

Abstract
CK2, a pleiotropic Ser/Thr kinase, is an important target for cancer therapy. We tested our novel tenfibgen-based nanocapsule for delivery of the inhibitor 2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole (DMAT) and an siRNA directed against both CK2α and α′ catalytic subunits to prostate cancer cells. We present data on the TBG nanocapsule itself and on CK2 inhibition or downregulation in treated cells, including effects on Nuclear Factor-kappa B (NF-κB) p65. By direct comparison of two CK2-directed cargos, our data provide proof that the TBG encapsulation design for delivery of drugs specifically to cancer cells has strong potential for small molecule- and nucleic acid-based cancer therapy.
Keywords: CK2, prostate cancer, nanocapsule, nanoparticle, DMAT, tenfibgen
1. Introduction
Among the many protein kinases proposed as anti-cancer targets, CK2 (formerly casein kinase II/2) is one of the most consistently elevated protein kinases associated with the oncogenic phenotype across various cancer types [1; 2]. CK2 is a protein serine/threonine kinase existing as a heterotetramer consisting of two catalytic subunits α and α′ linked through two regulatory β subunits. The kinase is localized both to the nuclear and cytoplasmic compartment and may be constitutively active; several modes of its regulation have been proposed, but their precise nature is not fully understood [2; 3]. Through its phosphorylation of numerous substrates, CK2 functions to influence the various biochemical and metabolic processes of cell growth and proliferation, as well as cell death and inflammation and stress signaling, among other processes. This kinase is a very highly conserved protein in nature, and cells or organisms cannot survive without CK2 expression [4; 5]. We originally proposed that the essential nature of CK2 for cell survival renders it a particularly attractive target for the goal of eradicating tumor cells by inhibition of CK2 activity or molecular downregulation of CK2 expression [6].
CK2 steady-state expression levels are distinct for various cell types, and its expression in different tissues under normal quiescent conditions remains stable. However, it has been found to be upregulated in all cancers that have been examined. In this regard, CK2 can be considered a “non-oncogene” whose dysregulated expression is critical for the maintenance of the cancer cell malignant phenotype [1; 7; 8]. This increase in CK2 expression in cancer cells is generally evident at the protein and activity levels and correlates with increased nuclear localization [1; 7]. Elevated CK2 expression, at either protein or RNA levels, or activity has been noted in human leukemias, breast, colorectal, gastric, head and neck, kidney, lung, multiple myeloma, and prostate cancers [1; 2; 9; 10; 11]. Also significant are the observations that CK2 upregulation in neoplasia reflects a complex state of dysplasia, not simply the enhanced proliferative state of the tumor cells [9]. In addition, dysregulation of CK2 expression may relate to the severity of disease and serve as a prognostic indicator [1; 2; 11; 12; 13; 14]. The more recently established function of CK2 in suppressing apoptosis supports its role in cell survival and firmly ties CK2 upregulation and function to the cancer cell phenotype [7; 15]. It is noteworthy that several lines of evidence point to nuclear associated CK2 as the most sensitive subcellular fraction in responding to alterations in cell growth and death [6; 16]. Thus, the combined pleiotropic functions of CK2 and the general dependence of cancer cells on increased CK2 expression offer a strong impetus for the development of anti-CK2 cancer therapeutics [6].
Downregulation of CK2 in numerous human cancer cell lines using siRNAs and antisense oligonucleotides directed against CK2 subunits has generally resulted in reduced cell viability or cell death [6; 10; 11; 16; 17; 18; 19; 20]. Xenograft tumor studies in nude mice have verified that antisense-mediated downregulation of CK2 expression promotes rapid and early loss of CK2 from the nuclear matrix and is associated with induction of apoptosis in both prostate and head neck squamous cell carcinoma (HNSCC) tumors in vivo [2; 21; 22; 23]. Likewise, numerous small molecule inhibitors of CK2 have been developed over the past 30 years. In cultured human cancer cells, these inhibitors effectively reduce cell viability and cause cell death [10; 11; 19; 20; 24; 25; 26; 27]. Distinctive features of the CK2 activation and catalytic site suggest that these characteristics of CK2 can be exploited for the design of inhibitors [28; 29]. There are four published studies to date using small molecule CK2 inhibitors in animal models of cancer and retinal neovascularization [30; 31; 32]. There are also animal and human studies utilizing a novel peptide that impairs CK2 substrate phosphorylation and demonstrates an anti-neoplastic effect in several cancers [33].
Based on the above information, it is clear that CK2 can be targeted using either small molecule inhibitors to affect kinase activity or using antisense and siRNA-mediated molecular downregulation of RNA and protein expression; however, as discussed subsequently, there are positive and negative aspects to both of these approaches. Regardless of whether a pharmacologic or molecular approach is used, a particularly important issue that must be addressed concerns the ubiquitous and essential nature of the CK2 signal. In order to avoid unwanted toxic side-effects in the host, it would be highly advantageous to administer the anti-CK2 drug in a delivery vehicle designed to specifically enter malignant cells while sparing the normal. Currently available delivery methods have certain limitations including in vivo protection of the cargo and bioavailability and specific targeting to tumor cells [34]; these limitations are overcome by our novel delivery technology, designated sub-50 nanometer (i.e., less than 50 nm size) nanocapsules (or s50 nanocapsules). As described subsequently, the s50 nanocapsule is composed entirely of a protein ligand (tenfibgen or TBG) designed to form a shell around the cargo (such as a small molecule inhibitor or condensed antisense or siRNA).
Here we have tested the TBG nanoencapsulated anti-CK2 small molecule inhibitor DMAT (2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole) as well as TBG nanoencapsulated siRNA directed against both catalytic subunits of CK2 α and α′ for their effect on the proliferation and viability of malignant prostate cancer cells compared with that of benign prostate cells. We present characterization data for our unique s50 TBG nanocapsule, including size, charge and morphology. We demonstrate the efficacy of CK2 inhibition or molecular downregulation in the cells and the resulting effects on the downstream CK2 target NF-κB p65. We show that TBG nanocapsules carrying anti-CK2 drug specifically target cancer cells and not the normal cells. Significantly, these data also provide proof, by direct comparison of two types of anti-CK2 cargos, that the TBG encapsulation design for delivery of drugs specifically to cancer cells has utility for both small molecule- and nucleic acid-based drugs, thus reinforcing the utility of CK2 as an important anti-cancer target.
2. Materials and Methods
2.1. Cell Lines and Cell Culture
PC3-LN4 cells, obtained as described [21], were maintained in monolayer culture containing modified Eagle’s MEM or RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM L-glutamine, and penicillin-streptomycin [35]. Immortalized BPH-1 cells were obtained from Dr. Simon Hayward (Vanderbilt University, Nashville, TN). BPH-1 cells were maintained in monolayer culture containing RPMI 1640 supplemented with 10% heat-inactivated FBS and 2 mM L-glutamine [36]. The human prostate epithelial cells (PrEC) were obtained along with their specific medium from Lonza (Walkersville, MD). C4-2 cells were obtained from MD Anderson Cancer Center and were maintained in monolayer culture containing RPMI 1640 supplemented with 10% heat-inactivated FBS and 2 mM L-glutamine. Cells are grown in an incubator at 37 °C with 5% CO2. All cells had undetectable levels of mycoplasma. These cell lines have not been tested or authenticated in this laboratory.
2.2. Oligonucleotides and DMAT
Standard chemistry siRNAs were obtained from Dharmacon (Lafayette, CO). The guide strand of the bispecific siCK2 sequence is 5′-auacaacccaaacuccacauuu-3′ and that for siRFP is 5′-agcuugacgggcuucuugguu-3′. The 2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole (DMAT, compound 2c) was synthesized as described previously [37]; it was suspended in DMSO at a concentration of 5 – 10 mg/ml prior to use.
