

Non-technical summary
The body adjusts the level of sympathetic nervous system activation to regulate blood pressure in health and disease. Therefore, knowing the level of sympathetic activation is important to treat patients with abnormal sympathetic nervous system function. Directly measuring sympathetic activation is difficult, however, and not possible in most clinical settings. Previous investigators have shown that the variability in blood pressure increases during sympathetic activation. To determine if this is a good non-invasive index of sympathetic activation, we compared blood pressure variability to the direct measurement of sympathetic activation in healthy humans. We activated the sympathetic nervous system by redistributing blood volume away from the head and brain, which also occurs with prolonged standing or haemorrhage. We show that the relationship between variability in blood pressure and sympathetic activation is not the same in all individuals, indicating that blood pressure variability cannot be reliably used to measure sympathetic activation.
Abstract
Abstract
The spectral power of low frequency oscillations of systolic arterial pressure (SAPLF) has been used as a non-invasive surrogate of muscle sympathetic nerve activity (MSNA) in both experimental and clinical situations. For SAPLF to be used in this way, a relationship must exist between SAPLF and MSNA within individuals during sympathetic activation. Using progressive central hypovolaemia to induce sympathetic activation, we hypothesised that SAPLF would correlate with MSNA in all subjects. ECG, beat-by-beat arterial pressure and MSNA were recorded in humans (n = 20) during a progressive lower body negative pressure (LBNP) protocol designed to cause presyncope in all subjects. Arterial pressure oscillations were assessed in the low frequency (LF; 0.04–0.15 Hz) domain using a Fourier transform. For the entire group, SAPLF, MSNA burst frequency, and total MSNA increased during LBNP. Values for coefficients of determination (r2) describing the linear associations of SAPLF with MSNA burst frequency and total MSNA were 0.73 and 0.84, but rose to 0.89 and 0.98 when curvilinear fits were used, indicating that the relationship is curvilinear rather than linear. Associations between SAPLF and MSNA within each individual subject, however, varied widely for both MSNA burst frequency and total MSNA, whether derived by linear (r2 range, 1.7 × 10−6 to 0.99) or polynomial (r2 range, 0.09 to 1.0) regression analysis. Similar results were obtained when relationships between low frequency oscillations in diastolic arterial pressure and MSNA were evaluated. These results do not support the use of low frequency oscillations in arterial pressure as a non-invasive measure of sympathetic outflow for individual subjects during sympathetic activation.
Introduction
Using haemorrhage as a stimulus, Guyton and Harris demonstrated in 1951 that profound sympathetic activation elicited increases in oscillations of the blood pressure waveform in dogs (Guyton & Harris, 1951). Because these oscillations were attenuated by sinoaortic denervation, the authors suggested that the oscillations were dependent on baroreflex activation of the sympathetic nervous system. Reports later emerged confirming that sympathetic activation induced by a variety of experimental perturbations was associated with increases in the amplitude of low frequency blood pressure oscillations in humans, while perturbations producing decreases in sympathetic activation decreased the amplitude of these oscillations (Malliani et al. 1991; Julien, 2006). These so-called Mayer waves occur at a frequency of about 0.1 Hz and may be produced by either the oscillatory activity of centrally located pacemakers controlling sympathetic nerve function or by a resonance within the baroreflex loop (Julien, 2006).
The application of power spectral techniques to interrogate physiological oscillatory patterns led to the quantification of the amplitude of these Mayer waves as systolic arterial pressure oscillations in the low frequency range (SAPLF; 0.04–0.15 Hz). The hypothesis that SAPLF could be used as a non-invasive surrogate for sympathetic vasomotor tone and nerve activation was first advanced by Pagani et al. (1986), who showed that SAPLF increased during head-up tilt in humans. SAPLF was subsequently shown to be increased during mental stress (Pagani et al. 1989) and daytime ambulatory activity (Furlan et al. 1990), both states characterised by sympathetic activation. While direct measurement of sympathetic activity was not performed in these early studies, linear relationships between SAPLF and muscle sympathetic nerve activity (MSNA) were later identified in human subjects during sympathetic activation induced by sodium nitroprusside infusion (Pagani et al. 1997) and head-up tilt (Furlan et al. 2000). On the basis of such observations, SAPLF has been used as a non-invasive metric for the determination of autonomic abnormalities in disease states such as hypertension (Lucini et al. 2002) and Type 1 diabetes (Lucini et al. 2009).
However, this viewpoint has been controversial (Parati et al. 1995, 2006; Malpas, 1998; Taylor & Studinger, 2006). Taylor et al. (1998) demonstrated in humans that resting levels of sympathetic activation vary across individuals of different ages and sexes, and that SAPLF does not correlate with these differences. Furthermore, results from animal studies suggest that SAPLF is more closely related to low frequency oscillatory patterns in sympathetic nerve activity (i.e. MSNALF) than to the absolute level of sympathetic nerve activation (Persson et al. 1992; Stauss et al. 1995). Sympathetic activation is also not accompanied by an increase in SAPLF under all physiological (e.g. heat stress: Cui et al. 2004) or pathophysiological (e.g. congestive heart failure: Radaelli et al. 1999) conditions.
Over the past five years, we have created a large database of human experiments (>170) using a protocol of lower body negative pressure (LBNP) that produces central hypovolaemia to the point of haemodynamic decompensation (i.e. presyncope). In the course of this work, we have performed microneurography in a subset of subjects to directly measure MSNA while simultaneously measuring beat-to-beat continuous blood pressure. Because these experiments uniquely provide multiple levels of MSNA in the same subject, we therefore used these data to test the hypothesis that SAPLF directly tracks increases in MSNA and MSNALF in all individuals during sympathetic activation induced by progressive central hypovolaemia.
Methods
Ethical approval
This study was conducted under a protocol reviewed and approved by the Brooke Army Medical Center Institutional Review Board and the US Army Medical Research and Materiel Command Institutional Review Board, and was conducted at the US Army Institute of Surgical Research, Fort Sam Houston, TX in accordance with the approved protocol and with the tenets of the Declaration of Helsinki. Subjects signed a written informed consent document before experimentation.
