptors in endosomes. SP can induce neurotoxicity by β-arrestin-, Raf- and ERK1/2-mediated activation of Nur77, a mediator of cell death (Castro-Obregon et al. 2004). From studies of cell lines, we reported that ECE-1 inhibitors cause remarkably sustained SP-induced activation of Nur77, which results in cell death (Cottrell et al. 2009). By terminating SP signalling in endosomes and attenuating activation of ERK1/2, ECE-1 may also protect against the SP-induced neurotoxicity in the enteric nervous system.
Physiological functions of ECE-1 in the enteric nervous system
ECE-1 is best known for its role in the extracellular and intracellular conversion of big endothelin to endothelin-1, which plays a major role in regulating blood pressure and sodium homeostasis (Kohan et al. 2011). ECE-1 can also generate endothelins in the enteric nervous system, which may control motility and secretion (Escrig et al. 1992). The prominent detection of ECE-1 in endosomes, and the realization that ECE-1 can degrade certain neuropeptides in endosomes and thereby regulate receptor trafficking and signalling (Padilla et al. 2007; Roosterman et al. 2007, 2008; Cattaruzza et al. 2009; Cottrell et al. 2009), have revealed new functions for endosomal ECE-1. By promoting recycling and resensitization of the NK1R in myenteric neurones, ECE-1 may allow the peristaltic reflex to be maintained. SP and the NK1R mediate ascending contraction during peristalsis, when intestinal distension triggers the release of SP and orad contraction of smooth muscle (Grider, 2003). Given that the contractile effects of SP in the intestine rapidly desensitize to repeated challenge (Gaddum, 1953), resensitization, possibly mediated by ECE-1-dependent NK1R recycling, may permit the maintenance of peristalsis, which is essential for normal digestion. It is also possible that ECE-1 protects against sustained β-arrestin-mediated signalling of the NK1R, which could cause neurotoxicity (Castro-Obregon et al. 2004). The endothelin system plays an essential role in the development of neural crest-derived tissues, including enteric neurones. Enteric neurones are absent from mice deficient in ECE-1, recapitulating the effects of deletion of the endothelin B receptor or endothelin-3 (Yanagisawa et al. 1998). Although these findings indicate that ECE-1-mediated processing of endothelin and activation of endothelin B receptors is required for normal development of enteric neurones, the absence of ECE-1 may also reveal unexpected toxic effects of neuropeptides in the enteric nervous system.
In addition to SP, endosomal ECE-1 can regulate endosomal trafficking of receptors for other neuropeptides that are ECE-1 substrates at endosomal pH, including calcitonin gene-related peptide and somatostatin-14, which control numerous gastrointestinal functions, including peristalsis (Grider, 2003). Additional studies are required to delineate the other functions of ECE-1 in the enteric nervous system that may be related to the control of endocytic trafficking and signalling of various neuropeptide receptors. Whether ECE-1 deficiency contributes to abnormal functions of the enteric nervous system due to abnormal endosomal signalling remains to be determined.
Acknowledgments
This work was supported by NIH grants DK39957, DK57850, DK43207 (N.W.B.), DK07573 (J.-C.P.), AR059402 (M.S.), CJ Martin Fellowship NHMRC 454858 (D.P.P.), and the British Heart Foundation FS/08/017/25027 (G.S.C.).
Glossary
Abbreviations
- 7TMD
7 transmembrane domain
- DMEM
Dulbecco's modified Eagle's medium
- ECE-1
endothelin-converting enzyme-1
- EEA1
early endosomal antigen 1
- ERK
extracellular signal-regulated kinase
- FBS
fetal bovine serum
- HBSS
Hank's balanced salt solution
- IR
immunoreactivity
- MAPK
mitogen-activated protein kinase
- ME-MP
muscularis externa myenteric plexus
- NK1R
neurokinin 1 receptor
- NHS
normal horse serum
- PDBu
phorbol 12,13-dibutyrate
- PLA
proximity ligation assay
- PSS
physiological salt solution
- SP
substance P
Author's present address
N. Bunnett: NHMRC Australia Fellow, Professor of Pharmacology and Medicine, Monash Institute of Pharmaceutical Sciences, 381 Royal Parade, Parkville, VIC 3052, Australia.