2.3. Cell Proliferation and Viability Assays
For [3H]-thymidine incorporation assays, PC3-LN4 cancer cells were plated in medium containing 0.01% FBS, 0.1% for C4-2 cells, at a density of 3000 cells per well in 96 plates containing nanofiber tissue engineering scaffolds (Surmodics Synthetic ECM-NAN) pre-coated overnight with human tenascin-C (Millipore CC065) and bovine fibronectin (Sigma F-1141) in a 3:1 ratio at a total concentration of 25 fg/cm2 (100 pg/ml) in PBS. Benign BPH-1 and PrEC cells were grown as described above and plated at a density of 1500 cells per well in 96 well Primaria™ plates pre-coated overnight with laminin (Sigma L-2020) at 0.5 μg/cm2 in PBS. For DMAT studies, cells were treated the following day at various concentrations with DMAT or DMSO, or TBG nanocapsules containing DMAT or the sugar trehalose. For siRNA studies, cells were treated the following day at various concentrations with TBG nanocapsules containing siCK2, siRFP or sugar. After 48 h for siRNA or 72 h for DMAT studies, the media was replaced with 100 μl fresh media containing 0.5 μCi [3H]-thymidine (NEN NET027). After an additional 24 h, the cells are harvested onto filters and placed into scintillation vials with scintillation fluid for measurement of radioactivity. Different batches of TBG-DMAT or TBG-siCK2 gave similar results.
For WST-1 assays, cells were plated in RPMI containing 10% heat-inactivated FBS at a density of 2500 cells per well in 96 well Primaria™ plates. The following day, transfection complexes of varying siRNA molarities containing 0.2 μl of Dharmafect 2 in a total volume of 100 μl were added to the cells. After 5 h incubation, an additional 100 μl of complete media was added to each well. WST-1 reagent (10 μl, Roche 11 644 807 001) was added to the cells at 72 h in a total volume of 100 μl media, the cells incubated at 37 °C for 1 h, and assayed according to the manufacturer’s instructions.
2.4. Immunoblot Analysis of DMAT Treated Cells
PC3-LN4 cells were plated onto 60 mm plates. The following day, the cells at 50% confluence were treated with naked DMAT (or equivalent dilution of DMSO) at 10, 1 and 0.1 μM. Cells were collected 24 h after treatment and the cell pellets were frozen at −20 °C immediately following collection. Processing of cell pellets and further analyses were performed as described [38]. Antibodies used were: CK2α and CK2α′ (Bethyl Laboratories A300-197A and A300-199A); CK2β (sc-12739), actin (sc-1616), p-NF-κB p65 Ser 529 (sc-101751) were from Santa Cruz Biotechnology; and NF-κB p65 from (BD Transduction Laboratories 610869).
2.5. TUNEL Analysis for Detection of Apoptosis
PC3-LN4 cells were plated at a density of 5 × 105 cells on 60 mm plates. The following day, siRNAs were transfected into the cells at various concentrations using 10 μl of Dharmafect 2 (Dharmacon, Inc.) per 600 μl final volume transfection mixture. The final volume of transfection mixture plus complete media was 3 ml. After 5 h, the transfection mixture was replaced with 5 ml fresh complete media. Cells were split as needed at 24 h, and then were plated onto cover slips at 48 h post-transfection. At 72 h, the cells were fixed with 3% paraformaldehyde for 30 min at 4°C, washed twice with phosphate-buffered saline (PBS), permeabilized with ice cold methanol for 10 min, and washed twice with water. TUNEL analysis was performed using the kit APO-BRDU (Phoenix Flow Systems, Inc.). Cell nuclei were counterstained for 30 s in 5 μg/ml bisbenzimide, the cells washed 1× PBS (pH 7.4) with 0.05% NP-40 (PBS-NP) and 1× water for 5 min each, and mounted onto a slide in aqueous no-fade solution. The cells were viewed using an Olympus BX60 fluorescent microscope 40× objective with images collected using a digital color Q Imaging Retiga 2000R Fast1394 camera. 100 cells were analyzed per transfection condition, and the experiment was performed twice. Antibodies used: BrdU from BD Transduction Laboratories (347580); biotinylated sheep anti-mouse (515-066-072) and streptavidin-Cy3 (016-030-084) from Jackson Labs.
2.6. Immunofluorescence Analysis of DMAT Effect
PC3-LN4 cells were plated onto glass coverslips pre-coated overnight with either poly-D-Lysine at 0.5 mg/ml (Sigma P6407) for naked DMAT treatment or with human tenascin-C (Millipore CC065) and bovine fibronectin (Sigma F-1141) in a 3:1 ratio at a total concentration of 0.25 μg/cm2 in PBS. The next day, the cells were treated with 0.1, 1 or 10 μM DMAT or an equivalent dilution of DMSO in complete media, or with TBG-DMAT or TBG-sugar nanocapsules at a final concentration of 210 nM. Coverslips were collected at 24, 48 or 72 h post-drug addition and the cells were fixed in 2% paraformaldehyde for 15–30 min at room temperature. The fixed cells were permeabilized either with 0.1% Triton X-100/PBS for 3 min for naked DMAT treatment or with cold ethanol for 10 min for TBG-DMAT treatment. One day before immunofluorescence processing, antibodies were incubated at 4 °C overnight in a 50:50 mixture of 0.05% Igepal CA630/PBS:Sea Block (Pierce 37527), then spun through a Spin-X® filter (Costar 8161) in a microcentrifuge for 5 min at 10,000 rpm. Naked DMAT treated cells were incubated with Sea Block containing 5 mg/ml normal donkey for 15 min at RT. TBG-DMAT treated cells were incubated with Sea Block containing 5 mg/ml normal donkey serum and 0.3% avidin for 15 min at RT, washed 1× PBS/NP, and incubated with 0.03% biotin in PBS for 15 min. All cells were then washed 1× PBS/NP, incubated with primary antibody cocktails at the indicated concentrations for 1 h at RT, washed 2× PBS/NP for 3 min, incubated with secondary antibody cocktail for 30 min at RT, then washed 2× PBS/NP for 3 min and processed for DNA staining and mounting as described for TUNEL. For TBG-DMAT treated cells, two consecutive secondary antibody incubations were performed: the first incubation with biotinylated antibody, and the second incubation with streptavidin antibody. Antibodies used: NF-κB p65 phospho-serine 529 (1:50, Santa Cruz ); D8E (1:20) [39]; Jackson Immunoresearch secondary antibodies goat anti-mouse IgM DyLight-549 (115-506-075) and donkey anti-rabbit F(ab′)2 DyLight-488 (711-486-152); biotinylated sheep anti-mouse IgM (515-066-072) and streptavidin-Cy3 (016-030-084). Naked DMAT coverslips were viewed and images captured as described for TUNEL analyses. The remaining images were captured with a Nikon C1si Laser Scanning Confocal Microscope using a 60x water immersion objective, with a minimum of three fields for each of the duplicate experiments captured.
2.7. Quantitative Real-Time RT-PCR Analysis
PC3-LN4 and BPH-1 cultured cells were plated at a density of 1 × 105 cells into 35 mm dishes containing polyamine surface nanofibers treated with matrix proteins as described above for proliferation and viability analyses, with the exception that 10% FBS was used for both cell lines. One day after plating the cells were treated with 105 nM DMAT or equivalent volume DMSO, or 10 μM TBG-siCK2 or equivalent volume TBG-sugar. Cells were collected by scraping into buffer (Qiagen) using a cell scraper at 2 h treatment with DMAT or DMSO, and 24 and 48 h treatment with TBG-siCK2 or -sugar. Total RNA was isolated using the RNeasy mini kit (Qiagen), including the on-column DNase digestion according to the manufacturer’s protocol, and quantitated using a NanoDrop spectrophotometer. The Superscript III kit (Invitrogen) was used to synthesize cDNA from total RNA (0.25 μg) using the oligo-dT primers according to the manufacturer’s protocol. The real-time reactions were run using RT2 qPCR™ primer assay (PA-042) SYBR®Green primers and reactions mix (SAbiosciences) and 96 well FAST plates on an Applied Biosystems, Inc. (ABI) 7900HT machine (Foster City, CA). Analyses were performed using the SDS 2.3 ABI software and changes calculated according to the 2(−ΔΔCt) method. Initial experiments were performed to determine the amount of total RNA (0.25 and 0.5 μg input to cDNA reaction) and cDNA (100 – 10−3 dilution used for 1 μl input to RT2 qPCR reaction) to use in order to remain in the linear range of results. HPRT-1 was used as the reference gene for normalization. All results are reported as the average of reactions run at 2 different dilutions of cDNA and a minimum of 2 different RNA isolations.