Subjects
MSNA recordings were maintained throughout LBNP to either the point of presyncope or to a high (–80 mmHg) LBNP level without a shift in baseline (thereby ensuring that electrode position had not changed) in 20 subjects (16 males and 4 females; age 30 ± 9 years; height 176 ± 10 cm; weight 81 ± 11 kg; means ± standard deviation). We have previously used data from subsets of these subjects to investigate relationships between MSNA and heart rate variability metrics during LBNP (Cooke et al. 2008) and to understand MSNA responses at the onset of presyncope (Cooke et al. 2009). All subjects first completed a medical history and physical examination by a physician to ensure that they had no previous or current medical conditions that would preclude their participation. Female subjects were not pregnant, as confirmed by a urine test performed within 24 h before experimentation. Subjects maintained their normal sleep patterns, refrained from exercise, and abstained from caffeine and other autonomic stimulants at least 24 h before the experiment. They received a verbal briefing along with written descriptions of all procedures and risks associated with the study, and were made familiar with the laboratory, the experimental protocol and all procedures. Subjects were encouraged to ask questions of the investigators and then signed an informed consent form.
Experimental protocol
Subjects were instrumented with a standard four-lead ECG to record cardiac electrical potentials, and an inflatable finger cuff to record beat-by-beat finger arterial pressure (Finometer Blood Pressure Monitor; TNO-TPD Biomedical Instrumentation, Amsterdam, The Netherlands). End-tidal CO2 was monitored on a breath-by-breath basis as subjects exhaled into a face mask (BCI Capnocheck Plus, Smiths Medical, Waukesha, WI, USA), and was used to derive respiratory rate. Multifibre recordings of MSNA were made directly from the peroneal nerve using the microneurography technique (Hagbarth & Vallbo, 1968). Nerve signals were band-pass filtered (100–2000 Hz) and integrated (time constant, 0.1 s) to obtain mean voltage neurograms.
Central hypovolaemia was induced by application of LBNP (Cooke et al. 2004). Subjects were positioned supine within an airtight chamber that was sealed at the level of the iliac crest by way of a neoprene skirt. Each subject was brought to the point of presyncope using progressive LBNP. The LBNP protocol consisted of a 5 min control period (baseline), followed by 5 min of chamber decompression at −15, −30, −45 and −60 mmHg, then additional increments of −10 mmHg every 5 min until the onset of haemodynamic decompensation (i.e. presyncope). Haemodynamic decompensation was identified by the attending investigator by a precipitous fall in systolic pressure greater than 15 mmHg, progressive diminution of systolic pressure to less than 70 mmHg, bradycardia, and/or voluntary subject termination caused by symptoms such as grey-out (loss of colour vision), tunnel vision, sweating, nausea, or dizziness. All experiments were conducted at room temperature (21.7–24.4°C) and ambient temperature did not change during the experiment (23.05 ± 0.19°C at baseline, 23.09 ± 0.17°C at recovery; P = 0.85).
Data analysis
Data were sampled at 500 Hz and digitized to computer (WINDAQ; Dataq Instruments, Akron, OH, USA). Using commercially available data analysis software (WinCPRS, Absolute Aliens, Turku, Finland), R-waves from the ECG as well as systolic (SAP) and diastolic (DAP) arterial pressures generated from the Finometer were detected. Mean arterial pressure (MAP) was calculated as the area under the arterial pressure waveform. Stroke volumes were estimated from the Finometer using the pulse contour method (Jansen et al. 1990). The software also detected bursts of MSNA based on two primary criteria: (1) pulse synchronous spontaneous bursts with signal-to-noise ratios of approximately 3:1, and (2) reflex latencies from preceding R-waves of about 1.3 s (Fagius & Wallin, 1980). Burst detection results were then manually edited by a single experienced microneurographer. As the amplitude and area of sympathetic bursts varies among subjects due to electrode position, MSNA was normalized by dividing the integral of all bursts by the number of bursts occurring during the baseline period. Subsequent burst areas were then divided by this number (i.e. burst areas that were equal to the average baseline area were assigned a value of 1.0) and then multiplied by the number of bursts occurring during a given time period for calculation of total MSNA during LBNP (Cooke et al. 2009). MSNA is reported as both burst frequency (bursts min−1) and total MSNA (arbitrary units).
Data for all parameters were averaged across the last 3 min of each LBNP stage. If a subject reached presyncope before collection of 3 min of data at a particular LBNP level, data for that level were not used; if a subject reached presyncope after collection of at least 3 min of data, data for that LBNP level were used. To quantify oscillatory rhythms, non-equidistant beat-to-beat data were first interpolated linearly and resampled at a frequency of 5 Hz. Data were then passed through a low-pass impulse response filter with a cut-off frequency of 0.5 Hz. Three-minute data sets were fast Fourier transformed with a Hanning window to obtain power spectra. Spectral power was expressed as the integrated area within the low-frequency (LF = 0.04 – 0.15 Hz) and high-frequency (HF = 0.15–0.4 Hz) ranges for arterial pressure and MSNA oscillations (Task Force of the European Society of Cardiology, 1996); individually identified burst areas were used for MSNA oscillation determination. We calculated coherence by dividing the cross-spectral densities of the two signals of interest (either SAP or DAP with MSNA) by the product of the individual autospectra, and then averaged coherence within the LF range (deBoer et al. 1987).
Statistical analysis
Analysis was accomplished using commercially available software (SigmaStat; Systat Software, San Jose, CA, USA). A one-way (LBNP level) randomized block (subjects) ANOVA for repeated measures was used for comparison of outcome variables across LBNP level. If statistical differences were found, Bonferroni-corrected comparisons with baseline measurements were performed. Regression analysis (linear and polynomial) was used to calculate coefficient of determination (r2) as a general summary statistic for describing the concurrency between the mean values of SAPLF and MSNA measures. Additionally, coefficients of determination were determined for individual data points from all subjects at baseline as well as for responses elicited by LBNP within each individual subject. All data are presented as means ± SEM unless otherwise specified and exact P values are presented for all comparisons.
Results
Resting sympathetic activation levels varied greatly among subjects, whether assessed by burst frequency (range: 4.3–35.7 bursts min−1) or total MSNA (range: 13–107 au). At baseline, low frequency oscillations in SAP (SAPLF) were not associated with MSNA burst frequency (r2 = 0.02), total MSNA (r2 = 0.02) or low frequency oscillations in MSNA (MSNALF; r2 = 0.04). Similarly, low frequency oscillations in DAP (DAPLF) were not associated with MSNA parameters (r2≤ 0.13). Furthermore, the coherence between DAP and MSNA was ≥0.5 in only 9 of our 20 subjects (45%) at baseline; similarly, the coherence between SAP and MSNA was ≥0.5 in 10 of 20 subjects (50%).
Average time to presyncope was 1774 ± 110 s. Presyncope occurred in two subjects at −30, three at −60, six at −70, three at −80, five at −90 and one at −100 mmHg. Haemodynamic, respiratory and oscillatory responses during LBNP are shown in Table 1. Stroke volume and SAP decreased progressively during LBNP, while DAP and respiratory rate were not significantly altered. Heart rate increased during LBNP. High frequency oscillations in both SAP and DAP increased, but only at high levels of LBNP (≥60 mmHg). Coherence between MSNA and both SAP and DAP was maintained at or above baseline levels throughout LBNP.
