

Abstract
ATP-sensitive potassium (KATP) channels are inhibited by ATP and activated by phosphatidylinositol-4,5-bisphosphate (PIP2). Both channel subunits Kir6.2 and sulfonylurea receptor 1 (SUR1) contribute to gating: while Kir6.2 interacts with ATP and PIP2, SUR1 enhances sensitivity to both ligands. Recently, we showed that a mutation, E128K, in the N-terminal transmembrane domain of SUR1 disrupts functional coupling between SUR1 and Kir6.2, leading to reduced ATP and PIP2 sensitivities resembling channels formed by Kir6.2 alone. We show here that when E128K SUR1 was coexpressed with Kir6.2 mutants known to disrupt PIP2 gating, the resulting channels were surprisingly stimulated rather than inhibited by ATP. To explain this paradoxical gating behavior, we propose a model in which the open state of doubly mutant channels is highly unstable; ATP binding induces a conformational change in ATP-unbound closed channels that is conducive to brief opening when ATP unbinds, giving rise to the appearance of ATP-induced stimulation.
Key words: KATP channel, sulfonylurea receptor 1, Kir6.2, gating, ATP, PIP2
Introduction
ATP-sensitive potassium (KATP) channels are central to the translation of metabolic status into cellular electrical excitability. In no place is this more evident than pancreatic β-cells where they couple blood glucose concentrations to insulin secretion. KATP channel dysfunction leads to a spectrum of insulin secretion disorders including congenital hyperinsulinism associated with loss of channel activity and neonatal diabetes associated with gain of channel activity. The β-cell ATP-sensitive potassium (KATP) channel is a hetero-octamer composed of two different subunit types.1–3 Four Kir6.2 subunits line the conduction pathway and are similar in topology and sequence to other members of the Kir channel family. Surrounding Kir6.2 are four sulfonylurea receptor 1 (SUR1) subunits. SUR1 is a member of the ATP binding cassestte (ABC) transporter superfamily and contains the canonical ABC transporter core domain [two 6-spanning transmembrane domains (TMDs) and two cytoplasmic nucleotide binding folds (NBF)] preceded by a unique 5-spanning N-terminal TMD (TMD0) connected to the core domain via a large cytosolic loop.4–6
Both Kir6.2 and SUR1 contribute to channel gating. ATP inhibits the channel by interacting with Kir6.2. Phosphatidylinositol-4,5-bisphosphate (PIP2) also interacts with Kir6.2 but stimulates channel activity and this interaction underlies the intrinsic open probability (Po) of Kir6.2.7,8 SUR1 increases the efficacy of both ATP and PIP2.9–11 In the absence of ATP, SUR1 increases the intrinsic Po and confers the bursting patterns of activity within a single channel. A substantial body of work exists that supports a role of TMD0 of SUR1 in modulating the intrinsic gating of Kir6.2.12,13 However, TMD0 is not involved in increasing channel sensitivity to ATP. “Mini-KATP” channels formed by TMD0 of SUR1 and a C-terminal deletion mutant Kir6.2 (Kir6.2ΔC) exhibit Po and bursting behavior similar to wild-type channels but have an ATP sensitivity that is lower than that of wild-type or Kir6.2ΔC channels. How TMD0 of SUR1 modulates the channel's intrinsic gating properties was unknown. Recently, we showed that a residue in the TMD0, E128, plays a critical role in the engagement between SUR1 and Kir6.2 by stabilizing Kir6.2 interactions with PIP2.14 Mutation of this residue to an oppositely charged amino acid, E128K, disrupts functional coupling between the two subunits leading to decreased Po and also decreased PIP2 and ATP sensitivity in full-length channels. In mini-KATP channels, E128K also diminishes the Po and bursting behavior conferred by TMD0 and gives rise to an ATP-sensitivity approaching that of Kir6.2ΔC channels. Moreover, another mutation at the same site, E128W, causes rapid destabilization of channel activity that is reversed by PIP2. The study has guided a new way of thinking about the mechanism by which SUR1 modulates Kir6.2 gating; namely, TMD0 controls intrinsic gating of Kir6.2 by modulating channel sensitivity to endogenous PIP2 and this interaction is, in turn, required for the ability of full-length SUR1 to confer increased channel sensitivity to ATP inhibition.
E128 is negatively charged therefore unlikely to contribute directly to PIP2 binding. We postulate that E128 may instead interact with Kir6.2 residues to enhance PIP2 affinity or efficacy. To begin to investigate this possibility, we performed a mutagenesis study of select positively charged residues in Kir6.2. Our simple hypothesis is that E128 interacts electrostatically with a positively charged Kir6.2 residue, which when mutated to a negatively-charged amino acid should suppress the defect brought about by the E128K mutation. Although we did not identify a residue that behaved as our hypothesis predicted in this study, we quite unexpectedly identified several mutations within Kir6.2 which, when coexpressed with E128K, were more active in the presence of ATP than in its absence. We present a model of KATP channel conformations in different liganded states to explain this unorthodox finding.
Results
Candidate Kir6.2 residues for our screen of potential SUR1 E128-interactors were chosen based upon their placement on the tetrameric Kir6.2 structural model for likelihood to physically interact with E128, which is predicted to lie close to the plasma membrane. Residues previously shown to not affect ATP or PIP2 sensitivity when mutated to neutral or negatively charged residues were excluded. Eight residues were examined, including K47, R50, R54, R176, R177, R192, R201 and R206 (Fig. 1A).