2.8. Nanocapsule Preparation and Characterization
For TBG nanocapsules, a dispersion atomization method was used to package DMAT or 20-mer siRNA oligonucleotides into nanocapsules composed of tenfibgen, the recombinant fibrinogen fragment of tenascin-C. Tenfibgen was prepared by the method of Aukhil [40]. All other reagents used were of the highest purity available. Briefly, 500 μg [5 μg/μl] of DMAT was first complexed with 25 μg of spermine [0.5 μg/μl] (Sigma-Aldrich) and dispersed using a water-insoluble surfactant system consisting of 10 μg of (2,4,7,9-tetramethyl-5-decyn-4,7-diol) [50% v/v] in DMSO, SE-30; (Air Products and Chemicals, Inc.). Following emulsification with a water-miscible solvent, DMSO, it was then inverted by dilution into suspension by addition of 750 μl of sterile PBS, pH 7.2. The resultant hydrophobic micelles were coated and adsorbed to 12.5 μg of TBG [1 μg/μl] dispersed into the solution prior to spray dispersion atomization (40) into a 25 ml LiCl salt solution (135 mM Li+, 9 mM Ca2+, 37.5 nM Sr2+, 12.5 nM Mg2+). Following incubation at 4 to 6°C with rotation in the salt solution for 14.5 h, the <50 nm nanocapsules were recovered by diafiltration (5000 MWCO) and buffer exchange into PBS containing 10% lactitol (w/v) at a concentration of 0.5 μg/μl for 0.2 μm filter sterilization prior to characterization. DMAT drug incorporation was quantified by 79Br neutron activation analysis. Neutron activation analysis (NAA) was performed at the University of Missouri-Columbia Research Reactor (MURR). The samples were analyzed in an aqueous solution of 10% lactitol, 150 mM Li-acetate, 10 mM Hepes buffer (pH 7.2), irradiated for 5 s, allowed to decay for 300 s, and live-time counted for 300 s. The Br from the analysis was quantified using the interactive peak area of the 617 keV gamma-ray from the decay of 80Br (t1/2 = 17.68 min), which was produced via the neutron capture reaction on 79Br. Pure Br and DMAT (5 mg/ml stock in DMSO) were used as comparative standards.
For TBG-siRNA nanocapsules, all conditions described above apply, except that 250 μg siRNA is complexed with 37.5 μg of spermine. The other difference is that following incubation at 4 to 6°C with rotation in the salt solution for 14.5 h, the < 50 nm nanocapsules were recovered by centrifugation at 20,000 × g for 2 h, and resuspended in PBS containing 10% lactitol (w/v) at a concentration of 1 μg/μl for 0.2 μm filter sterilization prior to characterization.
Physical characterization of particles including average particle size of <50 nm as measured by AFM, and surface charge determination were confirmed by published methods [41].
2.9. Statistical Analysis
The results of cell viability and proliferation assays are presented as means ± SEM. The statistical analyses were performed using GraphPad Prism 5.02 (GraphPad Software). Statistical differences between mean values for treatment versus the control (DMSO, TBG-Sugar, or Dharmafect) for the proliferation and viability assays were evaluated using the 1-tailed unpaired t-test with Welch’s correction. Differences were considered significant for P values less than 0.05.
3. Results
3.1. Effect of Naked DMAT on Cell Proliferation and CK2 Activity in Malignant and Benign Prostate Cells
The CK2 inhibitor DMAT has been shown to be relatively specific with respect to CK2 activity [42]. We first examined the effects of naked, or unformulated, DMAT on the proliferation of cultured prostate cells. PC3-LN4 (PC-3M-LN4) cells were derived from a fourth generation lymph node metastases of PC3M cells after orthotopic prostate injection in a nude mouse [35]. PC3M cells were, in turn, derived from liver metastases subsequent to intrasplenic injection of the androgen insensitive PC3 cells. These cells are highly tumorigenic, metastatic, and androgen insensitive [35]. Benign prostatic hyperplasia-1 (BPH-1) cells were established from SV40T-immortalized primary epithelial human prostate cells [36]. BPH-1 cells are not tumorigenic when injected into immune-incompetent mice [43]. Treatment of PC3-LN4 cells grown on a tenascin/fibronectin matrix with low nanomolar amounts of DMAT for 4 days resulted in a marked loss of cell proliferation (Fig. 1A), as measured by [3H]-thymidine incorporation during DNA synthesis. Similarly, DMAT treatment of BPH-1 cells grown on a laminin matrix also reduced cell proliferation (Fig. 1A). As has been observed previously, the benign cells were relatively less responsive to inhibition of CK2 activity compared to the malignant cells [18]. Thus, both malignant and benign prostate cells showed loss of cell proliferation in response to the introduction of naked DMAT.
Fig. 1.

Cellular effects of naked DMAT in benign and malignant prostate cancer cells. A. Reduced cellular proliferation following treatment with naked DMAT. PC3-LN4 and BPH-1 cells grown on tenascin-C/fibronectin or laminin protein matrix, respectively, in 96-well plates were treated with 26 and 13 nM DMAT. DMSO was used as the control. [3H]-thymidine was added after 72 h, and counts collected at 96 h following drug addition. Results are expressed relative to the DMSO controls, with treatments and cell lines used indicated below. Error bars indicate SEM (n = 4–6). * indicates significant difference from control (P < 0.05). B. Treatment with DMAT results in loss of phospho-Ser529, but not total, NF-κB p65, and no reduction in CK2 expression. PC3-LN4 cells were treated with various concentrations of DMAT as indicated and processed for immunoblot analysis. Treatments and concentration are shown above, with molecular weight markers and proteins detected indicated on the left and right, respectively. Removal of an intervening lane between untreated and 10 μM DMAT is indicated by black line. C. CK2 inhibition by DMAT results in loss of phospho-Ser529 NF-κB p65. PC3-LN4 cells grown on glass coverslips were treated with DMAT or DMSO and fixed 24 h later for indirect immunofluorescence analysis. The treatments applied are indicated on the left and the protein visualized by immunofluorescence or nuclei by DAPI staining is shown above the panels. Magnification, 400×.
Because treatment with an inhibitor will not necessarily translate to transcript or protein steady-state level effects, we chose to assess CK2 kinase activity in the cells by examining the effect of CK2 inhibition on the phosphorylation status of an endogenous CK2 substrate, Nuclear Factor-kappaB (NF-κB) RelA/p65. CK2 has been shown to phosphorylate NF-κB p65 Serine 529 (P-Ser529) and reduced NF-κB p65 P-Ser529 signal has been observed after inhibition or downregulation of CK2 expression in vivo [23; 44]. As is shown in Fig. 1B, a marked loss of NF-κB p65 P-Ser529 signal is detected in PC3-LN4 cells after 24 h treatment with 10, 1 and 0.1 μM DMAT, with signal reduced to 37%, 57% and 60% of the DMSO control, respectively. In contrast, the total amount of immunoreactive NF-κB p65 protein remained constant (Fig. 1B). Furthermore, no change in CK2α, CK2α′, or CK2β steady-state protein levels was observed following treatment with DMAT for up to 72 h (Fig. 1B and data not shown).
For confirmation, PC3-LN4 cells were grown on glass coverslips, treated with DMAT or DMSO for 24 h and processed for indirect immunofluorescence analysis. The results again demonstrated that treatment with DMAT reduced the number of cells with bright detectable punctate NF-κB p65 P-Ser529 signal (Fig. 1C. lower left panel). In further agreement with the immunoblot data, CK2 proteins in the cells showed no change after DMAT treatment (Fig. 1C, middle panels). Moreover, the cells were more rounded after treatment with DMAT as can be observed by the less defined cytosolic signal detectable by the CK2 antibody. This NF-κB p65 P-Ser529 detection strategy serves the purpose of assessing the inhibitor effect on CK2 activity because direct enzyme activity measurement in cells grown on nanofiber tissue engineering scaffolds as performed here was not feasible. These results establish that downregulation of NF-κB p65 Ser529 phosphorylation serves as a marker for loss of cellular CK2 activity.