Table 1.
Haemodynamic and oscillatory responses to LBNP
| Variable | Baseline (n = 20) | LBNP 15 (n = 20) | LBNP 30 (n = 20) | LBNP 45 (n = 18) | LBNP 60 (n = 17) | LBNP 70 (n = 13) | LBNP 80 (n = 7) | Recovery (n = 20) |
|---|---|---|---|---|---|---|---|---|
| HR (bpm) | 63 ± 2 | 64 ± 2 | 69 ± 3 | 76 ± 3* | 90 ± 5* | 97 ± 5* | 102 ± 8* | 59 ± 2 |
| SAP (mmHg) | 128 ± 2 | 126 ± 3 | 122 ± 3* | 120 ± 3* | 115 ± 3* | 110 ± 5* | 114 ± 5* | 132 ± 3* |
| DAP (mmHg) | 75 ± 2 | 75 ± 2 | 75 ± 2 | 77 ± 2 | 77 ± 2 | 76 ± 3 | 81 ± 3 | 81 ± 2* |
| MAP (mmHg) | 94 ± 2 | 93 ± 2 | 91 ± 2 | 92 ± 2 | 90 ± 2* | 88 ± 3* | 92 ± 3* | 101 ± 2* |
| SV (ml) | 96 ± 3 | 91 ± 3 | 79 ± 3* | 68 ± 3* | 55 ± 3* | 45 ± 3* | 37 ± 5* | 97 ± 4 |
| RR (breaths min−1) | 15 ± 1 | 14 ± 1 | 13 ± 1 | 13 ± 1 | 13 ± 1 | 13 ± 1 | 13 ± 2 | 15 ± 1 |
| SAPHF (mmHg2) | 2.4 ± 0.3 | 2.3 ± 0.3 | 2.4 ± 0.3 | 2.6 ± 0.3 | 3.7 ± 0.6 | 4.3 ± 0.7* | 5.3 ± 1.7* | 2.0 ± 0.3 |
| DAPHF (mmHg2) | 0.78 ± 0.12 | 0.84 ± 0.16 | 0.97 ± 0.19 | 1.11 ± 0.18 | 2.00 ± 0.46* | 2.14 ± 0.44* | 2.25 ± 0.72* | 1.56 ± 0.39 |
| DAPLF (mmHg2) | 3.7 ± 0.5 | 5.6 ± 0.8 | 6.5 ± 0.9 | 7.7 ± 1.1* | 12.1 ± 1.6* | 17.1 ± 2.5* | 24.9 ± 2.6* | 4.1 ± 0.9 |
| SAP-MSNA coherence | 0.46 ± 0.03 | 0.50 ± 0.02 | 0.50 ± 0.04 | 0.59 ± 0.04 | 0.60 ± 0.05 | 0.49 ± 0.05 | 0.66 ± 0.04* | 0.42 ± 0.03 |
| DAP-MSNA coherence | 0.47 ± 0.03 | 0.52 ± 0.03 | 0.52 ± 0.04 | 0.62 ± 0.04* | 0.60 ± 0.05 | 0.52 ± 0.05 | 0.68 ± 0.04* | 0.44 ± 0.04 |
HR, heart rate; SAP, systolic arterial pressure; DAP, diastolic arterial pressure; MAP, mean arterial pressure; SV, stroke volume; RR, respiratory rate; SAPHF and DAPHF, spectral power of high frequency (0.15–0.4Hz) oscillations in systolic and diastolic arterial pressure; DAPLF, spectral power of low frequency (0.04–0.15 Hz) oscillations in diastolic arterial pressure.
P ≤ 0.045 compared with baseline value.
Figure 1 depicts blood pressure and MSNA responses in a representative subject during progressive central hypovolaemia induced by LBNP. Reductions in stroke volume during LBNP (Table 1) were associated with increases in MSNA that were evident early in the course of LBNP (i.e. at −30 mmHg). By −60 mmHg, both MSNA and blood pressure demonstrated an increase in oscillatory patterns that became more pronounced at −80 mmHg (Fig. 1). Group averages for MSNA burst frequency, total MSNA, low frequency oscillations in SAP (SAPLF) and MSNA (MSNALF) during LBNP are presented in Fig. 2. MSNA increased progressively during LBNP, with a return to baseline levels upon cessation of LBNP. For the entire group of subjects, SAPLF and MSNALF increased from baseline only at high levels of LBNP (≥60 mmHg).
Figure 1.
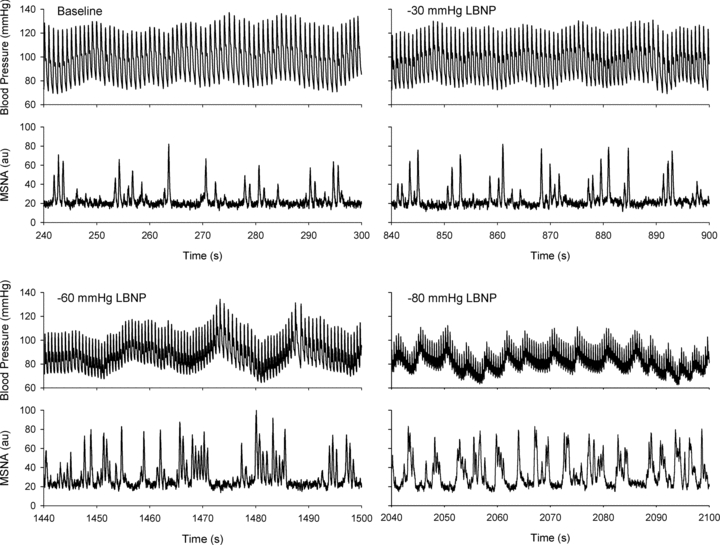
Blood pressure and muscle sympathetic nerve activity (MSNA) in a representative subject during baseline and progressive lower body negative pressure (−30, −60, −80 mmHg LBNP)
Figure 2. Muscle sympathetic nerve activity (MSNA; A and B), low frequency oscillations in systolic arterial pressure (SAPLF; C), and low frequency oscillations in MSNA (MSNALF; D) during progressive lower body negative pressure (LBNP) and recovery (REC).