Author contributions
J.-C.P. and D.P.P. designed, performed and analysed experiments and wrote the manuscript; G.S.C. performed experiments and provided critical scientific input; M.S. provided critical scientific input; N.W.B. designed and supervised the study and wrote the manuscript. All authors approved the final version.
References
- Ahn K, Sisneros AM, Herman SB, Pan SM, Hupe D, Lee C, et al. Novel selective quinazoline inhibitors of endothelin converting enzyme-1. Biochem Biophys Res Commun. 1998;243:184–190. doi: 10.1006/bbrc.1998.8081. [DOI] [PubMed] [Google Scholar]
- Allalou A, Wahlby C. BlobFinder, a tool for fluorescence microscopy image cytometry. Comput Methods Programs Biomed. 2009;94:58–65. doi: 10.1016/j.cmpb.2008.08.006. [DOI] [PubMed] [Google Scholar]
- Bunnett NW, Dazin PF, Payan DG, Grady EF. Characterization of receptors using cyanine 3-labeled neuropeptides. Peptides. 1995;16:733–740. doi: 10.1016/0196-9781(95)00042-i. [DOI] [PubMed] [Google Scholar]
- Castro-Obregon S, Rao RV, del Rio G, Chen SF, Poksay KS, Rabizadeh S, et al. Alternative, nonapoptotic programmed cell death: mediation by arrestin 2, ERK2, and Nur77. J Biol Chem. 2004;279:17543–17553. doi: 10.1074/jbc.M312363200. [DOI] [PubMed] [Google Scholar]
- Cattaruzza F, Cottrell GS, Vaksman N, Bunnett NW. Endothelin-converting enzyme 1 promotes re-sensitization of neurokinin 1 receptor-dependent neurogenic inflammation. Br J Pharmacol. 2009;156:730–739. doi: 10.1111/j.1476-5381.2008.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell GS, Padilla BE, Amadesi S, Poole DP, Murphy JE, Hardt M, et al. Endosomal endothelin-converting enzyme-1: a regulator ofβ-arrestin-dependent ERK signaling. J Biol Chem. 2009;284:22411–22425. doi: 10.1074/jbc.M109.026674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFea KA, Vaughn ZD, O'Bryan EM, Nishijima D, Dery O, Bunnett NW. The proliferative and antiapoptotic effects of substance P are facilitated by formation of aβ-arrestin-dependent scaffolding complex. Proc Natl Acad Sci U S A. 2000;97:11086–11091. doi: 10.1073/pnas.190276697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escrig C, Bishop AE, Inagaki H, Moscoso G, Takahashi K, Varndell IM, et al. Localisation of endothelin like immunoreactivity in adult and developing human gut. Gut. 1992;33:212–217. doi: 10.1136/gut.33.2.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaddum JH. Tryptamine receptors. J Physiol. 1953;119:363–368. doi: 10.1113/jphysiol.1953.sp004851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107–144. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- Garret C, Carruette A, Fardin V, Moussaoui S, Peyronel JF, Blanchard JC, Laduron PM. RP 67580, a potent and selective substance P non-peptide antagonist (in French) C R Acad Sci III. 1992;314:199–204. [PubMed] [Google Scholar]
- Grady EF, Baluk P, Bohm S, Gamp PD, Wong H, Payan DG, et al. Characterization of antisera specific to NK1, NK2, and NK3 neurokinin receptors and their utilization to localize receptors in the rat gastrointestinal tract. J Neurosci. 1996a;16:6975–6986. doi: 10.1523/JNEUROSCI.16-21-06975.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady EF, Gamp PD, Jones E, Baluk P, McDonald DM, Payan DG, Bunnett NW. Endocytosis and recycling of neurokinin 1 receptors in enteric neurones. Neuroscience. 1996b;75:1239–1254. doi: 10.1016/0306-4522(96)00357-0. [DOI] [PubMed] [Google Scholar]