3.2. Encapsulation of DMAT with Tenfibgen
In order to deliver DMAT specifically to cancer cells in a protected manner, we encapsulated the DMAT within a protein shell composed of tenfibgen (TBG). TBG is the fibrinogen-binding subdomain of tenascin-C (TN-C). TBG nanocapsules are typically smaller than 50 nm in size, and enter tumor cells via tenascin receptors using the lipid raft-mediated caveolar pathway [17]. TBG nanocapsules are specifically taken up by tumor cells and tumor-derived microvessels but not by normal cells or vessels [2; 17]. The ability of the TBG nanocapsule design with an anti-CK2 antisense or antisense-derived oligonucleotide (OGN) cargo to target and induce cell death of cancer cells has been demonstrated in prostate cancer and in HNSCC xenografts [2; 22; 23]. The characteristics and morphology of the TBG-DMAT nanocapsules are summarized in Table I and Fig. 2. Quantitation of DMAT encapsulation in TBG was accomplished using neutron activation analysis; in these assays, the decay of the 4 bromine (Br) radicals present in the DMAT structure from 80Br, produced via the neutron capture reaction, to 79Br was quantified to determine the amount of DMAT in TBG-DMAT. From this analysis, we calculated the concentration of the TBG-DMAT nanocapsules as 0.8 μg/ml (1.7 μM).
Table I.
Nanocapsule Characteristics and Information
| Batch | Shell Ligand | Particle Size (nm)1 | Zeta Potential (meV)2 | Morphology3 | Cargo4,5 | Sequence |
|---|---|---|---|---|---|---|
| 1 | Tenfibgen (TBG) 27 kDa |
17.1 ± 0.9 | −9.0 ± 4.4 | Uniform, Single Capsules | DMAT | Not Applicable |
| 2 | Tenfibgen (TBG) 27 kDa |
15.6 ± 1.1 | −7.7 ± 3.8 | Uniform, Single Capsules | DMAT | Not Applicable |
| 1 | Tenfibgen (TBG) 27 kDa |
10.7 ± 0.8 | −5.4 ± 5.8 | Uniform, Single Capsules | siCK2 | auacaacccaaacuccacau (antisense/guide strand) |
| 2 | Tenfibgen (TBG) 27 kDa |
11.5 ± 1.13 | −5.9 ± 5.7 | Uniform, Single Capsules | siCK2 | auacaacccaaacuccacau (antisense/guide strand) |
Mean ± SEM of the average elliptical diameter determined from AFM micrographs of 25 capsules from two different preparations after drying on mica at 0.5 ng/ml.
Average surface charge measured by DLS from two different preparations across a 20 volt potential in 1 mM KCl at 2 μg/ml. Data shown is the mean ± SEM of 15 independent measurements.
Morphology of all nanocapsules determined by visual AFM observation as uniform, single capsules.
TBG-siCK2 encapsulation 100% as measured by the Burton assay relative to unencapsulated OGN. Estimated 60 siRNA molecules per 12 nm nanocapsule.
TBG-DMAT stock 1.7 μM with incorporation of 0.08% as measured by stoichiometric analysis utilizing isotopic bromine neutron activation.
Fig. 2.

Atomic force micrographs of TBG-encapsulated DMAT and siCK2 show morphology and size of nanocapsules. TBG-DMAT and TBG-siCK2, prepared as described in Materials and Methods, were subjected to atomic force microscopy (AFM). Representative fields are shown, and 3 individual nanocapsules in each field are circled. The size of nanocapsules was determined by AFM image analysis using data collected in the tapping mode. Scale bar, 50 nm.
3.3. Effect of TBG Nanoencapsulated DMAT on Cell Proliferation and CK2 Activity in Malignant versus Benign Prostate Cells
We then evaluated whether delivery of TBG-DMAT specifically to cancer cells, avoiding the benign cells, decreases proliferation similar to non-encapsulated inhibitor. In experiments run in parallel to those of Fig. 1A, PC3-LN4 and BPH-1 cells were treated with nanomolar amounts of TBG-DMAT or -sugar nanocapsules or DMSO. After 3 days, [3H]-thymidine was added to the cells and after an additional 24 h its incorporation was quantitated. The specific reduction in cell proliferation for the malignant cells is shown in Fig. 3A, and is very similar to that observed using naked DMAT. Importantly, the viability of the benign cells was not changed by treatment with TBG-DMAT at doses resulting in great than 90% inhibition of the PCa cells, emphasizing that non-malignant cells do not take up TBG nanocapsules. Loss of CK2 activity in DMAT nanocapsule treated PC3-LN4 cells was confirmed by indirect immunofluorescence detection of NF-κB p65 P-Ser529 (Fig. 3B, lower left panel). Consistent with the “naked” DMAT treatment no change in CK2 protein abundance or cellular location was observed following TBG-DMAT treatment (Fig. 3B, middle panels).
Fig. 3.

Cellular effects of TBG-DMAT in benign and malignant prostate cancer cells. A. Reduced cellular proliferation following treatment with TBG-DMAT. PC3-LN4 and BPH-1 cells grown on tenascin-C/fibronectin or laminin protein matrix, respectively, in 96-well plates were treated with 210 and 105 nM TBG-DMAT. DMSO and TBG-Sugar were used as controls. [3H]-thymidine was added after 72 h, and counts collected at 96 h following drug addition. Results are expressed relative to the controls, with treatments and cell lines used shown below. Error bars indicate SEM (n = 4–6). * indicates significant difference from control (P < 0.05). B. CK2 inhibition by DMAT results in loss of NF-κB p65 phospho-Ser529 phosphorylation. PC3-LN4 cells grown on tenascin-C/fibronectin protein matrix were treated with TBG-DMAT or TBG-Sugar and fixed 48 h later for immunofluorescence analysis. TBG nanocapsules used for treatment are indicated to the left of the panels and the proteins detected and nuclear staining with bisbenzimide above. Magnification, 600×.
3.4. Effects of a Bispecific Anti-CK2αα′ siRNA on Cultured Prostate Cells
We have investigated TN-C or TBG encapsulated antisense OGNs targeting CK2 catalytic subunits [2; 18; 22; 23], as well as the TBG encapsulated CK2 inhibitor DMAT. Thus it remained to be determined whether a TBG encapsulated siRNA targeting CK2 would also effectively induce death in cancer cells. We designed a bi-specific siRNA which targets both CK2 α and α′ catalytic subunits, termed siCK2. This sequence has been used successfully in vivo as an antisense-based single stranded OGN [23]. The importance of simultaneously downregulating CK2α and α′ to avoid possible increased expression of CK2α ′ was recently demonstrated by us[38]. Here we tested the effectiveness of siCK2 in reducing cell viability and CK2αα′ protein steady-state levels specifically in prostate cancer PC3-LN4 and benign prostate BPH-1 cells as a “naked” molecule in cultured cells using a commercial transfection reagent. The data shown in Fig. 4A demonstrate that siCK2 caused loss of cell viability to a greater extent than transfection with siRNA pools directed against CK2α or CK2α′ alone in both the malignant and benign cell types. There was a dose-dependent effect on cell viability observed with different amount of siCK2 in these cells (data not shown). Transfection of siCK2 effectively reduced CK2αα′ protein levels in all prostate cell lines tested, both malignant (PC3-LN4, C4-2) and benign (BPH-1, PNT1a) (Fig. 3B, top panel, and data not shown). Also, in the C4-2 cells, downregulation of CK2α, whether using the bi-specific siCK2 or the CK2α siRNA pool, resulted in reduced CK2β protein levels, while loss of CK2α′ did not (Fig. 4B, middle panel). Furthermore, transfection of siCK2 also induced apoptosis as measured by TUNEL assay (Fig. 4C, lower left panel). TUNEL signal was quantitated at 72 h post-transfection in 100 cells for 2 experiments and found to be 37.7 ± 1.9% and 100 ± 0% positive signals for 10 and 20 nM siCK2, respectively. The TUNEL-positive signals for control cells were 4.3 ± 1.76% and 1.3 ± 0.44% for siCyclophilin B (10 nM) and Dharmafect reagent, respectively. These data demonstrate that both the benign and malignant cells are equally responsive to RNAi-mediated CK2 downregulation.
Fig. 4.