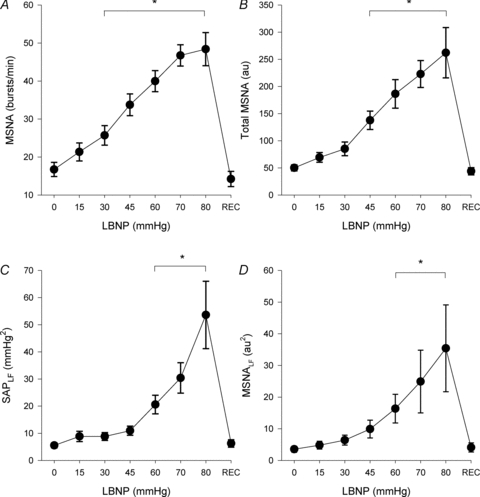
*For all points included within bars, P ≤ 0.021 compared with baseline (0 mmHg LBNP) level. Sample size (n) = 20 at 0, 15 and 30 mmHg LBNP and REC; n = 18 at 45 mmHg LBNP; n = 17 at 60 mmHg LBNP; n = 13 at 70 mmHg LBNP; n = 7 at 80 mmHg LBNP.
Relationships between mean values of SAPLF and MSNA parameters are shown in Fig. 3. Despite increases in sympathetic activation during even early levels of LBNP, SAPLF did not increase initially. Because of this, the association between SAPLF and direct measurements of MSNA (either MSNA frequency or total MSNA) were curvilinear rather than linear. Coefficients of determination (r2) assuming linear relationships were 0.73 and 0.84 for MSNA frequency and total MSNA, but rose to 0.89 and 0.98 when quadratic functions were used. In contrast, the relationship between SAPLF and MSNALF was highly linear (r2 = 0.97).
Figure 3. Associations between low frequency oscillations in systolic arterial pressure (SAPLF) and muscle sympathetic nerve activity (MSNA; A and B) and low frequency oscillations in MSNA (MSNALF; C) during progressive lower body negative pressure.
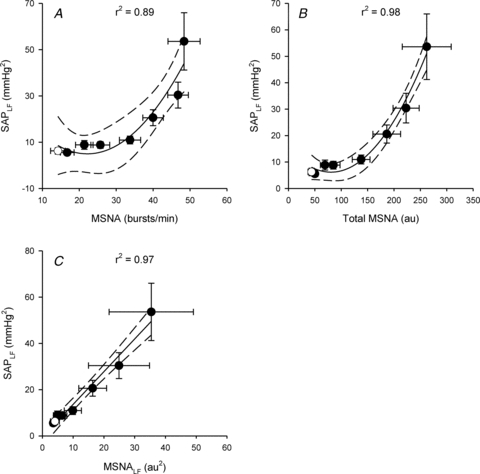
Open circle denotes recovery point; continuous line represents best fit with 95% confidence intervals (dashed lines). Coefficients of determination (r2) reflect polynomial (A and B) or linear (C) regression on mean values. Sample size (n) = 20 at 0, 15 and 30 mmHg LBNP and recovery; n = 18 at 45 mmHg LBNP; n = 17 at 60 mmHg LBNP; n = 13 at 70 mmHg LBNP; n = 7 at 80 mmHg LBNP.
While group mean data demonstrated a strong curvilinear relationship between SAPLF and MSNA, plotting of the association between these variables during LBNP for each individual subject revealed a great deal of inter-individual variability (Fig. 4). Furthermore, coefficients of determination (r2) describing relationships between SAPLF and MSNA within each subject varied widely between subjects, whether calculated using linear or quadratic equations (Table 2). Indeed, 7 of 20 subjects (35%) demonstrated r2 < 0.5 when linear regression was applied and 4 of 18 subjects (22%) demonstrated r2 <0.5 when quadratic equations were used (data from the 2 subjects who experienced presyncope at −30 mmHg could not be used because they had too few data points for valid polynomial regression analysis). Likewise, high individual variability characterised the relationship between SAPLF and MSNALF produced by LBNP (Table 2); 30% and 22% of subjects showed r2<0.5 using linear and quadratic regression analysis.
Figure 4. Individual subject associations between low frequency oscillations in systolic arterial pressure (SAPLF) and muscle sympathetic nerve activity (MSNA; A and B) and low frequency oscillations in MSNA (MSNALF; C) during progressive lower body negative pressure.
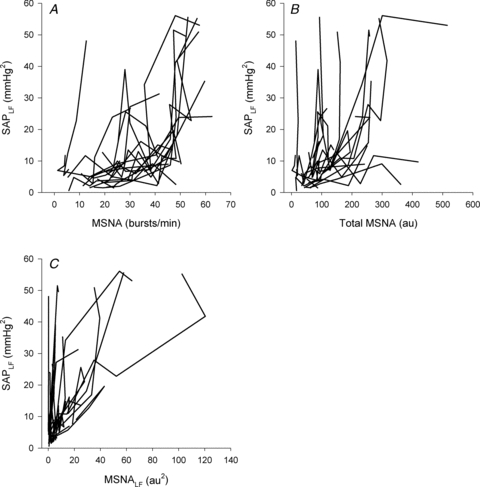
n = 20.
Table 2.
Coefficients of determination (r2) for associations between low frequency oscillations in systolic arterial pressure (SAPLF) and muscle sympathetic nerve activity (MSNA) and its oscillations (MSNALF) during LBNP for each individual subject
| Mean ± SEM | Range | |
|---|---|---|
| Linear fit (n = 20) | ||
| MSNA (b min−1) vs. SAPLF | 0.52 ± 0.07 | 1.62 × 10−6–0.98 |
| Total MSNA (au) vs. SAPLF | 0.54 ± 0.07 | 8.03 × 10−4–0.94 |
| MSNALFvs. SAPLF | 0.63 ± 0.07 | 7.87 × 10−6–0.99 |
| Quadratic Fit (n = 18) | ||
| MSNA (b min−1) vs. SAPLF | 0.67 ± 0.06 | 0.09–0.99 |
| Total MSNA (au) vs. SAPLF | 0.67 ± 0.07 | 0.09–0.96 |
| MSNALFvs. SAPLF | 0.72 ± 0.07 | 0.06–1.0 |
Because of the wide range of LBNP tolerances in our subjects, we subsequently divided subjects into either high tolerant (those who completed at least the −60 mmHg level of LBNP; n = 15) or low tolerant (those who did not complete the −60 mmHg level; n = 5) groups. The high level of individual variability in linear r2 values persisted in both high tolerant (burst frequency, 0.08–0.98; total MSNA, 0.12–0.94; MSNALF, 7.87 × 10−6–0.99) and low tolerant (burst frequency, 1.65 × 10−6–0.83; total MSNA, 8.00 × 10−4–0.70; MSNALF, 0.04–0.98) groups. Polynomial regression produced similar variability in r2 values in high tolerant (burst frequency, 0.09–0.99; total MSNA, 0.09–0.96; MSNALF, 0.30–1.0) and low tolerant (burst frequency, 0.16 × 10−6–0.94; total MSNA, 0.09–0.90; MSNALF, 0.06–0.63) groups.