- Grady EF, Garland AM, Gamp PD, Lovett M, Payan DG, Bunnett NW. Delineation of the endocytic pathway of substance P and its seven-transmembrane domain NK1 receptor. Mol Biol Cell. 1995;6:509–524. doi: 10.1091/mbc.6.5.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grider JR. Neurotransmitters mediating the intestinal peristaltic reflex in the mouse. J Pharmacol Exp Ther. 2003;307:460–467. doi: 10.1124/jpet.103.053512. [DOI] [PubMed] [Google Scholar]
- Hsu YH, Huang SC. Immunohistochemical localization of endothelin-converting enzyme-1 in neuroendocrine tumors and normal human tissue. Kaohsiung J Med Sci. 2003;19:555–562. doi: 10.1016/S1607-551X(09)70506-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohan DE, Rossi NF, Inscho EW, Pollock DM. Regulation of blood pressure and salt homeostasis by endothelin. Physiol Rev. 2011;91:1–77. doi: 10.1152/physrev.00060.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Figini M, Emanueli C, Geppetti P, Grady EF, Gerard NP, et al. The control of microvascular permeability and blood pressure by neutral endopeptidase. Nat Med. 1997;3:904–907. doi: 10.1038/nm0897-904. [DOI] [PubMed] [Google Scholar]
- McConalogue K, Corvera CU, Gamp PD, Grady EF, Bunnett NW. Desensitization of the neurokinin-1 receptor (NK1-R) in neurones: effects of substance P on the distribution of NK1-R, Gaq/11, G-protein receptor kinase-2/3, and β-arrestin-1/2. Mol Biol Cell. 1998;9:2305–2324. doi: 10.1091/mbc.9.8.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConalogue K, Dery O, Lovett M, Wong H, Walsh JH, Grady EF, Bunnett NW. Substance P-induced trafficking ofβ-arrestins. The role of β-arrestins in endocytosis of the neurokinin-1 receptor. J Biol Chem. 1999;274:16257–16268. doi: 10.1074/jbc.274.23.16257. [DOI] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Mann PT, Southwell BR, Furness JB. Internalization of the neurokinin 1 receptor in rat myenteric neurones. Neuroscience. 1999;91:353–362. doi: 10.1016/s0306-4522(98)00595-8. [DOI] [PubMed] [Google Scholar]
- Mantyh PW, DeMaster E, Malhotra A, Ghilardi JR, Rogers SD, et al. Receptor endocytosis and dendrite reshaping in spinal neurones after somatosensory stimulation. Science. 1995;268:1629–1632. doi: 10.1126/science.7539937. [DOI] [PubMed] [Google Scholar]
- Matsumura Y, Kuro T, Kobayashi Y, Umekawa K, Ohashi N, Takaoka M. Protective effect of SM-19712, a novel and potent endothelin converting enzyme inhibitor, on ischemic acute renal failure in rats. Jpn J Pharmacol. 2000;84:16–24. doi: 10.1254/jjp.84.16. [DOI] [PubMed] [Google Scholar]
- Murphy JE, Padilla BE, Hasdemir B, Cottrell GS, Bunnett NW. Endosomes: a legitimate platform for the signaling train. Proc Natl Acad Sci U S A. 2009;106:17615–17622. doi: 10.1073/pnas.0906541106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JE, Roosterman D, Cottrell GS, Padilla BE, Feld M, Brand E, et al. Protein phosphatase 2a mediates resensitization of the neurokinin 1 receptor. Am J Physiol Cell Physiol. 2011 doi: 10.1152/ajpcell.00096.2011. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Association ofβ-arrestin with G protein-coupled receptors during clathrin-mediated endocytosis dictates the profile of receptor resensitization. J Biol Chem. 1999;274:32248–32257. doi: 10.1074/jbc.274.45.32248. [DOI] [PubMed] [Google Scholar]
