

Abstract
Fuchs’ Endothelial Corneal Dystrophy is the most common corneal endotheliopathy, and a leading indication for corneal transplantation in the US. Relatively little is known about its underlying pathology. We created a cellular model of the disease focusing on collagen VIII alpha 2 (COL8A2), a collagen which is normally present in the cornea, but which is found in abnormal amounts and distribution in both early and late-onset forms of the disease. We performed cellular transfections using COL8A2 cDNAs including both wild-type and mutant alleles which are known to result in early-onset FECD. We used this cell model to explore the cellular production of wild-type and mutant monomeric and trimeric collagen VIII and measured production levels and patterns using Western blotting and immunofluorescence. We studied the thermal stability of the mutated collagen VIII helices using computer modeling, and further investigated these differences using collagen mimetic peptides. The Western blots demonstrated that similar amounts of wild-type and mutant collagen VIII monomers were produced in the cells. However, the levels of trimeric collagen peptide in the mutant-transfected cells were elevated. Intracellular accumulation of trimeric collagen VIII was confirmed on immunofluorescence studies. Both the computer model and the collagen mimetic peptides demonstrated that the L450W mutant was less thermally stable than either the Q455K or wild-type collagen VIII. Thus, although both mutant collagen VIII peptides were retained intracellularly, the biochemical reasons for the retention varied between genotypes. Collagen VIII mutations, which clinically result in Fuchs’ Dystrophy, are associated with abnormal cellular accumulation of collagen VIII. Different collagen VIII mutations may act via distinct biochemical mechanisms to produce the FECD phenotype.
Keywords: Fuchs’ Endothelial Corneal Dystrophy, Collagen VIII, Corneal Endothelium, Collagen Mimetic Peptides
1. Introduction
Fuchs’ endothelial corneal dystrophy [FECD] is the most common primary disorder of the corneal endothelium (Suh et al., 2008). End-stage disease results in severely decreased visual acuity with transplant surgery representing the only definitive treatment and Fuchs’ endothelial corneal dystrophy is now a leading indication for corneal transplantation in the United States. However, the cellular pathophysiology of FECD has only recently become an area of focused investigation (Jurkunas et al., 2010; Vithana et al., 2008). Essentially, the study of FECD has been hampered by the scarcity of suitable tissue for research, as the only tissues available for research were from patients with severe disease undergoing surgery. Among the characteristic histologic changes noted in such specimens are thickening of Descemet’s membrane [the basement membrane secreted by the corneal endothelium] and the development of corneal endothelial excresences termed guttae (Hogan et al., 1974; Waring et al., 1982). Both of these features result from the presence of disorganized and excessive collagen VIII (Levy et al., 1996). In vivo collagen VIII is secreted by the corneal endothelium and forms an extracellular lattice subjacent to the corneal endothelium (Kapoor et al., 1986; Levy et al., 1996; Sawada et al., 1990). The exact function of this collagen VIII lattice in healthy individuals and its pathogenic role in FECD is poorly understood.
In general, the inheritance of late-onset FECD appears to be autosomal dominant with genetic and environmental modifiers and affected individuals become symptomatic in their sixth decade (Suh et al., 2008). However, several families with an early-onset and severe form of FECD have been identified. They were discovered to have collagen VIII mutations (Biswas et al., 2001; Gottsch et al., 2005; Liskova et al., 2007). Specifically, they were found to have single amino acid mutations in the collagen VIII α2 chain i.e. COL8A2. These missense mutations specify a glutamine to lysine substitution [Q455K] and a leucine to tryptophan mutation [L450W]. Histochemical studies from patients with the L450W mutation revealed that the FECD disease severity correlated with intra-endothelial cell and extracellular matrix collagen VIII accumulation, whilst the cellular pathology underlying the Q455K mutation has not yet been studied (Zhang et al., 2006). Thus, collagen VIII accumulation is noted in patients with late-onset FECD and collagen VIII mutations result in an early onset form of FECD, making collagen VIII an attractive starting point both in elucidating the mechanism of disease in FECD and designing novel treatments.
Collagens are trimeric proteins created by the association of three individual protein strands (Ramshaw et al., 1998). The hallmark structural feature of collagen is the triple helix which can be seen in part, or all, of the collagen molecule. The triple helical domain of the collagen monomers are typically composed of repeating Gly-X-Y tri-peptides, with glycine [Gly] found every third residue, as it is the only amino acid sufficiently small to fit on the inside of the collagen triple helix. Proline [Pro] and hydroxyproline [Hyp], which frequently occur in the X and Y positions respectively, play a critical role in triple helical folding. In the cell, trimers are formed by non-covalent association of the non-collagenous C-terminal domains. This is soon followed by triple helix folding and secretion from the cell after completion of the folding process.
Type VIII collagen, a non-fibrillar collagen which has a restricted distribution in the body, is a member of the short chain class of collagens (Shuttleworth, 1997). Two similar chains have been identified, designated α1(VIII) and α2(VIII) respectively, and both monomers have a molecular weight of approximately 80 kDa (Illidge et al., 1998 a; Muragaki et al., 1991). Early studies supported a heterotrimeric form of collagen VIII in an (α1)2(α2)1 ratio (Kapoor et al., 1986). However, Greenhill et al. reported that the α1 and α2 chains form homotrimeric proteins in vivo and exist in tissues as two distinct proteins (Greenhill et al., 2000). Alpha-2 collagen VIII consists of a collagenous domain of 457 amino acids, flanked by a short non-collagenous N-terminal domain [NC2; 72 amino acids] and a longer non-collagenous C-terminal domain [NC1; 167 amino acids] (Illidge et al., 2001; Muragaki et al., 1991).
The COL8A2 FECD mutations, namely L450W and Q455K, are both found within the collagenous domain of collagen VIII (Biswas et al., 2001; Gottsch et al., 2005; Muragaki et al., 1991). Collagen X, the collagen most closely related structurally to collagen VIII, is known to have several single-amino acid disease-causing mutations (Bateman et al., 2004; Wilson et al., 2005; Yamaguchi et al., 1991). However, these mutations are all found in the C-terminal non-collagenous domain of the protein, and are likely to interfere primarily with initiation of the collagen triple helix. Changes in the amino acid composition within the collagenous domain, as associated with FECD, are more likely to lead to alterations in the helical microenvironment surrounding the mutations and result in variations in the collagen super-helix twist and/or alter the biochemical stability of collagen helices (Baum et al., 1999; Kramer et al., 1999).
The purpose of the study presented herein is to explore the alterations in the cellular and biochemical behavior of mutant collagen VIII. Our initial investigations centered on the creation of a cellular model of FECD to assess the biological behavior of the mutant collagen VIII. To better understand the biochemical implications of the amino acid substitutions, we used both a collagen modelling algorithm and collagen mimetic peptides, to explore the changes in thermal stability consequent to the FECD mutations (Brodsky and Baum, 2008; Persikov et al., 2005).