Low frequency oscillations in diastolic arterial pressure (DAPLF) also increased during LBNP (Table 1). Linear relationships between mean values of DAPLF and MSNA parameters were described by higher coefficients of determination (0.83 for burst frequency and 0.91 for total MSNA) than those of SAPLF and MSNA, although quadratic fits of the data were even better (0.94 for burst frequency and 0.99 for total MSNA). As with SAPLF, the relationship between mean values of low frequency oscillations in both DAP and MSNA were highly linear (r2 = 0.99). However, interrogation of relationships between DAPLF and MSNA parameters within each individual subject again revealed very large ranges of r2 values, whether linear or polynomial regressions were used (Table 3). Just as with SAPLF, 7 of 20 subjects (35%) demonstrated r2 < 0.5 when linear regression was applied to relationships between DAPLF and MSNA parameters and 4 of 18 subjects (22%) demonstrated r2 <0.5 when quadratic equations were used. For the relationship between DAPLF and MSNALF, 35% and 28% of subjects demonstrated r2 < 0.5 using linear and polynomial (quadratic) regression analysis. Inter-individual variability was not dependent on the ability to tolerate LBNP (data not shown).
Table 3.
Coefficients of determination (r2) for associations between low frequency oscillations in diastolic arterial pressure (DAPLF) and muscle sympathetic nerve activity (MSNA) and its oscillations (MSNALF) during LBNP for each individual subject
| Mean ± SEM | Range | |
|---|---|---|
| Linear Fit (n = 20) | ||
| MSNA (b min−1) vs. DAPLF | 0.55 ± 0.06 | 4.93 × 10−3–0.95 |
| Total MSNA (au) vs. DAPLF | 0.58 ± 0.07 | 1.28 × 10−3–0.97 |
| MSNALFvs. DAPLF | 0.68 ± 0.07 | 1.31 × 10−4–1.0 |
| Quadratic Fit (n = 18) | ||
| MSNA (b min−1) vs. DAPLF | 0.67 ± 0.06 | 0.22–0.98 |
| Total MSNA (au) vs. DAPLF | 0.70 ± 0.07 | 7.21 × 10−3–1.0 |
| MSNALFvs. DAPLF | 0.75 ± 0.06 | 0.30–1.0 |
Discussion
There are two primary findings from this study. First, when considering the population as a whole, the relationship between SAPLF and direct measurement of sympathetic nerve activity is curvilinear rather than linear during progressive central hypovolaemia. More importantly, responses in SAPLF to increasing sympathetic activation are highly variable among individuals. These results indicate that SAPLF cannot be used universally as a non-invasive metric of sympathetic outflow in individual subjects in either the resting condition or during sympathetic activation.
Analysis using group means first demonstrated that the relationship between SAPLF and MSNA during sympathetic activation was not a direct linear relationship but was instead curvilinear. This was not unexpected, as we had previously observed that low frequency oscillations in arterial pressure increase significantly only at high levels of LBNP (Cooke et al. 2009) while MSNA burst frequency and total MSNA increased at lower levels (Cooke et al. 2009). Furlan et al. (2000), however, had previously shown that orthostatic stress induced by progressive head up tilt produced a 2-fold increase in MSNA burst frequency but a 12-fold increase in SAPLF. This profound increase in SAPLF occurred despite only a doubling of MSNALF (Furlan et al. 2000). In the present data set, however, MSNA was approximately doubled at −45 mmHg LBNP and was accompanied by doubling of both SAPLF and MSNALF (Fig. 2); in fact, group means of SAPLF were highly correlated (r2 = 0.97) with MSNALF at all levels of sympathetic activation throughout LBNP (Fig. 3). Despite these quantitative differences in SAPLF, our results are consistent with the conclusion reached by Furlan et al. (2000) that, when group mean values are considered, sympathoexcitation increases the coupling between low frequency spectral components of MSNA and SAP. While our group mean results suggest that SAPLF might be used as a non-invasive surrogate of MSNALF, it is clear that SAPLF is not linearly related to MSNA and therefore should not be used in this fashion as a non-invasive surrogate for MSNA. This conclusion is in agreement with the results from numerous animal studies (Persson et al. 1992; Malpas, 1995; Stauss et al. 1995; Tsai et al. 2009).
The existence of a curvilinear relationship between SAPLF and MSNA could, however, be useful as a non-invasive surrogate of sympathetic nerve activity as long as (1) the curvilinearity of the relationship is recognized and appropriate equations are used; and (2) this relationship is consistently strong among individual subjects. We therefore sought to determine whether SAPLF reliably tracked MSNA metrics on an individual subject basis. In our 20 subjects at baseline, neither MSNA nor MSNALF was correlated with SAPLF. Additionally, coherence values were <0.5 in approximately 50% of our subjects, indicating a lack of concordance between the oscillatory patterns in SAPLF and MSNALF in many subjects. These results confirm those of Taylor et al. (1998) acquired during the resting state in healthy subjects of differing sexes and ages and therefore different baseline levels of sympathetic activation. Our study extends this previous work by demonstrating very high inter-individual variability in the relationships between SAPLF and MSNA during profound sympathetic activation elicited by LBNP. This high level of inter-individual variability persisted with linear and polynomial analyses and was not dependent on the ability of the individual to tolerate LBNP. Importantly, r2 values characterising the associations between these variables were <0.5 in 22% of our subjects even when the curvilinear nature of the association was taken into account. It is therefore clear that the use of SAPLF to indicate the level of sympathetic activation is not valid in all subjects. Because of the wide inter-individual variability and the very weak SAPLF–MSNA association in a substantial proportion of the population, the use of SAPLF as a non-invasive surrogate of MSNA is not reliable and therefore not warranted.
These results are in contrast to the conclusion drawn by Pagani et al. (1997), who used infusions of sodium nitroprusside and phenylephrine to induce sympathoexcitation and sympathoinhibition. When plotted as individual points, these investigators demonstrated highly significant P values for correlations between SAPLF and MSNA metrics, but did not provide correlation coefficients (Pagani et al. 1997). Taylor et al. subsequently used the reported data of Pagani et al. to calculate correlation coefficients (r) of ≤0.46 for the relationships between SAPLF and MSNA (Taylor et al. 1998). The results from our study eliciting sympathoexcitation using nonpharmacological means suggest that the weak association between these variables in the previous study may reflect profound individual variability in the response of SAPLF to sympathetic activation, with some subjects demonstrating strong correlations and others demonstrating remarkably weak correlations. Taken together, these results indicate that the use of SAPLF as a non-invasive surrogate of MSNA either at rest or during sympathetic activation cannot be justified in individual subjects. Assessment of autonomic function using SAPLF during a clinical test involving sympathoexcitation therefore may not be appropriate, as even healthy individuals do not reliably demonstrate associations between SAPLF and direct measurement of sympathetic nerve activity.