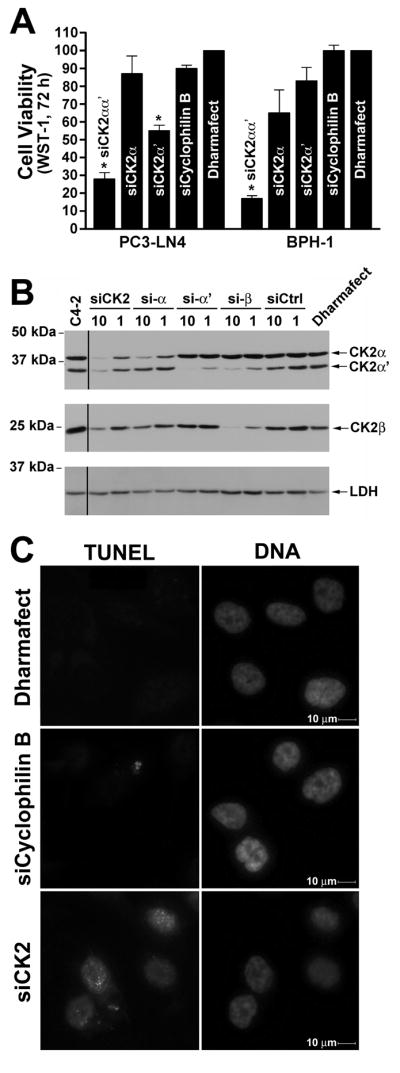
Characterization of cellular effect in benign and malignant prostate cancer cells following transfection of siCK2. A. Transfection of the bi-specific siCK2 siRNA into PC3-LN4 and BPH-1 cells results in greatly reduced cellular viability. WST-1 assays were performed 72 h after transfection of siRNAs (20 nM) to CK2αα′ (siCK2), CK2α, CK2α′, and cyclophilin B as well as Dharmafect reagent alone. Results are expressed relative to the Dharmafect reagent controls with the siRNA used for treatment shown on the graph and the cell line displayed below. Error bars indicate SEM (n = 3–5). * indicates significant difference from control (P < 0.05). B. Transfection of siCK2 siRNA downregulates both CK2α and α′ in cultured C4-2 prostate cancer cells. C4-2 cells were transfected with of the different siRNAs and analyzed 48 h post-transfection by immunoblot for CK2α, α′, and β expression. Lactate dehydrogenase (LDH) expression was used to evaluate protein loading. The siRNA used and concentration in nM are indicated above the lanes, the proteins detected and molecular weight markers shown on the right and left, respectively. Lane to the left of black line is from a separate blot and was replaced due to poor transfer of actin and CK2αα′. C. siCK2 induces apoptosis in cultured PC3-LN4 prostate cancer cells. PC3-LN4 cells were transfected with various concentrations of siRNA and the cells plated onto cover slips 48 h later. Cells were fixed at 72 h post-transfection, and analyzed using the TUNEL staining kit APO-BRDU (Phoenix Flow Systems, Inc.). The treatment is indicated to the left of the panels and the TUNEL or DAPI staining of the treated cells above. Magnification, 400×.
3.5. TBG Encapsulation of siCK2 Reduces Cell Proliferation, CK2 Expression, and CK2 Activity Specifically in Cancer Cells
Next we evaluated whether siCK2 in s50 TBG nanocapsules would selectively affect proliferation of PCa but not benign cells. The characteristics and morphology of the TBG-siCK2 nanocapsules are described in Table I and Fig. 2. PC3-LN4, C4-2, BPH-1 and PrEC cells grown on extra-cellular matrix protein were treated with TBG-siCK2, TBG-siRFP (red fluorescent protein) or TBG-sugar nanocapsules. After 2 days, [3H]thymidine was added to the media and its incorporation in the cells was quantitated 24 h later. As is illustrated in Fig. 5, treatment with the TBG-siCK2 nanocapsules specifically reduced proliferation for the malignant PC3-LN4 and C4-2 cells, but not for the benign BPH-1 and PrEC cells. Because treatment with TBG-siCK2 nanocapsules is likely to result in reduction of CK2 mRNA steady-state levels in cancer cells, we determined the amount of CK2α and α′ mRNA in PC3-LN4 and BPH-1 cells grown on extra-cellular matrix protein and treated with TBG-siCK2 or TBG-sugar. Results from quantitative real-time RT-PCR are shown for BPH-1 cells at 72 h; however, we were not able to recover enough RNA from treated PC3-LN4 cells at 72 h, thus the experiments were repeated so that RNA could be purified at 24 and 48 h post-treatment as shown in Table II. The reduction of both CK2α and α′ mRNA levels in PC3-LN4 and not in BPH-1 cells treated with TBG encapsulated siCK2 demonstrated the specificity of the malignant cell targeting as well as the expected effect of delivery of siCK2 into the cancer cells.
Fig. 5.

Cellular effects of s50 TBG encapsulated siCK2 in benign and malignant prostate cancer cells. A. Reduced cellular proliferation following treatment with TBG-siCK2. PC3-LN4 and C4-2 grown on tenascin-C/fibronectin matrix and PrEC and BPH-1 cells grown on laminin matrix in 96-well plates were treated with 5 μM TBG-siCK2. TBG-siRFP and -Sugar were used as controls. [3H]-thymidine was added after 48 h, and counts collected at 72 h post-nanocapsule addition. Results are expressed relative to the controls. The TBG nanocapsule cargo and cell lines used are indicted below. Error bars indicate SEM (n = 2–4). * indicates significant difference from control (P < 0.05).
Table II.
CK2αα′ mRNA Steady-State Expression Levels in TBG-siCK2 Treated Cells
| Cell Line | Treatment (10 μM) | Time Point (h) | CK2α | CK2α′ |
|---|---|---|---|---|
| PC3-LN4 | TBG-siCK2 | 24 h | 0.641 ± 0.102 | 0.868 ± 0.055 |
| PC3-LN4 | TBG-siCK2 | 48 h | 0.672 ± 0.085 | 0.874 ± 0.025 |
| PC3-LN4 | TBG-Sugar | 24 & 48 h | 1 | 1 |
| BPH-1 | TBG-siCK2 | 72 h | 0.828 ± .0162 | 1.115 ± 0.031 |
| BPH-1 | TBG-Sugar | 72 h | 1 | 1 |
All values normalized to HPRT mRNA expression and expressed relative to TBG-Sugar control treatment.
4. Discussion
Subsequent to our original proposal [6], the potential of targeting CK2 for cancer therapy has gained general recognition. However, even though there may be a small pharmacological window for general targeting of CK2 as suggested in some recent reports [30; 45], we believe it would be important to target CK2 in a cancer specific manner. This is particularly important since CK2 is a ubiquitous and essential cell survival signal, and thus its potent downregulation in normal cells could provoke serious toxicity in the host. These concerns would apply to both the small molecule inhibitors and antisense or siRNA based therapeutics. When delivering unprotected drug systemically, both types of therapeutics would suffer from the issue of drug stability in circulation and lack of targeted delivery. In the present studies, we have demonstrated the utility of our novel cancer specific delivery system to target CK2 in cancer cells and not the normal cells. We have shown that TBG nanocapsule design is suitable for cancer-specific delivery of either a small molecule protein kinase inhibitor or siRNA.
Potential problems of the pharmacological approach using cell-permeable small molecule inhibitors include the issue of targeting different subcellular populations of the CK2 holoenzyme and, importantly, the development of cellular resistance to the inhibitor as observed with other kinase targeted inhibitors [46]. Similarly, possible considerations associated with the molecular approach to CK2 downregulation include the relative abundance and long protein half-life of CK2 [20; 47], as well as the potential for disparate cellular expression of components that are involved in antisense or siRNA processing in different cell types. Because of the critical importance of drug delivery to only malignant cells, the use of nanocapsules or nanoparticles to safely and specifically deliver either siRNA or small molecule drugs is a burgeoning area of research although thus far there has been limited success in specifically targeting cancer cells thorough the currently available strategies. In the case of systemic delivery of small molecule drugs to tumors, the advantages of encapsulating or otherwise modifying the drug into a nanoparticle formulation would include improved water solubility, protection of the drug during circulation, tumor-specific biodistribution, and better antitumor efficacy [48]. An important additional advantage of this form of drug delivery is the potential for reduced nonspecific host toxicity. Here, we are the first to report nanoencapsulation of a small molecule kinase inhibitor against CK2 to specifically induce death of cancer cells without affecting the benign cells.