2. Materials and Methods
2.1 Reagents & Antibodies
The endotoxin-free plasmid purification kit was purchased from Qiagen [Valencia, CA]. PcDNA3.1[+], One Shot TOP10 Electrocomp E. coli, Lipofectamine 2000, Nu-PAGE LDS Sample Buffer, Opti-MEM, trypsin and molecular grade water were purchased from Invitrogen [Carlsbad, CA]. Ascorbic acid, EDTA, protease inhibitor cocktail, 2-mercaptoethanol, collagenase type III, and calcium chloride were purchased from Sigma-Aldrich [St. Louis, MO]. Quick start BSA standard set, Quick Start Bradford dye reagent, 7.5% Tris-HCl gels, Precision Plus pre-stained protein standard, nitrocellulose membranes, Blotting Grade Blocker and Tween-20 were purchased from BioRad [Hercules, CA]. M-PER mammalian protein extraction reagent was purchased from Thermo Scientific [Rockford, IL]. Phosphate buffered saline was purchased from Quality Biological [Gaithersburg, MD]. Kpn, XhoI and HindIII restriction enzymes were obtained from New England Biolabs [Ipswich, MA]. F12K cell culture medium was purchased from ATCC [Manassas, VA].
Primary antibodies used were polyclonal anti-FLAG antibody [Sigma-Aldrich], anti-actin [Sigma-Aldrich] and anti-neomycin phosphotransferase II [Millipore, Temecula, CA]. A polyclonal antibody to trimeric collagen α2(VIII) was kindly provided by Dr. P. Davies, New Zealand. Although reactive in immunofluorescence experiments, this antibody was unreactive under the Western blotting conditions used in this study. Secondary antibodies used were anti-mouse IgG HRP-linked whole antibody, anti-rabbit IgG HRP-linked whole antibody [both GE Healthcare, Piscataway NJ] and Alexa-fluor anti-rabbit IgG [Invitrogen].
2.2 COL8A2 Expression Constructs
A full-length wild-type [WT] human COL8A2 cDNA clone was obtained from OriGene [Rockville, MD]. The full-length identity of this clone was sequence confirmed using primers described by Biswas et al (Biswas et al., 2001). PCR amplification of the full-length [2.1 kb] COL8A2 cDNA clone was performed using forward primer COL8A2cDNA_KPNF [5′-TATAGGTACCGGACGCCATGCTGGG-3′] containing a KpnI restriction enzyme site [italics] and the COL8A2 start codon [underline], and reverse primer COL8A2cDNA_XHOR [5′-Relative TATACTCGAGGAAAAGGTCGCTCTACCAC-3′] containing an XhoI restriction enzyme site [italics] and with its 3′ end corresponding to 42 nucleotides downstream of the putative COL8A2 stop codon. PCR amplification was performed using an annealing temperature of 62 °C, 2 minutes extension time, and 35 cycles. PCR product was column-purified, restriction enzyme digested with KpnI and XhoI, gel-purified, and cloned into pcDNA3.1[+]. Fidelity of the full-length WT cDNA clone was sequence verified.
L450W and Q455K mutant cDNA clones were produced from the WT clone using the QuikChange Site-Directed Mutagenesis Kit [Agilent Technologies, Santa Clara, CA]. For the L450W mutation, forward primer COL8A2_cDNA_450F [5′-GCAGAAAGGTGACTGGGGGCTCCCTGGGC-3′] containing the T to G transversion [underline] specifying the L450W mutation and the homologous reverse primer COL8A2_cDNA_450Fr [5′-GCCCAGGGAGCCCCCAGTCACCTTTCTGC-3′] were used. For the Q455K mutation, forward primer COL8A2_cDNA_455F [5′-GGGGCTCCCTGGGAAGCCTGGCCTGAG-3′] containing the C to A transversion [underline] specifying the Q455K mutation and the homologous reverse primer COL8A2_cDNA_455Fr [5′-CTCAGGCCAGGCTTCCCAGGGAGCCCC-3′] were used. The double mutant (DM) cDNA clone containing both the L450W and Q455K mutations was produced from the L450W clone using the QuikChange Kit with COL8A2_cDNA_455F and COL8A2_cDNA_455Fr as primers. Confirmation of the expected change[s] for each mutant cDNA clone [L450W, Q455K, and DM] was performed by full-length sequencing.
To optimize efficient intracellular translation of COL8A2 cDNA clones, site-directed mutagenesis of WT and mutant cDNA clones was performed to introduce a Kozak consensus base at position +4 of the COL8A2 clones (Kozak, 1986). Using each COL8A2 cDNA clone as a template, PCR amplification of the full length cDNA was performed using forward primer 8A2cDNAKOZAKKPNF [5′-TATAGGTACCGGACGCCATGGTGGG-3′] containing a KpnI restriction enzyme site [italics], the COL8A2 start codon [underline], and the adjacent C to G sequence change [bold] establishing the Kozak consensus sequence. Reverse primer COL8A2cDNA_XHOR [see above] was used. PCR amplification was performed using an annealing temperature of 62 °C, 2 minutes extension time, and 35 cycles. PCR product was column-purified, restriction enzyme digested with KpnI and XhoI, gel-purified, and cloned into pcDNA3.1[+]. Fidelity of the cDNA clones was verified by full-length sequencing.
At the time this study was conducted no commercially available COL8A2 antibodies were available. The COL8A2 antibody which we utilized recognized only trimeric collagen VIII, so the pCMV-3Tag-1C Epitope-Tagging Mammalian Expression Vector [Agilent Technologies] was used for Western blotting experiments. Kozak sequence-containing WT, L450W, Q455K, and DM cDNAs were subcloned from the pcDNA3.1[+] vector into the pCMV-3Tag-1C vector with the proper orientation using the HindIII and XhoI restriction enzyme sites within both vectors. Fidelity and frame of the cDNA clones was confirmed by full-length sequencing. The proteins produced, which have three copies of an N-terminal ‘FLAG-tag’, were identified on Western blotting using commercially available anti-FLAG antibodies.
2.3 Bacterial Transformation
TOP10 E. coli were transformed with the pCMV plasmids and colonies were grown on kanamycin agar at 37 °C. Of note, E. coli colonies transformed with the DM plasmid grew only when cultured at 30 °C. Correct plasmid composition was verified by means of restriction enzyme digestion using XhoI and HindIII, and DNA sequencing.
2.4 Culture of Cells
Human corneal endothelial cells (HCEC) are arrested in the G1-phase but can be induced to divide in culture with mitogenic stimulation (Joyce N.C., 2003). Human corneas for research are in limited supply, and the rate successful establishment of HCEC cell lines from donor corneas may be as low as 29% (Engler, C. et al. 2009). In addition, the slow doubling time of HCEC (46–92 hours) makes these cells less suitable for transient transfections experiments. (Zhu, C. et al. 2008). Thus, due to the substantial difficulties associated with culturing corneal endothelial cells, CHO-K1 cells [ATCC, Manassas, VA] were chosen for protein-production experiments. Additionally CHO-K1 cells have intrinsically high levels of prolyl-4-hydroxylase and proline hydroxylation is essential for collagen triple helix stability. Both reverse transcriptase PCR and immunofluorescence studies demonstrated no native collagen VIII production by CHO-K1 cells. The cells were maintained in F12K medium [ATCC, Manassas, VA] supplemented with 10% FBS and 1 mM ascorbic acid. Ascorbic acid promotes essential intracellular hydroxylation of prolyl and lysyl residues during collagen synthesis.