Although our main intent was to investigate the proposed use of SAPLF as a non-invasive surrogate of MSNA, diastolic pressure is more closely related to sympathetic nerve activity than systolic pressure (Sundlof & Wallin, 1978). Indeed, relationships between DAPLF and MSNA parameters demonstrated a higher degree of linearity than did SAPLF in our study. However, further analysis revealed extremely high variability in the strength of the relationships between DAPLF and MSNA among individual subjects, just as in the associations between SAPLF and MSNA. Hence, low frequency oscillations in arterial blood pressure, whether systolic or diastolic, cannot be used as non-invasive surrogates of sympathetic nerve activity.
The lack of reliable associations between low frequency oscillations in blood pressure and MSNA should not be surprising. Low frequency oscillations in arterial pressure have been suggested to be produced by either the action of central oscillating pacemaker neurons or by a resonance in the baroreflex (Julien, 2006). In either case, low frequency rhythms of sympathetic nerve activity would be produced via central mechanisms, and it is these rhythms rather than an absolute level of sympathetic activation that act to produce low frequency oscillations in blood pressure (Malpas, 1998). The observation that heat stress produces profound sympathetic activation without an increase in SAPLF can therefore be understood because MSNALF did not concomitantly increase (Cui et al. 2004). However, it would be simplistic to assume that oscillations in sympathetic nerve activity would be directly reflected as a proportional oscillation in blood pressure, as the ability of the nerve signal to be transduced into vascular resistance (Myers et al. 2001) and intrinsic vasomotor rhythmicity of blood vessels (Stauss, 2007) must also contribute to the genesis of blood pressure oscillations. Indeed, low frequency oscillations in blood pressure persist, albeit at lower levels, following complete ganglionic blockade in humans (Zhang et al. 2002). Furthermore, O'Leary et al. (2004) demonstrated in an elegant study that the increased blood pressure variability associated with head up tilt was the result of both baroreflex regulation through the sympathetic nervous system and a non-baroreflex component presumed to be a myogenic response. It is possible that summation of myogenic responses with input from sympathetic nerves at the level of the vasculature might underlie the lack of strong relationships between MSNALF and SAPLF in individual subjects either at baseline or during sympathetic activation. Our results therefore provide an additional caution against the use of SAPLF to provide information about MSNALF in individual subjects.
While the use of group mean data for insight into physiological processes is necessary and appropriate, further investigation is required before advocating the use of a non-invasive surrogate measure in clinical settings. Such non-invasive measurements as heart rate variability have been suggested for use in tracking compensatory autonomic responses to physiological challenges based on group mean data, but have failed to provide reliable information in individual subjects (Rickards et al. 2010; Ryan et al. 2010). In this study, we applied the same ‘next step’ approach to the data presented herein, moving from the group mean level to the individual level. With this approach, we determined that neither SAPLF nor DAPLF can be reliably used as a non-invasive surrogate of MSNA in individuals, despite its convenience of use. We therefore strongly advocate the use of this ‘next step’ approach for investigation of non-invasive surrogates to ensure clinical utility in individual patients.
This study is not without potential methodological limitations. First, arterial blood pressure oscillations were determined by the use of non-invasive photoplethysmography rather than via a direct arterial catheter. However, measurement of blood pressure by finger photoplethysmography has been shown to be highly accurate compared with direct arterial measurement during a variety of physiological manoeuvres including orthostatic stress (Imholz et al. 1988; Parati et al. 1989; Imholz et al. 1990). More germane to this study, use of the Finometer technology has been validated against direct intra-arterial recordings of blood pressure for use in power spectral analysis of blood pressure variability (Omboni et al. 1993). While it is recognized that blood pressure measurement using this technique may be affected when temperature of the finger or body is altered (Imholz et al. 1998), all experiments in this study were conducted within a small range of ambient temperature, suggesting little to no temperature-induced alteration in vasomotor tone during the experiments. Additionally, measurement of MSNA might not be reflective of sympathetic nerve activity to other vascular beds (Malpas, 2010). We therefore cannot discount the possibility that direct measurement of sympathetic nerve activity in other vascular beds might yield improved relationships with low frequency oscillations in arterial pressure.
Validation of a non-invasive metric that could be used as a surrogate for direct measurement of sympathetic nerve activity would be useful both for clinical investigation of dysautonomias as well as for laboratory investigations. Measurement of SAPLF has been suggested as such a metric based primarily on group mean data contained within earlier studies. While curvilinear relationships exist between SAPLF or DAPLF and MSNA on a group level, neither SAPLF nor DAPLF is reliably correlated with absolute levels of MSNA either at rest or during central hypovolaemia in individual subjects. These data therefore do not support the use of low frequency oscillations in arterial pressure as a non-invasive surrogate of MSNA.
Acknowledgments
We thank the research volunteers for their cheerful participation and Mr Gary Muniz for his excellent laboratory assistance. This study was funded by the United States Army, Medical Research and Materiel Command.
Glossary
Abbreviations
- DAP
diastolic arterial pressure
- DAPHF
high frequency oscillations of diastolic arterial pressure
- DAPLF
low frequency oscillations of diastolic arterial pressure
- HF
high frequency
- HR
heart rate
- LBNP
lower body negative pressure
- LF
low frequency
- MAP
mean arterial pressure
- MSNA
muscle sympathetic nerve activity
- MSNALF
low frequency oscillations of muscle sympathetic nerve activity
- RR
respiratory rate
- SAP
systolic arterial pressure
- SAPHF
high frequency oscillations of systolic arterial pressure
- SAPLF
low frequency oscillations of systolic arterial pressure
- SV
stroke volume
Author contributions
All experiments were performed in the laboratory of the Advanced Capabilities for Emergency Medical Monitoring Task Area at the US Army Institute of Surgical Research, Fort Sam Houston, TX, USA. The authors, K.L.R., C.A.R., C.H.-L., W.H.C. and V.A.C. contributed to: conception and design of the experiments; collection, analysis and interpretation of data; and drafting the article and revising it critically for important intellectual content. All authors approved the final version of the manuscript.
Disclaimer
The opinions or assertions contained herein are the private views of the authors, and are not to be construed as official or as reflecting the views of the Department of the Army or the Department of Defense.