Systemic delivery of siRNA through the use of nanocapsules or nanoparticles possesses many of the same advantages as detailed above for small molecule drugs, and we are the first to report on the efficacy of nanoencapsulated cancer cell-targeted delivery of siRNA to CK2α and α′. There are only a few examples of attempted systemic siRNA targeted delivery to cancer cells; these targeted delivery methods include aptamer-based prostate-specific membrane antigen (PSMA) targeting for prostate cancer and transferrin receptor targeting for numerous solid tumors including melanoma [49; 50]. Moreover, supporting the further development of siRNA-based drugs for cancer treatment, there are several reports confirming either the RNAi-based mRNA cleavage mechanism or the reduction of transcript steady-state levels following systemic delivery of siRNA to tumors in mice and humans [49; 50]. However, these approaches have produced limited success and the issues of effective cancer specific delivery remain a major concern. We believe that the advantage of the nanocapsule developed by us relates to our ability to customize the nanocapsules with different ligand shells which allows us to design the nanocapsules to target specific cells via receptor-mediated uptake. In the present study, the nanocapsule shell was constructed of tenfibgen (TBG). TBG is the fibrinogen-binding domain of tenascin-C (TN-C). TN-C is a branched, 225-kDa, fibronectin-like extracellular matrix protein prominent in specialized embryonic tissues, wound healing, and tumors. In addition to many other cancer types, stroma rich in TN-C has been observed consistently adjacent to nests of prostatic cancer cells [51]. Normal adult cells, outside of wound healing, do not migrate on tenascin [52]. TN-C also has been linked to the vascularization of tumor tissue [53]. The angiogenic behavior of TN-C has been localized to the TBG domain that contains a binding site for important receptors, including the angiogenic αvβ3 integrin [54]. Importantly, these TN-C/TBG cell receptors are elevated in cancer cells, but are minimal in normal cells [55]. The use of TBG for cancer cell targeting as well as the use of hyaluronan or asialoorosomucoid as ligands for liver cell targeting have been successfully achieved [23; 41]. Thus, the TBG nanocapsule is capable of intracellular delivery of large-molecule cargos (such as nucleic acid-based products) in a specific tumor-targeted manner [23]. Here, we have shown that the TBG nanocapsule facilitated the delivery of siRNA to cultured cells and produced the expected reduction of CK2α and α′ mRNA steady-state levels specifically in malignant but not benign cells.
Our future goal is to use TBG nanoencapsulated DMAT and siCK2 to treat prostate tumor bearing mice through inhibition or downregulation of our chosen therapeutic target CK2. There are only a handful of animal studies using CK2 inhibitors. For example, the effect of CK2 inhibitors emodin, DRB, 3,3′,4′,5,7-pentahydroxyflavone (quercetin), 4,5,6,7-tetrabromobenzotriazole (TBBt), 4,5,6,7-tetrabromobenzimidazole (TBBz), DMAT, and tetrabromocinnamic acid (TBCA) has been examined in mouse models of retinal neovascularization. These studies demonstrated the requirement for CK2 activity in the process of retinal neovascularization and in the recruitment of hematopoietic stem cells after oxygen-induced retinopathy [31]. Siddiqui-Jain et. al. have reported that oral administration of CX-4945, an ATP-competitive inhibitor of CK2, inhibited growth of mouse BxPC-3 prostate and BT-474 breast xenograft tumors by 88 to 97%, and resulted in complete eradication of the pancreatic tumors at 75 mg/kg given twice per day for 35 days in 3 mice [30]. Finally, a recent report demonstrated reduced growth in hepatocellular carcinoma xenograft tumors after treatment with DMAT via interference with NK-κB activation and Wnt signaling [32]. However, the possible concern of affecting the normal cells on prolonged use of such inhibitors remains, especially if these therapies were to enter clinical application. These studies using a non-targeted and unprotected drug would suggest that TBG nanoencapsulation of a CK2 small molecule inhibitor or siRNA would hold an even greater potential for elimination of tumor in a targeted manner and with minimal potential of host toxicity.
In summary, the present work demonstrates several important features relating to targeting CK2 for cancer therapy. The nanocapsule drug delivery utilized by us (i.e., s50 TBG-DMAT and s50 TBG-siCK2) has numerous positive features for its future potential in therapy because of its ability to specifically target cancer cells avoiding the normal cells and to deliver the drug in a protected manner without concerns relating to drug inactivation in circulation. As shown here, this nanocapsule is highly flexible with application for small molecules (such as DMAT) and nucleic acid molecules (such as siRNA or antisense to CK2). Another important observation presented here is that targeting both the α and α′ subunits of CK2 is much more effective than targeting individual subunits; interestingly, the bi-specific siCK2 employed by us demonstrated some downregulation of the β subunit as well. Based on our results, it appears that siRNA targeting of CK2 is a more effective approach than using kinase inhibitors for inducing cell death. Regardless, whether the nanocapsule cargo is siRNA or a small molecule inhibitor directed against CK2 signaling or an established pharmacological agent targeting other pathways, we propose that TBG nanoencapsulation holds enormous promise for the cancer cell-directed treatment of malignancy.
Acknowledgments
The authors thank Ms. Lindsey M. Watch and Mr. Omar Cespedes-Gomez for excellent technical assistance. Confocal microscopy was performed at the: College of Biological Sciences’ Imaging Center at the University of Minnesota (http://www.cbs.umn.edu/ic/).
Grant Sponsors: Department of Veterans Affairs Medical Research Merit Review Funds (KA); National Cancer Institute grant numbers UO1-CA15062 (KA) and RO1-CA150182 (KA); Ministry of Science and Higher Education of Poland, grant N N209 371439 (ZK); AIRC, Associazione Italiana per la Ricerca sul Cancro (LAP); National Institute of Health grant number R01-DK067436 (BTK).
Abbreviations
- CK2
official acronym for former casein kinase 2 or II
- d
day(s)
- DMAT
2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole
- h
hour(s)
- HNSCC
head and neck squamous cell carcinoma
- FBS
fetal bovine serum
- i.p
intraperitoneal
- i.v
intravenous
- ID
injected dose
- IHC
immunohistochemical or immunohistochemistry
- kDa
kiloDalton
- min
minutes
- NF-κB
Nuclear Factor kappa B
- NP
NP-40
- OGN
oligonucleotide
- PBS
phosphate buffered saline
- PCa
prostate cancer
- PCR
polymerase chain reaction
- P-Ser529
phosphorylated at serine residue 529
- Q-RT-PCR
quantitative reverse-transcriptase PCR
- RFP
red fluorescent protein
- s.c
subcutaneous
- Ser
serine
- siCK2
siRNA to CK2αα′
- siRNA
small interfering RNA
- TBG
tenfibgen
- Thr
threonine
- TN-C
tenascin-C
Footnotes
Disclaimer: The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the U.S. Department of Veterans Affairs or the U.S. government.