2.5 Recombinant Expression of COL8A2
The pCMV plasmids were introduced into the CHO-K1 cells using Lipofectamine 2000 transfection reagent using manufacturer’s recommendations. Twenty-four hours after transfection, cells were lysed by vigorous shaking after the addition of 500 μl of lysis buffer [M-PER mammalian protein extraction reagent, 1% EDTA, 1% protease inhibitor cocktail]. The amount of protein in the cell lysate was measured using Bradford dye and standards. Thirty-five μg of cellular protein was heated at 65 °C for 5 minutes with loading buffer [Nu-PAGE LDS Sample Buffer with 5% 2-mercaptoethanol].
Samples were run at 120 V on 7.5% Tris-HCl gels. Samples resolved on SDS-PAGE gels were transferred to nitrocellulose membranes. Transfer of proteins to the membranes was confirmed by visualization of pre-stained protein standards. The nitro-cellulose membranes were blocked in blocking buffer [5% blotting grade blocker and 0.1% Tween-20 in PBS] for one hour. Membranes were incubated overnight at 4 °C with primary antibodies diluted in blocking buffer. Membranes were washed three times with wash buffer [0.1% Tween-20 in PBS] and then incubated with secondary antibody for one hour. Membranes were washed three times in wash buffer before addition of HyGLO ECL Spray and exposure to HyBlot CL autoradiography film [both Denville Scientific, Metuchen, NJ]. This procedure was performed using cells grown at both 30 and 37 °C. Densitometry was performed on Western blots using Adobe Photoshop [http://www.lukemiller.org/journal/2007/08/quantifying-western-blots-without.html].
2.6 Collagenase Digestion
Aliquots of lysed cells from transfection experiments were digested with collagenase type III supplemented with 5 mM calcium chloride at 37 °C for one hour. The reaction was stopped by the addition of 2X SDS-PAGE loading buffer containing 5 mM EDTA. Western blotting was then performed as previously described.
2.7 Immunofluorescence
The pCMV COL8A2 plasmids were introduced into the CHO-K1 cells using Lipofectamine 2000 transfection reagent. Twenty-four hours after transfection, the cells were washed briefly with PBS. The cells were then incubated in ice cold methanol for one hour at room temperature. The cells were washed briefly with PBS and then incubated with 10% goat serum and 0.5% Triton-X 100 in PBS for one hour at 37 °C. The cells were left overnight in primary antibody diluted in 2% goat serum in PBS at 4 °C. After three brief washes with PBS, the cells were incubated with secondary antibody diluted in 2% goat serum in PBS. Confocal microscopy was carried out on a Zeiss LSM 510 Meta Confocal Microscope [Thornwood, NY].
2.8 Stability Profile Generator
Brodsky et al. developed a collagen relative stability profile generator, which predicts thermal stability profiles for perfect collagen sequences, i.e. those without interruptions in their triple helix [http://compbio.cs.princeton.edu/csc/]. This calculation is based upon the triple helical stabilization tendencies of individual Gly-X-Y tri-peptide units, their intermolecular and intramolecular interactions, the thermal stability of neighboring tri-peptides and the presence of any nearby stabilizing sequences. Using this algorithm, we predicted the relative stability of a 177-amino acid sequence of uninterrupted triple helix surrounding the sites of the collagen VIII mutations. KGE [Lys-Gly-Glu] and KGD [Lys-Gly-Asp] sequences are also capable of electrostatically stabilizing collagen triple helices; the presence of these stabilizing sequences was also ascertained.
2.9 Collagen Mimetic Peptides
Given the substantial length of COL8A2 [740 amino acids], we elected to use collagen mimetic peptides [39 amino acids] for thermal melting studies. Our collagen mimetic peptides [CMPs] employ a ‘guest-host’ design, in which triple helix-stabilizing Gly-Pro-Hyp [GPO] sequences flank a ‘region of interest’ (Shah et al., 1996). In our case, the ‘region of interest’ was a 15 amino acid region centered on known collagen VIII mutations. The CMPs used were designated WT-P [wild-type], Q455K-P, L450W-P and DM-P, a double mutant containing both the Q455K and the L450W changes. Any proline resides found in the ‘Y’ position within the ‘region of interest’ were assumed to have been post-translationally modified to hydroxyproline, as is typical in nature (Baum and Brodsky, 1999; Ramshaw et al., 1998) The CMPs, synthesized by Tufts University Core Facility [Boston, MA], were: WT-P (GPO)4GQKGDLGLOGQOGLR(GPO)4, Q455K-P (GPO)4GQKGDLGLOGKOGLR(GPO)4, L450W-P (GPO)4GQKGDWGLOGQOGLR(GPO)4 and DM-P (GPO)4GQKGDWGLOGKOGLR(GPO)4. CMPs had an acetylated N-terminus, an amidated C-terminus and were purified by HPLC.
2.10 Circular Dichroism Spectroscopy
Circular dichroism [CD] measurements were taken using a JASCO 710 spectropolarimeter [JASCO Inc., Easton, MD]. Prior to CD measurements, each 150 μM sample was incubated at 4 °C overnight to allow formation of a triple helical structure. All the CMPs studied formed stable triple-helices, as indicated by their characteristic circular dichroism spectrum with a maximum near 225 nm and a minimum near 198 nm. The characteristic triple-helix CD maximum at 225 nm was used to monitor the thermal melting transitions, by measuring the mean residue molar ellipticity, as the samples were continuously heated from 5 °C to 60 °C at a rate of 1 °C/min. Temperature was ramped using the JASCO temperature controller. The melting temperature, Tm, of the sigmoidal melting transition was defined as the minimum of the first derivative of the ellipticity versus temperature curve (Shat et al., 1996).
2.11 Statistics
Statistical analysis was performed using GraphPad Prism 4 software [La Jolla, CA]. Data are expressed as the mean +/− standard error. Significant differences (p<0.05) were determined using the two-tailed Mann Whitney test.