- Okamoto A, Lovett M, Payan DG, Bunnett NW. Interactions between neutral endopeptidase (EC 3.4.24.11) and the substance P (NK1) receptor expressed in mammalian cells. Biochem J. 1994;299:683–693. doi: 10.1042/bj2990683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla BE, Cottrell GS, Roosterman D, Pikios S, Muller L, Steinhoff M, Bunnett NW. Endothelin-converting enzyme-1 regulates endosomal sorting of calcitonin receptor-like receptor andβ-arrestins. J Cell Biol. 2007;179:981–997. doi: 10.1083/jcb.200704053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole DP, Castelucci P, Robbins HL, Chiocchetti R, Furness JB. The distribution of P2X3 purine receptor subunits in the guinea pig enteric nervous system. Auton Neurosci. 2002;101:39–47. doi: 10.1016/s1566-0702(02)00179-0. [DOI] [PubMed] [Google Scholar]
- Poole DP, Matsuyama H, Nguyen TV, Eriksson EM, Fowler CJ, Furness JB. Inflammation and inflammatory agents activate protein kinase Cɛ translocation and excite guinea-pig submucosal neurones. Gastroenterology. 2007;133:1229–1239. doi: 10.1053/j.gastro.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Roosterman D, Cottrell GS, Padilla BE, Muller L, Eckman CB, Bunnett NW, Steinhoff M. Endothelin-converting enzyme 1 degrades neuropeptides in endosomes to control receptor recycling. Proc Natl Acad Sci U S A. 2007;104:11838–11843. doi: 10.1073/pnas.0701910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roosterman D, Cottrell GS, Schmidlin F, Steinhoff M, Bunnett NW. Recycling and resensitization of the neurokinin 1 receptorInfluence of agonist concentration and Rab GTPases. J Biol Chem. 2004;279:30670–30679. doi: 10.1074/jbc.M402479200. [DOI] [PubMed] [Google Scholar]
- Roosterman D, Kempkes C, Cottrell GS, Padilla BE, Bunnett NW, Turck CW, Steinhoff M. Endothelin-converting enzyme-1 degrades internalized somatostatin-14. Endocrinology. 2008;149:2200–2207. doi: 10.1210/en.2007-1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidlin F, Dery O, DeFea KO, Slice L, Patierno S, Sternini C, et al. Dynamin and Rab5a-dependent trafficking and signaling of the neurokinin 1 receptor. J Biol Chem. 2001;276:25427–25437. doi: 10.1074/jbc.M101688200. [DOI] [PubMed] [Google Scholar]
- Soderberg O, Leuchowius KJ, Gullberg M, Jarvius M, Weibrecht I, Larsson LG, Landegren U. Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods. 2008;45:227–232. doi: 10.1016/j.ymeth.2008.06.014. [DOI] [PubMed] [Google Scholar]
- Southwell BR, Seybold VS, Woodman HL, Jenkinson KM, Furness JB. Quantitation of neurokinin 1 receptor internalization and recycling in guinea-pig myenteric neurones. Neuroscience. 1998;87:925–931. doi: 10.1016/s0306-4522(98)00176-6. [DOI] [PubMed] [Google Scholar]
- Sternini C, Su D, Gamp PD, Bunnett NW. Cellular sites of expression of the neurokinin-1 receptor in the rat gastrointestinal tract. J Comp Neurol. 1995;358:531–540. doi: 10.1002/cne.903580406. [DOI] [PubMed] [Google Scholar]
- Sturiale S, Barbara G, Qiu B, Figini M, Geppetti P, Gerard N, et al. Neutral endopeptidase (EC 3.4.24.11) terminates colitis by degrading substance P. Proc Natl Acad Sci U S A. 1999;96:11653–11658. doi: 10.1073/pnas.96.20.11653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umekawa K, Hasegawa H, Tsutsumi Y, Sato K, Matsumura Y, Ohashi N. Pharmacological characterization of a novel sulfonylureid-pyrazole derivative, SM-19712, a potent nonpeptidic inhibitor of endothelin converting enzyme. Jpn J Pharmacol. 2000;84:7–15. doi: 10.1254/jjp.84.7. [DOI] [PubMed] [Google Scholar]
- Yanagisawa H, Yanagisawa M, Kapur RP, Richardson JA, Williams SC, Clouthier DE, et al. Dual genetic pathways of endothelin-mediated intercellular signaling revealed by targeted disruption of endothelin converting enzyme-1 gene. Development. 1998;125:825–836. doi: 10.1242/dev.125.5.825. [DOI] [PubMed] [Google Scholar]