References
- Cooke WH, Rickards CA, Ryan KL, Convertino VA. Autonomic compensation to simulated hemorrhage monitored with heart period variability. Crit Care Med. 2008;36:1892–1899. doi: 10.1097/CCM.0b013e3181760d0c. [DOI] [PubMed] [Google Scholar]
- Cooke WH, Rickards CA, Ryan KL, Kuusela TA, Convertino VA. Muscle sympathetic nerve activity during intense lower body negative pressure to presyncope in humans. J Physiol. 2009;587:4987–4999. doi: 10.1113/jphysiol.2009.177352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke WH, Ryan KL, Convertino VA. Lower body negative pressure as a model to study progression to acute hemorrhagic shock in humans. J Appl Physiol. 2004;96:1249–1261. doi: 10.1152/japplphysiol.01155.2003. [DOI] [PubMed] [Google Scholar]
- Cui J, Zhang R, Wilson TE, Crandall CG. Spectral analysis of muscle sympathetic nerve activity in heat-stressed humans. Am J Physiol Heart Circ Physiol. 2004;286:H1101–1106. doi: 10.1152/ajpheart.00790.2003. [DOI] [PubMed] [Google Scholar]
- deBoer RW, Karemaker JM, Strackee J. Hemodynamic fluctuations and baroreflex sensitivity in humans: a beat-to-beat model. Am J Physiol Heart Circ Physiol. 1987;253:H680–689. doi: 10.1152/ajpheart.1987.253.3.H680. [DOI] [PubMed] [Google Scholar]
- Fagius J, Wallin BG. Sympathetic reflex latencies and conduction velocities in normal man. J Neurol Sci. 1980;47:433–448. doi: 10.1016/0022-510x(80)90098-2. [DOI] [PubMed] [Google Scholar]
- Furlan R, Guzzetti S, Crivellaro W, Dassi S, Tinelli M, Baselli G, Cerutti S, Lombardi F, Pagani M, Malliani A. Continuous 24-hour assessment of the neural regulation of systemic arterial pressure and RR variabilities in ambulant subjects. Circulation. 1990;81:537–547. doi: 10.1161/01.cir.81.2.537. [DOI] [PubMed] [Google Scholar]
- Furlan R, Porta A, Costa F, Tank J, Baker L, Schiavi R, Robertson D, Malliani A, Mosqueda-Garcia R. Oscillatory patterns in sympathetic neural discharge and cardiovascular variables during orthostatic stimulus. Circulation. 2000;101:886–892. doi: 10.1161/01.cir.101.8.886. [DOI] [PubMed] [Google Scholar]
- Guyton AC, Harris JW. Pressoreceptor-autonomic oscillation: A probable cause of vasomotor waves. Am J Physiol. 1951;165:158–166. doi: 10.1152/ajplegacy.1951.165.1.158. [DOI] [PubMed] [Google Scholar]
- Hagbarth KE, Vallbo AB. Pulse and respiratory grouping of sympathetic impulses in human muscle-nerves. Acta Physiol Scand. 1968;74:96–108. doi: 10.1111/j.1748-1716.1968.tb04218.x. [DOI] [PubMed] [Google Scholar]
- Imholz BP, Settels JJ, van der Meiracker AH, Wesseling KH, Wieling W. Non-invasive continuous finger blood pressure measurement during orthostatic stress compared to intra-arterial pressure. Cardiovasc Res. 1990;24:214–221. doi: 10.1093/cvr/24.3.214. [DOI] [PubMed] [Google Scholar]
- Imholz BP, van Montfrans GA, Settels JJ, van der Hoeven GM, Karemaker JM, Wieling W. Continuous non-invasive blood pressure monitoring: reliability of Finapres device during the Valsalva manoeuvre. Cardiovasc Res. 1988;22:390–397. doi: 10.1093/cvr/22.6.390. [DOI] [PubMed] [Google Scholar]
- Imholz BP, Wieling W, van Montfrans GA, Wesseling KH. Fifteen years experience with finger arterial pressure monitoring: assessment of the technology. Cardiovasc Res. 1998;38:605–616. doi: 10.1016/s0008-6363(98)00067-4. [DOI] [PubMed] [Google Scholar]
- Jansen JR, Wesseling KH, Settels JJ, Schreuder JJ. Continuous cardiac output monitoring by pulse contour during cardiac surgery. Eur Heart J. 1990;11(Suppl I):26–32. doi: 10.1093/eurheartj/11.suppl_i.26. [DOI] [PubMed] [Google Scholar]
- Julien C. The enigma of Mayer waves: Facts and models. Cardiovasc Res. 2006;70:12–21. doi: 10.1016/j.cardiores.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Lucini D, Mela GS, Malliani A, Pagani M. Impairment in cardiac autonomic regulation preceding arterial hypertension in humans: insights from spectral analysis of beat-by-beat cardiovascular variability. Circulation. 2002;106:2673–2679. doi: 10.1161/01.cir.0000039106.89299.ab. [DOI] [PubMed] [Google Scholar]
- Lucini D, Zuccotti G, Malacarne M, Scaramuzza A, Riboni S, Palombo C, Pagani M. Early progression of the autonomic dysfunction observed in pediatric type 1 diabetes mellitus. Hypertension. 2009;54:987–994. doi: 10.1161/HYPERTENSIONAHA.109.140103. [DOI] [PubMed] [Google Scholar]
- Malliani A, Pagani M, Lombardi F, Cerutti S. Cardiovascular neural regulation explored in the frequency domain. Circulation. 1991;84:482–492. doi: 10.1161/01.cir.84.2.482. [DOI] [PubMed] [Google Scholar]
- Malpas SC. A new model for the generation of sympathetic nerve activity. Clin Exp Pharmacol Physiol. 1995;22:11–15. doi: 10.1111/j.1440-1681.1995.tb01911.x. [DOI] [PubMed] [Google Scholar]
- Malpas SC. The rhythmicity of sympathetic nerve activity. Prog Neurobiol. 1998;56:65–96. doi: 10.1016/s0301-0082(98)00030-6. [DOI] [PubMed] [Google Scholar]
- Malpas SC. Sympathetic nervous system overactivity and its role in the development of cardiovascular disease. Physiol Rev. 2010;90:513–557. doi: 10.1152/physrev.00007.2009. [DOI] [PubMed] [Google Scholar]
- Myers CW, Cohen MA, Eckberg DL, Taylor JA. A model for the genesis of arterial pressure Mayer waves from heart rate and sympathetic activity. Auton Neurosci. 2001;91:62–75. doi: 10.1016/S1566-0702(01)00289-2. [DOI] [PubMed] [Google Scholar]