3. Results
3.1 Recombinant Expression of COL8A2
Immunoreactive bands at 75 and 250 kDa were detected in the transfected cells; these molecular weights correspond to collagen VIII monomers and trimers [Figure 1(a)]. In contrast, collagen VIII was not detected in the empty pCMV vector control cells, confirming the specificity of the antibody signal. These bands disappeared following collagenase treatment [results not shown]. Densitometry was performed to quantify the relative amounts of collagen VIII [Table 1], and demonstrated that the amount of monomeric collagen VIII was similar in all cells [Relative density WT 1.00+/− 0.20; Q455K 0.72+/−0.09; L450W 1.38+/−0.29; DM 1.25+/−0.03 Mean+/−S.E.M.; p>0.05 for all three mutant genotypes compared to WT]. Thus, translation of monomeric collagen VIII was similar in all genotypes. However, significantly more trimeric collagen VIII was present within the mutant transfected cells [Relative density WT 1.00+/−0.18; Q455K 2.32+/−0.79; L450W 5.76+/−1.67; DM 4.86+/−0.77 Mean+/−S.E.M.; p<0.03 for all three mutant genotypes compared to WT]. The ratio of trimeric to monomeric collagen VIII is shown in Figure 1(b). Of note, there was no difference in the amounts of actin found in the different genotypes, indicating that the cellular proteins had been successfully quantified and normalized [Figure 1(a)]. Neomycin phosphotransferase II [NPTII] is the antibiotic resistance gene contained on the pCMV plasmid. Western blotting for NPTII showed an increased signal from the empty vector transfected cells, as compared to the wild-type and mutant transfected cells. However, there was no significant difference in expression of NPTII between the wild-type and mutant transfected cells, indicating consistent levels of transfection across COL8A2 genotypes [Figure 1(a)].
Figure 1. Recombinant Expression of COL8A2 Protein by Transfected CHO Cells.
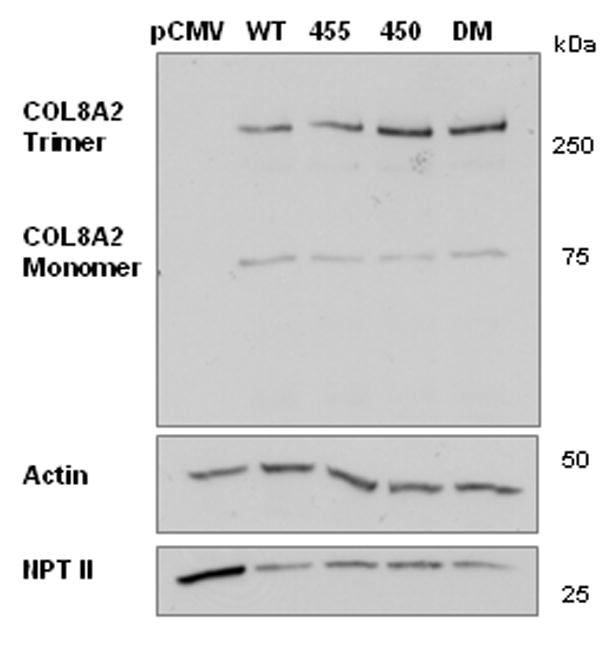

CHO cells were transfected with wild-type and mutant COL8A2 plasmids [n=5]. [A], levels of monomeric 75kD FLAG-tagged collagen VIII, normalized to actin levels, were similar in all genotypes. Levels of trimeric collagen VIII [250kD], normalized to actin levels, were significantly higher in the mutant genotypes. There was no significant difference in the levels of NPTII between the COL8A2 genotypes, indicating consistent levels of transfection across genotypes. [B], the ratio of trimeric to monomeric collagen VIII was calculated and found to be significantly higher in the mutant genotypes. *p<0.02. Error bars, S.E.M.
Table 1.
Cellular production (densitometry units) of FLAG-tagged collagen VIII at 37 °C.
| Genotype | Exp*. 1 | Exp. 2 | Exp. 3 | Exp. 4 | Exp. 5 | Average | Standard Error Mean | p Value |
|---|---|---|---|---|---|---|---|---|
| Monomeric Collagen VIII | ||||||||
| WT | 134,539 | 309,160 | 82,781 | 246,966 | 246,345 | 203,958 | 41,362 | |
| Q455K | 133,031 | 105,430 | 218,326 | 130,506 | 145,476 | 146,554 | 19,084 | NS |
| L450W | 295,280 | 138,825 | 432,239 | 155,859 | 388,520 | 282,145 | 59,374 | NS |
| DM | 270,311 | 231,465 | 258,043 | 256,663 | 260,655 | 255,427 | 6,448 | NS |
| Trimeric Collagen VIII | ||||||||
| WT | 68,145 | 134,940 | 129,533 | 225,656 | 179,045 | 147,464 | 26,345 | |
| Q455K | 295,974 | 143,861 | 794,924 | 186,311 | 288,852 | 341,984 | 116,958 | 0.028 |
| L450W | 351,620 | 766,371 | 1,785,611 | 575,158 | 768,903 | 849,533 | 246,258 | 0.004 |
| DM | 463,827 | 912,386 | 1,050,361 | 634,261 | 523,828 | 716,933 | 113,482 | 0.004 |
| Trimer/Monomer Collagen VIII Ratio | ||||||||
| WT | 0.5065 | 0.4364 | 1.5647 | 0.9137 | 0.7268 | 0.830 | 0.202 | |
| Q455K | 2.2248 | 1.3645 | 3.6410 | 1.4276 | 1.9855 | 2.129 | 0.411 | 0.016 |
| L450W | 1.1908 | 5.5204 | 4.1310 | 3.6902 | 1.9790 | 3.302 | 0.773 | 0.008 |
| DM | 1.7159 | 3.9417 | 4.0704 | 2.4711 | 2.0096 | 2.842 | 0.490 | 0.004 |
Experiment
3.2 Immunofluorescence of Transfected Cells
In order, to confirm the Western blot results we performed immunofluorescence of transfected cells using a collagen VIII antibody which recognizes only trimeric collagen VIII. Non-transfected CHO-K1 cells and pCMV empty-vector transfected cells did not demonstrate any immuno-staining, confirming the antibody specificity. All other cells showed staining with an anti-COL8A2 antibody [Representative cells are shown in Figure 2]. In the wild-type transfected cells the staining was mainly present in a discrete peri-nuclear fashion, but with some general cytoplasmic staining. The Q455K-transfected cells demonstrated a similar pattern but showed an increased density of staining. In contrast, the L450W- and DM-transfected cells both showed substantially more cytoplasmic staining. These findings confirmed the results of the cell-lysate Western blot experiments, indicating that there was an increase in the level of intracellular trimeric COL8A2 in FECD mutant genotypes.
Figure 2. Cellular Collagen VIII Immunofluorescence.