Conflict of Interest Statement
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ruzzene M, Pinna LA. Addiction to protein kinase CK2: a common denominator of diverse cancer cells? Biochim Biophys Acta. 2010;1804:499–504. doi: 10.1016/j.bbapap.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 2.Trembley JH, Chen Z, Unger G, Slaton J, Kren BT, Van Waes C, Ahmed K. Emergence of protein kinase CK2 as a key target in cancer therapy. Bio Factors. 2010;36:187–195. doi: 10.1002/biof.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Niefind K, Issinger OG. Conformational plasticity of the catalytic subunit of protein kinase CK2 and its consequences for regulation and drug design. Biochim Biophys Acta. 2010;1804:484–492. doi: 10.1016/j.bbapap.2009.09.022. [DOI] [PubMed] [Google Scholar]
- 4.Seldin DC, Lou DY, Toselli P, Landesman-Bollag E, Dominguez I. Gene targeting of CK2 catalytic subunits. Mol Cell Biochem. 2008;316:141–147. doi: 10.1007/s11010-008-9811-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buchou T, Vernet M, Blond O, Jensen HH, Pointu H, Olsen BB, Cochet C, Issinger OG, Boldyreff B. Disruption of the regulatory b subunit of protein kinase CK2 in mice leads to a cell-autonomous defect and early embryonic lethality. Mol Cell Biol. 2003;23:908–915. doi: 10.1128/MCB.23.3.908-915.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang H, Davis A, Yu S, Ahmed K. Response of cancer cells to molecular interruption of the CK2 signal. Mol Cell Biochem. 2001;227:167–174. [PubMed] [Google Scholar]
- 7.Tawfic S, Yu S, Wang H, Faust R, Davis A, Ahmed K. Protein kinase CK2 signal in neoplasia. Histol Histopathol. 2001;16:573–582. doi: 10.14670/HH-16.573. [DOI] [PubMed] [Google Scholar]
- 8.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faust RA, Gapany M, Tristani P, Davis A, Adams GL, Ahmed K. Elevated protein kinase CK2 activity in chromatin of head and neck tumors: association with malignant transformation. Cancer Lett. 1996;101:31–35. doi: 10.1016/0304-3835(96)04110-9. [DOI] [PubMed] [Google Scholar]
- 10.Piazza FA, Ruzzene M, Gurrieri C, Montini B, Bonanni L, Chioetto G, Di Maira G, Barbon F, Cabrelle A, Zambello R, Adami F, Trentin L, Pinna LA, Semenzato G. Multiple myeloma cell survival relies on high activity of protein kinase CK2. Blood. 2006;108:1698–1707. doi: 10.1182/blood-2005-11-013672. [DOI] [PubMed] [Google Scholar]
- 11.Kim JS, Eom JI, Cheong JW, Choi AJ, Lee JK, Yang WI, Min YH. Protein Kinase CK2α as an Unfavorable Prognostic Marker and Novel Therapeutic Target in Acute Myeloid Leukemia. Clinical Cancer Research. 2007;13:1019–1028. doi: 10.1158/1078-0432.CCR-06-1602. [DOI] [PubMed] [Google Scholar]
- 12.Gapany M, Faust RA, Tawfic S, Davis A, Adams GL, Ahmed K. Association of elevated protein kinase CK2 activity with aggressive behavior of squamous cell carcinoma of the head and neck. Mol Med. 1995;1:659–666. [PMC free article] [PubMed] [Google Scholar]
- 13.Lin KY, Fang CL, Chen Y, Li CF, Chen SH, Kuo CY, Tai C, Uen YH. Overexpression of nuclear protein kinase CK2 Beta subunit and prognosis in human gastric carcinoma. Annals of Surgical Oncology. 2010;17:1695–1702. doi: 10.1245/s10434-010-0911-9. [DOI] [PubMed] [Google Scholar]
- 14.Lin KY, Tai C, Hsu JC, Li CF, Fang CL, Lai HC, Hseu YC, Lin YF, Uen YH. Overexpression of Nuclear Protein Kinase CK2 alpha Catalytic Subunit (CK2alpha) as a Poor Prognosticator in Human Colorectal Cancer. PLoS One. 2011;6:e17193. doi: 10.1371/journal.pone.0017193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahmed K, Gerber DA, Cochet C. Joining the cell survival squad: an emerging role for protein kinase CK2. Trends Cell Biol. 2002;12:226–230. doi: 10.1016/s0962-8924(02)02279-1. [DOI] [PubMed] [Google Scholar]
- 16.Faust RA, Tawfic S, Davis AT, Bubash LA, Ahmed K. Antisense oligonucleotides against protein kinase CK2-α inhibit growth of squamous cell carcinoma of the head and neck in vitro. Head Neck. 2000;22:341–346. doi: 10.1002/1097-0347(200007)22:4<341::aid-hed5>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 17.Unger GM, Davis AT, Slaton JW, Ahmed K. Protein kinase CK2 as regulator of cell survival: implications for cancer therapy. Curr Cancer Drug Targets. 2004;4:77–84. doi: 10.2174/1568009043481687. [DOI] [PubMed] [Google Scholar]
- 18.Wang G, Unger G, Ahmad KA, Slaton JW, Ahmed K. Downregulation of CK2 induces apoptosis in cancer cells--a potential approach to cancer therapy. Mol Cell Biochem. 2005;274:77–84. doi: 10.1007/s11010-005-3077-1. [DOI] [PubMed] [Google Scholar]
- 19.Hamacher R, Saur D, Fritsch R, Reichert M, Schmid RM, Schneider G. Casein kinase II inhibition induces apoptosis in pancreatic cancer cells. Oncol Rep. 2007;18:695–701. [PubMed] [Google Scholar]
- 20.Zhu D, Hensel J, Hilgraf R, Abbasian M, Pornillos O, Deyanat-Yazdi G, Hua XH, Cox S. Inhibition of protein kinase CK2 expression and activity blocks tumor cell growth. Mol Cell Biochem. 2010;333:159–167. doi: 10.1007/s11010-009-0216-0. [DOI] [PubMed] [Google Scholar]
- 21.Slaton JW, Unger GM, Sloper DT, Davis AT, Ahmed K. Induction of apoptosis by antisense CK2 in human prostate cancer xenograft model. Mol Cancer Res. 2004;2:712–721. [PubMed] [Google Scholar]
- 22.Ahmad KA, Wang G, Slaton J, Unger G, Ahmed K. Targeting CK2 for cancer therapy. Anticancer Drugs. 2005;16:1037–1043. doi: 10.1097/00001813-200511000-00001. [DOI] [PubMed] [Google Scholar]
- 23.Brown MS, Diallo OT, Hu M, Ehsanian R, Yang X, Arun P, Lu H, Korman V, Unger G, Ahmed K, Van Waes C, Chen Z. CK2 Modulation of NF-κB, TP53, and the Malignant Phenotype in Head and Neck Cancer by Anti-CK2 Oligonucleotides In vitro or In vivo via Sub–50-nm Nanocapsules. Clinical Cancer Research. 2010;16:2295–2307. doi: 10.1158/1078-0432.CCR-09-3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yde CW, Frogne T, Lykkesfeldt AE, Fichtner I, Issinger OG, Stenvang J. Induction of cell death in antiestrogen resistant human breast cancer cells by the protein kinase CK2 inhibitor DMAT. Cancer Lett. 2007;256:229–237. doi: 10.1016/j.canlet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 25.Lawnicka H, Kowalewicz-Kulbat M, Sicinska P, Kazimierczuk Z, Grieb P, Stepien H. Anti-neoplastic effect of protein kinase CK2 inhibitor, 2-dimethylamino-4,5,6,7-tetrabromobenzimidazole (DMAT), on growth and hormonal activity of human adrenocortical carcinoma cell line (H295R) in vitro. Cell Tissue Res. 2010;340:371–379. doi: 10.1007/s00441-010-0960-1. [DOI] [PubMed] [Google Scholar]
- 26.Pagano MA, Meggio F, Ruzzene M, Andrzejewska M, Kazimierczuk Z, Pinna LA. 2-Dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole: a novel powerful and selective inhibitor of protein kinase CK2. Biochem Biophys Res Commun. 2004;321:1040–1044. doi: 10.1016/j.bbrc.2004.07.067. [DOI] [PubMed] [Google Scholar]
- 27.Prudent R, Moucadel V, Lopez-Ramos M, Aci S, Laudet B, Mouawad L, Barette C, Einhorn J, Einhorn C, Denis JN, Bisson G, Schmidt F, Roy S, Lafanechere L, Florent JC, Cochet C. Expanding the chemical diversity of CK2 inhibitors. Mol Cell Biochem. 2008;316:71–85. doi: 10.1007/s11010-008-9828-z. [DOI] [PubMed] [Google Scholar]
- 28.Niefind K, Raaf J, Issinger OG. Protein kinase CK2 in health and disease: Protein kinase CK2: from structures to insights. Cell Mol Life Sci. 2009;66:1800–1816. doi: 10.1007/s00018-009-9149-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mazzorana M, Pinna LA, Battistutta R. A structural insight into CK2 inhibition. Mol Cell Biochem. 2008;316:57–62. doi: 10.1007/s11010-008-9822-5. [DOI] [PubMed] [Google Scholar]