- O'Leary DD, Shoemaker JK, Edwards MR, Hughson RL. Spontaneous beat-by-beat fluctuations of total peripheral and cerebrovascular resistance in response to tilt. Am J Physiol Regul Integr Comp Physiol. 2004;287:R670–679. doi: 10.1152/ajpregu.00408.2003. [DOI] [PubMed] [Google Scholar]
- Omboni S, Parati G, Frattola A, Mutti E, Di Rienzo M, Castiglioni P, Mancia G. Spectral and sequence analysis of finger blood pressure variability. Comparison with analysis of intra-arterial recordings. Hypertension. 1993;22:26–33. doi: 10.1161/01.hyp.22.1.26. [DOI] [PubMed] [Google Scholar]
- Pagani M, Furlan R, Pizzinelli P, Crivellaro W, Cerutti S, Malliani A. Spectral analysis of R-R and arterial pressure variabilities to assess sympatho-vagal interaction during mental stress in humans. J Hypertension Suppl. 1989;7:S14–15. doi: 10.1097/00004872-198900076-00004. [DOI] [PubMed] [Google Scholar]
- Pagani M, Lombardi F, Guzzetti S, Rimoldi O, Furlan R, Pizzinelli P, Sandrone G, Malfatto G, Dell'Orto S, Piccaluga E, Turiel M, Baselli G, Cerutti S, Malliani A. Power spectral analysis of heart rate and arterial pressure variabilities as a marker of sympatho-vagal interaction in man and conscious dog. Circ Res. 1986;59:178–193. doi: 10.1161/01.res.59.2.178. [DOI] [PubMed] [Google Scholar]
- Pagani M, Montano N, Porta A, Malliani A, Abboud FM, Birkett C, Somers VK. Relationship between spectral components of cardiovascular variabilities and direct measures of muscle sympathetic nerve activity in humans. Circulation. 1997;95:1441–1448. doi: 10.1161/01.cir.95.6.1441. [DOI] [PubMed] [Google Scholar]
- Parati G, Casadei R, Groppelli A, Di Rienzo M, Mancia G. Comparison of finger and intra-arterial blood pressure monitoring at rest and during laboratory testing. Hypertension. 1989;13:647–655. doi: 10.1161/01.hyp.13.6.647. [DOI] [PubMed] [Google Scholar]
- Parati G, Mancia G, Di Rienzo M, Castiglioni P. Point: cardiovascular variability is/is not an index of autonomic control of circulation. J Appl Physiol. 2006;101:676–678. doi: 10.1152/japplphysiol.00446.2006. discussion 678–682. [DOI] [PubMed] [Google Scholar]
- Parati G, Saul JP, Di Rienzo M, Mancia G. Spectral analysis of blood pressure and heart rate variability in evaluating cardiovascular regulation. A critical appraisal. Hypertension. 1995;25:1276–1286. doi: 10.1161/01.hyp.25.6.1276. [DOI] [PubMed] [Google Scholar]
- Persson PB, Stauss H, Chung O, Wittmann U, Unger T. Spectrum analysis of sympathetic nerve activity and blood pressure in conscious rats. Am J Physiol Heart Circ Physiol. 1992;263:H1348–1355. doi: 10.1152/ajpheart.1992.263.5.H1348. [DOI] [PubMed] [Google Scholar]
- Radaelli A, Perlangeli S, Cerutti MC, Mircoli L, Mori I, Boselli L, Bonaita M, Terzoli L, Candotti G, Signorini G, Ferrari AU. Altered blood pressure variability in patients with congestive heart failure. J Hypertens. 1999;17:1905–1910. doi: 10.1097/00004872-199917121-00020. [DOI] [PubMed] [Google Scholar]
- Rickards CA, Ryan KL, Ludwig DA, Convertino VA. Is heart period variability associated with the administration of lifesaving interventions in individual prehospital trauma patients with normal standard vital signs? Crit Care Med. 2010;38:1666–1673. doi: 10.1097/CCM.0b013e3181e74cab. [DOI] [PubMed] [Google Scholar]
- Ryan KL, Rickards CA, Ludwig DA, Convertino VA. Tracking central hypovolemia with ECG in humans: cautions for the use of heart period variability in patient monitoring. Shock. 2010;33:583–589. doi: 10.1097/SHK.0b013e3181cd8cbe. [DOI] [PubMed] [Google Scholar]
- Stauss HM. Identification of blood pressure control mechanisms by power spectral analysis. Clin Exp Pharmacol Physiol. 2007;34:362–368. doi: 10.1111/j.1440-1681.2007.04588.x. [DOI] [PubMed] [Google Scholar]
- Stauss HM, Mrowka R, Nafz B, Patzak A, Unger T, Persson PB. Does low frequency power of arterial blood pressure reflect sympathetic tone? J Auton Nerv Syst. 1995;54:145–154. doi: 10.1016/0165-1838(94)00000-a. [DOI] [PubMed] [Google Scholar]
- Sundlof G, Wallin BG. Human muscle nerve sympathetic activity at rest. Relationship to blood pressure and age. J Physiol. 1978;274:621–637. doi: 10.1113/jphysiol.1978.sp012170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology. Heart rate variability: standards of measurement, physiological interpretation and clinical use. Circulation. 1996;93:1043–1065. [PubMed] [Google Scholar]
- Taylor JA, Studinger P. Counterpoint: cardiovascular variability is not an index of autonomic control of the circulation. J Appl Physiol. 2006;101:678–681. doi: 10.1152/japplphysiol.00446.2006. discussion 681. [DOI] [PubMed] [Google Scholar]
- Taylor JA, Williams TD, Seals DR, Davy KP. Low-frequency arterial pressure fluctuations do not reflect sympathetic outflow: gender and age differences. Am J Physiol Heart Circ Physiol. 1998;274:H1194–1201. doi: 10.1152/ajpheart.1998.274.4.H1194. [DOI] [PubMed] [Google Scholar]
- Tsai ML, Tseng WT, Yen CT, Chen RF. The correlation of mean sympathetic activity with low-frequency blood pressure and sympathetic variability. Clin Exp Hypertens. 2009;31:615–624. doi: 10.3109/10641960902929461. [DOI] [PubMed] [Google Scholar]
- Zhang R, Iwasaki K, Zuckerman JH, Behbehani K, Crandall CG, Levine BD. Mechanism of blood pressure and R-R variability: insights from ganglion blockade in humans. J Physiol. 2002;543:337–348. doi: 10.1113/jphysiol.2001.013398. [DOI] [PMC free article] [PubMed] [Google Scholar]