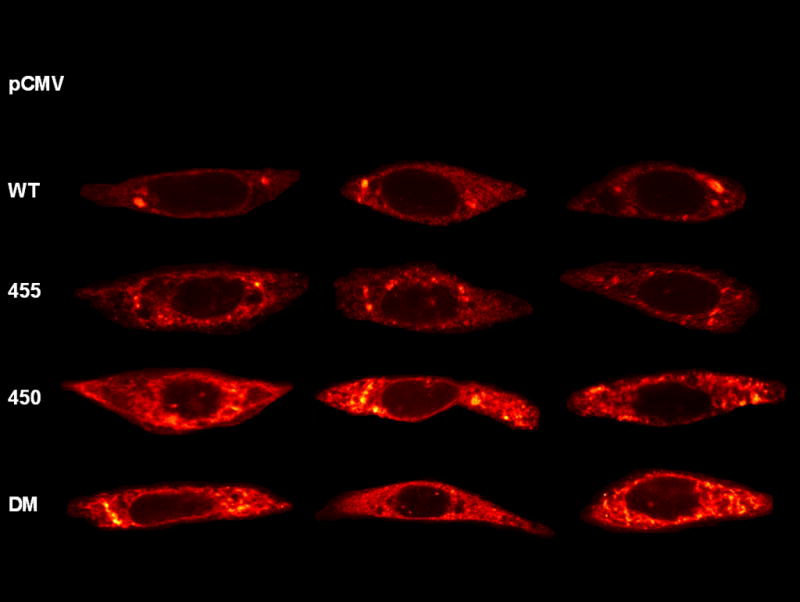
CHO cells were transfected with wild-type and mutant COL8A2 plasmids. After 24 hours, they were incubated with anti-COL8A2 antibody and confocal microscopy was performed. These representative micrographs demonstrate intracellular accumulation of collagen VIII in mutant transfected cells.
3.3 Relative Stability Profile Generator
In an effort to better understand the atypical cellular behavior of the mutant collagen VIII, we probed the biochemical differences induced in the collagen by the amino acid substitutions. We commenced by examining the thermal stability of the collagen molecules using an on-line program, and then used collagen mimetic peptides to further investigate any biochemical differences.
The relative thermal stability of the native and mutated collagen VIII triple-helical sequences, as predicted using the Brodsky collagen relative stability profile generator, varied between 32 and 46 °C [Figure 3(a)]. The plot of WT [black line] has three broadly defined unstable domains. Of note, according to the relative stability plot, the FECD mutations were located at a relatively unstable portion of the collagen triple helix. The Q455K mutation [black line with diamonds] gave a similar plot to that of the WT plot, but the 450 [light grey line] and DM [dashed grey line] gave a plot demonstrating an area with a further decrease in thermal stability [approximately 2 °C] in the region of the mutations [Figure 3(b)].
FIGURE 3. Collagen Relative Profile Stability Generator.


[A], the online Brodsky collagen relative stability profile generator [http://compbio.cs.princeton.edu/csc/] was used to generate an in silico prediction of the thermal stability of a collagenous region surrounding the FECD collagen VIII mutations. This prediction, which is based upon the collagen amino acid composition, showed that the FECD mutations are located at one of the more thermally unstable regions of the triple helix. The locations of the L450W and Q455K mutations are indicated with arrows. [B], a magnified view of the area containing the mutations. Both the L450W and DM mutations were predicted to decrease the relative thermal stability of the collagen triple helix by several degrees.
In addition, there was a KGD sequence immediately upstream of the L450W site [Table 2]. It is likely that the insertion of a tryptophan residue in close proximity to this stabilizing sequence contributed to the predicted lower melting temperature predicted in the L450W and DM substitutions compared with the Q455K and WT alleles.
Table 2.
The sites of pathogenic COL8A2 mutations. Both mutations [underlined] lie in close proximity to a stabilizing KGD [bold] sequence.
| Amino Acid Position | WT | Q455K | L450W | DM |
|---|---|---|---|---|
| 445–447 | GQK | GQK | GQK | GQK |
| 448–450 | GDL | GDL | GDW | GDW |
| 451–453 | GLO | GLO | GLO | GLO |
| 454–456 | GQO | GKO | GQO | GKO |
3.4 Circular Dichroism Spectroscopy and Thermal Melting of CMPs
The CD melting experiments indicated the major melting temperatures for WT and all mutant CMPs. In order to pinpoint more exact melting temperatures for all the CMPs, we plotted the first derivative of the CD signal against the temperature [Figure 4]. The melting temperatures of the CMPs as determined from the minimum of these plots were WT-P 44.5°C, Q455K-P 44.7°C, L450W-P 39.0°C and DM-P 40.5°C [Figure 4]. As predicted by the in silico algorithm, the triple helical domain of the WT-P and Q455K-P exhibited similar melting temperatures, whereas the L450W-P and DM-P were less thermally stable than the WT-P.
Figure 4. Circular Dichroism Data for the First Derivative of the Melting Curve for WT and Mutant CMPs.

The collagen mimetic peptides were found to have melting temperatures of WT-P 44.5°C, Q455K-P 44.7°C, L450W-P 39.0°C and DM-P 40.5°C. Thus, the triple helix in the L450W and DM CMPs was less thermally stable than that in the WT and Q455K CMPs.
3.5 Low Temperature Cell Culture & Recombinant Expression of COL8A2
Based on our results, which suggested that there exists an alteration in the thermal stability of the mutated collagen VIII, we hypothesized that local thermal instability could contribute to the cellular phenotype. Thermally unstable collagen could partially unfold at 37 °C and collagen secretion from the cell could be hindered by this alteration in the collagen conformation. We postulated that a lower CHO cell cultivation temperature would reduce the observed phenotype by means of decreased collagen unfolding. Consistent with this possibility was our observation [during preparation of COL8A2 cDNAs] that DM-transformed E. coli grew at 30 °C but not at 37 °C. Thus, CHO cells were cultured at 30 °C, the recombinant COL8A2 transfection experiments were repeated and levels of intra-cellular collagen VIII were ascertained [Table 3]. The amount of monomeric collagen VIII was similar in all genotypes [Relative Density WT 1.0+/−0.30; Q455K 1.28+/−0.86; L450W 1.15+/−0.55; DM 0.8+/−0.36 Mean+/−S.E.M.]. The amount of trimeric collagen VIII was elevated in the mutant genotypes although this did not reach statistical significance [Relative Density WT 1.0+/−0.56; Q455K 2.25+/−0.24; L450W 3.13+/−0.67; DM 3.01+/−1.13 Mean+/−S.E.M.] The ratio of trimeric to monomeric collagen VIII is shown in Figure 5 (a) & (b); [Trimer/Monomer Relative Density WT 1.0+/−0.45; Q455K 4.66 +/−2.64; L450W 3.76+/−1.0; DM 3.91+/−0.19 Mean+/−S.E.M]. The trimer/monomer ratio was higher with borderline significance in the L450W and DM genotypes compared with WT, p=0.05. Thus, aberrant trafficking of the mutant collagen VIII was not corrected by cultivation of the cells at a lower temperature. This implies that thermal instability of the collagen triple helix is not solely responsible for the intracellular accumulation of the collagen.
Table 3.