- 30.Siddiqui-Jain A, Drygin D, Streiner N, Chua P, Pierre F, O’Brien SE, Bliesath J, Omori M, Huser N, Ho C, Proffitt C, Schwaebe MK, Ryckman DM, Rice WG, Anderes K. CX-4945, an Orally Bioavailable Selective Inhibitor of Protein Kinase CK2, Inhibits Prosurvival and Angiogenic Signaling and Exhibits Antitumor Efficacy. Cancer Research. 2010;70:10288–10298. doi: 10.1158/0008-5472.CAN-10-1893. [DOI] [PubMed] [Google Scholar]
- 31.Kramerov A, Saghizadeh M, Caballero S, Shaw L, Li Calzi S, Bretner M, Montenarh M, Pinna L, Grant M, Ljubimov A. Inhibition of protein kinase CK2 suppresses angiogenesis and hematopoietic stem cell recruitment to retinal neovascularization sites. Molecular and Cellular Biochemistry. 2008;316:177–186. doi: 10.1007/s11010-008-9831-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sass G, Klinger N, Sirma H, Hashemolhosseini S, Hellerbrand C, Neureiter D, Wege H, Ocker M, Tiegs G. Inhibition of experimental HCC growth in mice by use of the kinase inhibitor DMAT. International journal of oncology. 2011;39:433–442. doi: 10.3892/ijo.2011.1037. [DOI] [PubMed] [Google Scholar]
- 33.Solares AM, Santana A, Baladron I, Valenzuela C, Gonzalez CA, Diaz A, Castillo D, Ramos T, Gomez R, Alonso DF, Herrera L, Sigman H, Perea SE, Acevedo BE, Lopez-Saura P. Safety and preliminary efficacy data of a novel casein kinase 2 (CK2) peptide inhibitor administered intralesionally at four dose levels in patients with cervical malignancies. BMC Cancer. 2009;9:146. doi: 10.1186/1471-2407-9-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fattal E, Barratt G. Nanotechnologies and controlled release systems for the delivery of antisense oligonucleotides and small interfering RNA. Br J Pharmacol. 2009;157:179–194. doi: 10.1111/j.1476-5381.2009.00148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pettaway CA, Pathak S, Greene G, Ramirez E, Wilson MR, Killion JJ, Fidler IJ. Selection of highly metastatic variants of different human prostatic carcinomas using orthotopic implantation in nude mice. Clinical Cancer Research. 1996;2:1627–1636. [PubMed] [Google Scholar]
- 36.Hayward S, Dahiya R, Cunha G, Bartek J, Deshpande N, Narayan P. Establishment and characterization of an immortalized but non-transformed human prostate epithelial cell line: BPH-1. In Vitro Cellular & Developmental Biology - Animal. 1995;31:14–24. doi: 10.1007/BF02631333. [DOI] [PubMed] [Google Scholar]
- 37.Pagano MA, Andrzejewska M, Ruzzene M, Sarno S, Cesaro L, Bain J, Elliott M, Meggio F, Kazimierczuk Z, Pinna LA. Optimization of protein kinase CK2 inhibitors derived from 4,5,6,7-tetrabromobenzimidazole. J Med Chem. 2004;47:6239–6247. doi: 10.1021/jm049854a. [DOI] [PubMed] [Google Scholar]
- 38.Trembley JH, Unger GM, Tobolt DK, Korman VL, Wang G, Ahmad KA, Slaton JW, Kren BT, Ahmed K. Systemic administration of antisense oligonucleotides simultaneously targeting CK2alpha and alpha’ subunits reduces orthotopic xenograft prostate tumors in mice. Molecular and Cellular Biochemistry. 2011 doi: 10.1007/s11010-011-0943-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goueli SA, Davis AT, Arfman E, Vessella R, Ahmed K. Monoclonal antibodies against nuclear casein kinase NII (PK-N2) Hybridoma. 1990;9:609–618. doi: 10.1089/hyb.1990.9.609. [DOI] [PubMed] [Google Scholar]
- 40.Aukhil I, Joshi P, Yan Y, Erickson HP. Cell- and heparin-binding domains of the hexabrachion arm identified by tenascin expression proteins. J Biol Chem. 1993;268:2542–2553. [PubMed] [Google Scholar]
- 41.Kren BT, Unger GM, Sjeklocha L, Trossen AA, Korman V, Diethelm-Okita BM, Reding MT, Steer CJ. Nanocapsule-delivered Sleeping Beauty mediates therapeutic Factor VIII expression in liver sinusoidal endothelial cells of hemophilia A mice. J Clin Invest. 2009;119:2086–2099. doi: 10.1172/JCI34332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pagano MA, Bain J, Kazimierczuk Z, Sarno S, Ruzzene M, Di Maira G, Elliott M, Orzeszko A, Cozza G, Meggio F, Pinna LA. The selectivity of inhibitors of protein kinase CK2: an update. Biochem J. 2008;415:353–365. doi: 10.1042/BJ20080309. [DOI] [PubMed] [Google Scholar]
- 43.Hayward SW, Wang Y, Cao M, Hom YK, Zhang B, Grossfeld GD, Sudilovsky D, Cunha GR. Malignant Transformation in a Nontumorigenic Human Prostatic Epithelial Cell Line. Cancer Research. 2001;61:8135–8142. [PubMed] [Google Scholar]
- 44.Wang D, Westerheide SD, Hanson JL, Baldwin AS., Jr Tumor necrosis factor alpha-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J Biol Chem. 2000;275:32592–32597. doi: 10.1074/jbc.M001358200. [DOI] [PubMed] [Google Scholar]
- 45.Perea SE, Reyes O, Baladron I, Perera Y, Farina H, Gil J, Rodriguez A, Bacardi D, Marcelo JL, Cosme K, Cruz M, Valenzuela C, Lopez-Saura PA, Puchades Y, Serrano JM, Mendoza O, Castellanos L, Sanchez A, Betancourt L, Besada V, Silva R, Lopez E, Falcon V, Hernandez I, Solares M, Santana A, Diaz A, Ramos T, Lopez C, Ariosa J, Gonzalez LJ, Garay H, Gomez D, Gomez R, Alonso DF, Sigman H, Herrera L, Acevedo B. CIGB-300, a novel proapoptotic peptide that impairs the CK2 phosphorylation and exhibits anticancer properties both in vitro and in vivo. Mol Cell Biochem. 2008;316:163–167. doi: 10.1007/s11010-008-9814-5. [DOI] [PubMed] [Google Scholar]
- 46.Stegmeier F, Warmuth M, Sellers WR, Dorsch M. Targeted Cancer Therapies in the Twenty-First Century: Lessons From Imatinib. Clin Pharmacol Ther. 2010;87:543–552. doi: 10.1038/clpt.2009.297. [DOI] [PubMed] [Google Scholar]
- 47.Seeber S, Issinger OG, Holm T, Kristensen LP, Guerra B. Validation of protein kinase CK2 as oncological target. Apoptosis. 2005;10:875–885. doi: 10.1007/s10495-005-0380-y. [DOI] [PubMed] [Google Scholar]
- 48.Chen Z. Small-molecule delivery by nanoparticles for anticancer therapy. Trends in Molecular Medicine. 2010;16:594–602. doi: 10.1016/j.molmed.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davis ME, Zuckerman JE, Choi CHJ, Seligson D, Tolcher A, Alabi CA, Yen Y, Heidel JD, Ribas A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dassie JP, Liu XY, Thomas GS, Whitaker RM, Thiel KW, Stockdale KR, Meyerholz DK, McCaffrey AP, McNamara JO, 2nd, Giangrande PH. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat Biotechnol. 2009;27:839–849. doi: 10.1038/nbt.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tuxhorn JA, Ayala GE, Rowley DR. Reactive stroma in prostate cancer progression. J Urol. 2001;166:2472–2483. [PubMed] [Google Scholar]
- 52.Erickson HP, Bourdon MA. Tenascin: an extracellular matrix protein prominent in specialized embryonic tissues and tumors. Annu Rev Cell Biol. 1989;5:71–92. doi: 10.1146/annurev.cb.05.110189.000443. [DOI] [PubMed] [Google Scholar]
- 53.Chiquet-Ehrismann R, Chiquet M. Tenascins: regulation and putative functions during pathological stress. The Journal of pathology. 2003;200:488–499. doi: 10.1002/path.1415. [DOI] [PubMed] [Google Scholar]
- 54.Yokoyama K, Erickson HP, Ikeda Y, Takada Y. Identification of amino acid sequences in fibrinogen gamma -chain and tenascin C C-terminal domains critical for binding to integrin alpha vbeta 3. J Biol Chem. 2000;275:16891–16898. doi: 10.1074/jbc.M000610200. [DOI] [PubMed] [Google Scholar]
- 55.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nature reviews. Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]