Cellular production (densitometry units) of FLAG-tagged collagen VIII at 30 °C.
| Genotype | Experiment 1 | Experiment 2 | Experiment 3 | Average | Standard Error Mean | p Value |
|---|---|---|---|---|---|---|
| Monomeric Collagen VIII | ||||||
| WT | 492,667 | 255,676 | 186,560 | 311,634 | 92,689 | |
| Q455K | 91,644 | 932,101 | 175,581 | 399,775 | 267,263 | 0.35 |
| L450W | 269,533 | 691,803 | 117,887 | 359,741 | 171,705 | 0.50 |
| DM | 137,277 | 472,574 | 142,727 | 250,859 | 110,868 | 0.20 |
| Trimeric Collagen VIII | ||||||
| WT | 435,640 | 17,228 | 193,125 | 215,331 | 121,294 | |
| Q455K | 585,116 | 449,475 | 416,106 | 483,566 | 51,681 | 0.10 |
| L450W | 671,311 | 923,245 | 426,224 | 673,593 | 143,482 | 0.10 |
| DM | 324,355 | 1,219,692 | 399,961 | 648,002 | 286,676 | 0.20 |
| Trimer/Monomer Collagen VIII Ratio | ||||||
| WT | 0.8842 | 0.0673 | 1.0351 | 0.662 | 0.30 | |
| Q455K | 6.3846 | 0.4822 | 2.3698 | 3.079 | 1.74 | 0.20 |
| L450W | 2.4906 | 1.3345 | 3.6155 | 2.480 | 0.66 | 0.05 |
| DM | 2.3627 | 2.5809 | 2.8022 | 2.582 | 0.13 | 0.05 |
Figure 5. Recombinant Expression of COL8A2 Protein by Transfected CHO Cells Cultured at 30°C.
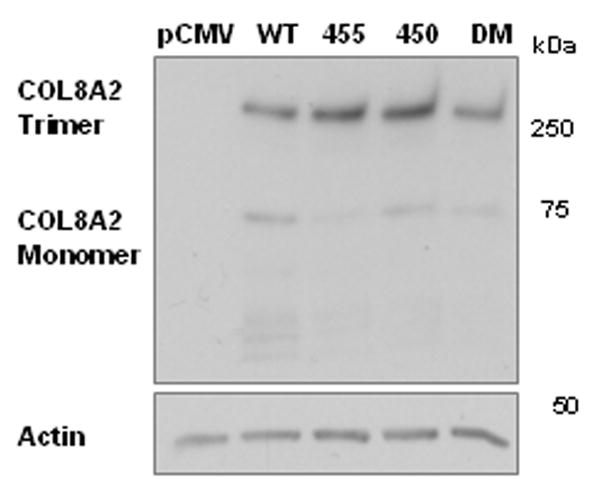

CHO cells were transfected with wild-type and mutant COL8A2 plasmids [n=3]. The cells were cultured at 30 °C, in an effort to ameliorate the intracellular accumulation of trimeric collagen VIII by mutant-transfected cells. [A], there was no significant difference in the levels of intracellular trimeric collagen VIII [250kD]. [B], the ratio of trimeric/monomeric collagen VIII was calculated and was borderline significantly higher in the L450W and DM gentotypes. *p=0.05. Error bars, S.E.M.
4. Discussion
Collagen VIII is present in pathologic guttae and thickened Descemet’s membrane in late-onset FECD, and mutations in the COL8A2 gene cause early-onset FECD. Thus, this gene provides a biological link between early and late onset FECD and represents an attractive starting point from which to probe the pathobiology of this disease.
Initially, we used a cell culture model. We discovered that, whilst similar amounts of monomeric collagen VIII were produced by all the transfected cells, the mutant transfected cells had increased amounts of trimeric collagen VIII. Trimeric collagen VIII is formed by the association of three monomeric strands. We believe that the disparity between the levels of single-stranded and triple-stranded collagen VIII within the cells is explained by comparable production of collagen monomers but either delayed secretion or impaired degradation of aberrant trimeric collagen VIII in mutant-transfected cells.
We were conscious that the presence of FLAG-tags could alter the collagen post-translational processing, but the FLAG-tags were located at the N-terminal. Collagen folding proceeds from the C terminal so we felt that their impact on collagen folding would be minimal. We observed increased intracellular accumulation of homotrimeric collagen with mutant COL8A2 genotypes compared with WT alleles. In addition, intracellular retention of incorrectly folded/abnormal collagen has previously been described in a variety of other collagen diseases, as well as in vivo in patients with COL8A2-related FECD (Fritsch et al., 2009; Wilson et al., 2002; Zhang et al., 2006).
The COL8A2 mutations investigated in this study [L450W and Q455K] are both located within the triple helical portion of the molecule and thus both have the ability to perturb the local helical microenvironment. Of interest, both mutations were located at a pre-existing area of decreased thermal stability. Thermally unstable regions of collagen are known to partially unfold, are widespread in collagen and most importantly are frequently areas of biological importance (Brodsky and Baum, 2008; Persikov et al., 2005). Of note, a study examining collagen VII also noted an association between increased intracellular retention of aberrant collagen peptides and decreasing collagen thermal stability (Fritsch et al., 2009).
We predicted that the L450W mutation would disrupt the collagen triple helix by several means. Although the L450W mutation represents the substitution of a neutral, non-polar amino acid [leucine] for another, tryptophan is a bulky amino acid rarely found in collagens and is likely to interfere sterically with helical/protein folding (Ramshaw et al., 1998). The presence of tryptophan is also likely to disrupt the nearby stabilizing KGD sequence. Our predictions were confirmed with the finding that the L450W mutation demonstrated significantly decreased thermal stability, but that intracellular retention was not resolved with low temperature cell cultivation.
In contrast, the Q455K mutation represents the substitution of an evolutionarily conserved polar, neutral amino acid [glutamine] with a polar, positive amino acid [lysine] (Biswas et al., 2001). Lysine is of particular importance in collagen biology as many of these residues are hydroxylated during post-translational modification. Lysine and hydroxy-lysine residues subsequently participate in the formation of covalent bonds resulting in collagen cross-linking (Biswas et al., 2001; Raghunath et al., 1994). Thus, the Q455K mutation may result in a more stable triple helix and may also influence inter-strand cross-linking (Persikov et al., 2005).
Of interest, both mutations, although displaying differing biochemical properties, resulted in intracellular retention of collagen VIII. Chronic intracellular accumulation of collagen VIII is likely to be injurious to the fragile corneal endothelium and the unfolded protein stress response and increased endothelial cell apoptosis have previously been noted in patients with FECD (Borderie et al., 2000; Engler et al., 2010; Joyce, 2003; Li et al., 2001). Collagen VIII turnover in the cornea is very limited, so retained intracellular collagen VIII remaining after endothelial apoptosis would likely result in collagen VIII accumulation within Descemet’s membrane as noted histologically in FECD (Kabsova et al., 2007)
We have concluded that aberrant collagen VIII is retained intracellularly, although we propose the mechanisms underlying the accumulation in the Q455K and L450W mutations differed. Fritsch et al. (2009) examined COL7A1 mutations associated with epidermolysis bullosa and, as with the L450W mutation, discovered a relationship between mutant collagen VII thermal instability and intracellular retention. In their study, over-expression of WT collagen resulted in the formation of collagen heterotrimers (i.e. collagen trimers formed with WT:mutant monomers in a 1:2 or 2:1 ratio) and stabilization of the collagen melting temperaure. A similar approach could be useful in the treatment of L450W-associated FECD. Similarly, mutant allele-specific knock-down may represent another useful strategy.
The mutations studied result clinically in early onset FECD and could be used potentially as the basis for producing an animal model of FECD. Although COL8A2 mutations are not associated with the more common, late-onset form of FECD, the pathology of both forms of the disease are similar in having progressive, specific loss of endothelial cells associated with characteristic excrescences and thickening of Descemet’s membrane. Thus, ongoing and future studies to elucidate the cellular pathophysiology of both early and late onset FECD through cell culture and animal models may yield valuable insights into endothelial cell biology and FECD pathogenesis with potentially therapeutic implications.
Highlights.
Collagen VIII accumulation is noted in the cornea in Fuchs Endothelial Dystophy
Mutated collagen VIII results in early onset FED.
Two mutations were examined and found to have different biochemical and cellular properties, although both cause FED in vivo.
Given their differing biochemical characteristics, animal models using these mutations may be useful both in assessing the pathobiology of this disease and investigating potential therapies.
Acknowledgments
The authors thank Rhonda Grebe, MS, for microscopy assistance and Yang Li for help with collagen mimetic peptide design. Supported by grants from the National Institutes of Health: R01EY11654 (SC), KO8EY15523 (ASJ), RO1EY19874 (ASJ), R01AR060484 (SMY), EY0001765 (Microscopy & Imaging Module of the Wilmer Core Grant), National Science Foundation DMR0645411 (MSY), Research to Prevent Blindness (Career Development and Special Scholar Awards, both to ASJ), Wilmer Professors’ Research Fund (ASJ), Medical Illness Counseling Center (Chevy Chase, MD to ASJ), Eye Bank Association of America (CK), and Wilmer Fellows’ Research Fund (CK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Clare Kelliher, Email: clarekelliher@yahoo.ie.
Shukti Chakravarti, Email: schakra1@jhmi.edu.
Neeraj Vij, Email: nvij1@jhmi.edu.
Steve Mazur, Email: lemazur@gmail.com.
Patrick J. Stahl, Email: pstahl@jhu.edu.
Christoph Engler, Email: christoph.engler1@gmail.com.
Mario Matthaei, Email: mmattha1@jhmi.edu.
S. Michael Yu, Email: yu@jhu.edu.
Albert S. Jun, Email: aljun@jhmi.edu.
References
- Bateman JF, Freddi S, McNeil R, Thompson E, Hermanns P, Savarirayan R, Lamande SR. Identification of four novel COL10A1 missense mutation in schmid metaphyseal chondrodysplasia: further evidence that collagen X NC1 mutations impair trimer assembly. Hum Mutat. 2004;23:396. doi: 10.1002/humu.9222. [DOI] [PubMed] [Google Scholar]
- Baum J, Brodsky B. Folding of peptide models of collagen and misfolding in disease. Curr Opin Struct Biol. 1999;9:122–128. doi: 10.1016/s0959-440x(99)80016-5. [DOI] [PubMed] [Google Scholar]
- Biswas S, Munier FL, Yardley J, Hart-Holden N, Perveen R, Cousin P, Sutphin JE, Noble B, Batterbury M, Kielty C, Hackett A, Bonshek R, Ridgway A, McLeod D, Sheffield VC, Stone EM, Schorderet DF, Black GCM. Missense mutations in COL8A2, the gene encoding the α2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum Mol Genet. 2001;10:2415–2423. doi: 10.1093/hmg/10.21.2415. [DOI] [PubMed] [Google Scholar]
- Borderie VM, Baudrimont M, Vallee A, Ereau TL, Gray F, Laroche L. Corneal endothelial cell apoptosis in patients with Fuchs’ dystrophy. Invest Ophthalmol Vis Sci. 2000;41:2501–2505. [PubMed] [Google Scholar]
- Brodsky B, Baum J. Modelling collagen diseases: structural biology. Nature. 2008;453:998–999. doi: 10.1038/453998a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler C, Kelliher C, et al. Comparison of non-viral methods to genetically enrich populations of primary human corneal endothelial cells. Mol Vis. 2009;15:629–637. [PMC free article] [PubMed] [Google Scholar]
- Engler C, Kelliher C, Spitze AR, Speck CL, Eberhart CG, Jun AS. Unfolded protein response in Fuchs’ endothelial dystrophy: a unifying pathogenic pathway? Am J Ophthalmol. 2010;149:194–202.e2. doi: 10.1016/j.ajo.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch A, Spassov S, Elfert S, Schlosser A, Gache Y, Meneguzzi G, Bruckner-Tuderman L. Dominant-negative effects of COL7A1 mutations can be rescued by controlled overexpression of normal collagen VII. JBiol Chem. 2009;44:30248–30256. doi: 10.1074/jbc.M109.045294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottsch JD, Sundin OH, Liu SH, Jun AS, Broman KW, Stark WJ, Vito ECL, Narang AK, Thompson JM, Magovern M. Inheritance of a novel COL8A2 mutation defines a distinct early-onset subtype of Fuchs corneal dystrophy. Invest Ophthalmol Vis Sci. 2005;46:1934–1939. doi: 10.1167/iovs.04-0937. [DOI] [PubMed] [Google Scholar]
- Greenhill NS, Ruger BM, Hasan Q, Davis PF. The α1(VIII) and α2(VIII) collagen chains form two distinct homotrimeric proteins in vivo. Matrix Biol. 2000;19:19–28. doi: 10.1016/s0945-053x(99)00053-0. [DOI] [PubMed] [Google Scholar]
- Hogan MJ, Wood I, Fine M. Fuchs’ endothelial dystophy of the cornea. 29th Sanford Gifford memorial lecture. Am J Ophthalmol. 1974;78:363–383. doi: 10.1016/0002-9394(74)90224-4. [DOI] [PubMed] [Google Scholar]
- Joyce NC. Proliferative capacity of the corneal endothelium. Prog Ret Eye Res. 2003;22:359–389. doi: 10.1016/s1350-9462(02)00065-4. [DOI] [PubMed] [Google Scholar]
- Jurkunas UV, Bitar MS, Funaki T, Azizi B. Evidence of oxidative stress in the pathogenesis of Fuchs endothelial corneal dystrophy. Am J Pathol. 2010;177:2278–2289. doi: 10.2353/ajpath.2010.100279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor R, Bornstein P, Sage EH. Type VIII collagen from bovine Descemet’s membrane: structural characterization of a triple-helical domain. Biochemistry. 1986;25:3930–3937. doi: 10.1021/bi00361a029. [DOI] [PubMed] [Google Scholar]
- Illidge C, Kielty CM, Shuttleworth CA. Stability of type VIII collagen homotrimers: comparison with α1(X) Biochem Soc Trans. 1998a;26:S18. doi: 10.1042/bst026s018. [DOI] [PubMed] [Google Scholar]
- Illidge C, Kielty C, Shuttleworth A. The α1(VIII) and α2(VIII) chains of type VIII collagen can form stable homotrimeric molecules. JBC. 1998b;1998:22091–22095. doi: 10.1074/jbc.273.34.22091. [DOI] [PubMed] [Google Scholar]
- Illidge C, Kielty C, Shuttleworth A. Type VIII collagen: heterotrimeric chain association. Int J Biochem Cell Biol. 2001;33:521–529. doi: 10.1016/s1357-2725(01)00013-9. [DOI] [PubMed] [Google Scholar]
- Joyce NC. Proliferative capacity of the corneal endothelium. Prog Retin Eye Res. 2003;22:359–389. doi: 10.1016/s1350-9462(02)00065-4. [DOI] [PubMed] [Google Scholar]
- Kabosova A, Azar DT, Bannikov GA, Campbell KP, Durbeej M, Ghohestani RF, Jones JCR, Kenney MC, Koch M, Ninomiya Y, Patton BL, Paulsson M, Sado Y, Sage EH, Sasaki T, Sorokin LM, Steiner-Champliaud MF, Sun TT, SundarRaj N, Timpl R, Virtanen I, Ljubimov AV. Compositional differences between infant and adult human corneal basement membranes. Invest Ophthalmol Vis Sci. 2007;48:4989–4999. doi: 10.1167/iovs.07-0654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44:283–292. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- Kramer RZ, Bella J, Mayville P, Brodsky B, Berman HM. Sequence dependent conformation variations of collagen triple-helical structure. Nat Struct Biol. 1999;6:454–457. doi: 10.1038/8259. [DOI] [PubMed] [Google Scholar]
- Lee TY, Hsu JBK, Chang WC, Wang TY, Hsu PC, Huang HD. A comprehensive resource ofr integrating and displaying protein post-translational modifications. BMC Res Notes. 2009;2:111. doi: 10.1186/1756-0500-2-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy SG, Moss J, Sawada H, Dopping-Hepenstal PJ, McCartney AC. The composition of wide-spaced collagen in normal and diseased Descemet’s membrane. Curr Eye Res. 1996;15:45–52. doi: 10.3109/02713689609017610. [DOI] [PubMed] [Google Scholar]
- Li QJ, Ashraf MF, Shen DF, Green WR, Stark WJ, Chan CC, O’Brien TP. The role of apoptosis in the pathogenesis of Fuchs endothelial dystrophy of the cornea. Arch Ophthalmol. 2001;119:1597–1604. doi: 10.1001/archopht.119.11.1597. [DOI] [PubMed] [Google Scholar]
- Liskova P, Prescott Q, Bhattacharya SS, Tuft SJ. British family with early-onset Fuchs’ endothelial corneal dystrophy associated with pL450W mutation in the COL8A2 gene. Br J Ophthalmol. 2007;91:1717–1718. doi: 10.1136/bjo.2007.115154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muragaki Y, Jacenko O, Apte S, Mattei MG, Ninomiya Y, Olsen BR. The α2(VIII) collagen gene. J Biol Chem. 1991;266:7721–7727. [PubMed] [Google Scholar]
- Persikov AV, Ramshaw JAM, Brodsky B. Prediction of collagen stability from amino acid sequence. J Biol Chem. 280:19343–19349. doi: 10.1074/jbc.M501657200. [DOI] [PubMed] [Google Scholar]
- Raganuth M, Bruckner P, Steinmann B. Delayed triple helix formation of mutant collagen from patients with Osteogenesis Imperfecta. J Mol Biol. 1994;236:940–949. doi: 10.1006/jmbi.1994.1199. [DOI] [PubMed] [Google Scholar]
- Ramshaw JAM, Shah NK, Brodsky B. Gly-X-Y tripeptide frequencies in collagen: a context for host-guest triple-helical peptides. J Struct Biol. 1998;122:86–91. doi: 10.1006/jsbi.1998.3977. [DOI] [PubMed] [Google Scholar]
- Sawada H, Konomi H, Hirosawa K. Characterization of the collagen in the hexagonal lattice of Descemet’s membrane; its relation to type VIII collagen. J Cell Biol. 1990;110:219–227. doi: 10.1083/jcb.110.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NK, Ramshaw JA, Kirkpatrick A, Shah C, Brodsky B. A host-guest set of triple-helical peptides: stability of Gly-X-Y triplets containing common nonpolar residues. Biochemistry. 1996;35:10262–10268. doi: 10.1021/bi960046y. [DOI] [PubMed] [Google Scholar]
- Shuttleworth CA. Type VIII collagen. Int J Biochem Cell Biol. 1997;29:1145–1148. doi: 10.1016/s1357-2725(97)00033-2. [DOI] [PubMed] [Google Scholar]
- Suh LH, Emerson MV, Jun AS. Fuchs Endothelial Dystrophy: Pathogenesis and Management. In: Reinhard T, Larkin F, editors. Cornea and External Eye Disease. Springer Verlag; Berlin Heidelberg: 2008. pp. 1–13. [Google Scholar]
- Vithana EN, Morgan PE, Ramprasad V, Tan DTH, Yong VHK, Venkataraman D, Venkatraman A, Yam GHF, Nagasamy S, Law RWK, Rajagopal R, Pang CP, Kumaramanickevel G, Casey JR, Aung T. SLC4A11 mutations in Fuchs endothelial corneal dystrophy. Hum Mol Genet. 2008;17:656–666. doi: 10.1093/hmg/ddm337. [DOI] [PubMed] [Google Scholar]
- Waring GO, 3rd, Bourne WM, Edelhauser HF, Kenyon KR. The corneal endothelium. Normal & pathological structure and function. Ophthalmology. 1982;89:531–590. [PubMed] [Google Scholar]
- Wilson R, Freddi S, Bateman JF. Collagen X chains harboring schmid metaphyseal chondrodysplasia NC1 domain mutations are selectively retained and degraded in stably tranfected cells. J Biol Chem. 2002;277:12516–12524. doi: 10.1074/jbc.M112044200. [DOI] [PubMed] [Google Scholar]
- Wilson R, Freddi S, Chan D, Cheah KSE, Bateman JF. Misfolding of collagen X chains harboring schmid metaphyseal chondrodysplasia mutations results in aberrant disulfide bond formation, intracellular retention, and activation of the unfolded protein response. J Biol Chem. 2005;280:15544–15552. doi: 10.1074/jbc.M410758200. [DOI] [PubMed] [Google Scholar]
- Yamaguchi N, Mayne R, Ninomiya Y. The alpha1(VIII) collagen gene is homologous to the alpha1(X) collagen gene and contains a large exon encoding the entire triple helical and carboxyl-terminal non-triple helical domains of the alpha1(VIII) polypeptide. J Biol Chem. 1991;266:4508–4513. [PubMed] [Google Scholar]
- Zhang C, Bell R, Sundin OH, De La Cruz Z, Stark WJ, Green WR, Gottsch JD. Immunohistochemistry and electron microscopy of early-onset Fuchs corneal dystrophy in three cases with the same L450W COL8A2 mutation. Trans Am Ophthalmol Soc. 2006;104:85–97. [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Rawe I, Joyce NC. Differential protein expression in human corneal endothelial cells cultured from young and older donors. Mol Vis. 2008;14:1805–1814. [PMC free article] [PubMed] [Google Scholar]