Figure 1.

(A) Amino acid sequence of rat Kir6.2 showing the residues mutated in this study. (B) Response to application of ATP (1, 0.1 and 0.01 mM) of mutant channels assessed by inside-out patch voltage-clamp recording. Recording was performed in symmetrical Kint/EDTA solutions, with differing concentrations of ATP added to the bath solution. Error bars represent SEM, except R50E//E128K 1 mM ATP which is the difference between the individual values from the two patches tested; the number of patches tested (n) for each condition is given below the x-axis. WT: wild-type; EK: E128K.
Each of the eight Kir6.2 residues were mutated to negatively charged glutamic acid (and R206 to aspartic acid also). Next, COSm6 cells were co-transfected with mutant Kir6.2 and either WT or E128K SUR1 plasmids. Inside-out patch, voltage-clamp recordings were performed to determine the ATP-sensitive inhibition of single and double mutants (1, 0.1 and 0.01 mM ATP were tested in most cases). The prediction is that a residue involved in direct electrostatic interaction with E128 of SUR1 would cause significant reduction in ATP-sensitivity when mutated to a negatively charged amino acid and co-expressed with WT SUR1, but would have WT-like ATP sensitivity when co-expressed with E128K SUR1 as the ionic interaction would be re-established. E128K mutation prevents efficient surface expression of the channel; we have previously shown that this defect is partially corrected by treating cells with a KATP channel antagonist, tolbutamide, which acts as a chemical chaperone.15 Cells expressing the E128K mutation were therefore pre-treated with 300 µM tolbutamide overnight to facilitate surface expression. Tolbutamide was then washed out for 2 hours prior to recording to remove its inhibitory effect on channel activity.
Figure 1B shows results for ATP inhibition studies. Of the eight residues examined, none demonstrated the predicted pattern of activity that supports paired electrostatic interaction with E128. Three of the mutations including K47E, R50E and R201E did indeed decrease ATP sensitivity when expressed with WT SUR1; but when co-expressed with E128K, even greater insensitivity to ATP inhibition was observed, indicating the Kir6.2 and SUR1 mutations possibly cause ATP-insensitivity through different, additive mechanisms. For two of these residues, R50 and R201, there is evidence that they are involved directly in ATP binding.16 Another mutant, R54E, had WT-like ATP-sensitivity as a single mutant and when it was co-expressed with E128K was still inhibited by 1 mM ATP although the currents were very small (smaller than E128K alone even with 300 µM tolbutamide pre-treatment) making it difficult to properly assess ATP sensitivity. The R54 residue has previously been implicated in PIP2 affinity or efficacy as mutation of this residue led to decreased channel open probability that could be rectified by exogenous PIP2.17,18 Two mutants, R177E and R206D, had no detectable currents when co-expressed with either WT SUR1 or E128K SUR1 despite pre-treatment with 300 µM tolbutamide.
Strikingly, three Kir6.2 mutants, R176E, R192E and R206E, when coexpressed with E128K SUR1 (denoted as R176E//E128K, R192E//E128K and R206E//E128K hereinafter) displayed increased activity with the application of ATP (Fig. 2A and B). The locations of the three Kir6.2 mutations that lead to ATP-activation when combined with E128K SUR1 are shown in a Kir6.2 homology model (Fig. 2C). All three residues have previously been implicated in PIP2 efficacy.7,8,19–21 Two of them, R176 and R206, lie near the plasma membrane on the cytoplasmic C-terminus of Kir6.2 and may be involved in interactions with the negatively charged phosphate groups in PIP2.22 The R176E//E128K and R206E//E128K “ATP-activation” mutations showed no or small currents (usually distinguishable single channel openings with conductance consistent with KATP channels) in nucleotide-free Kint/EDTA solution. Exposure to 1 or 5 mM ATP led to increased channel openings in a reversible manner (Fig. 2A, top and bottom). Channel activity was quantified and expressed as N*Po where N represents the number of channels in the patch and Po represents the probability of an open channel. N*Po values in control and ATP solutions for R176E//E128K were 0.004 ± 0.002 and 0.029 ± 0.009, respectively, and 0 and 0.306 ± 0.132 for R206E//E128K, respectively (Fig. 2B). Exposure of R206E//E128K to 5 µM PIP2 also caused a small increase in channel opening (two patches were tested) but the extent of stimulation was far less than that seen with exposure to 5 mM ATP (Fig. 2A, bottom). The third residue R192 lies further within the cell at a Kir6.2-Kir6.2 subunit interface (Fig. 2C). Previous studies have shown that mutation of R192 to A or E causes an inactivation phenotype that can be reversed by exogenous PIP2.20,23 R192 has been proposed to interact with E272 of an adjacent Kir6.2 subunit to stabilize channel activity.23 The R192E//E128K pair usually had current on isolation of the inside-out membrane patch but rapidly inactivated in nucleotide-free Kint/EDTA and only became active again in high concentrations of ATP (1 mM). Subsequent removal of ATP recovered channels from inactivation, resulting in another round of channel opening and inactivation (Fig. 2A and middle). R192E//E128K records were not analyzed for N*Po values due to their more complicated inactivation phenotype.
Figure 2.
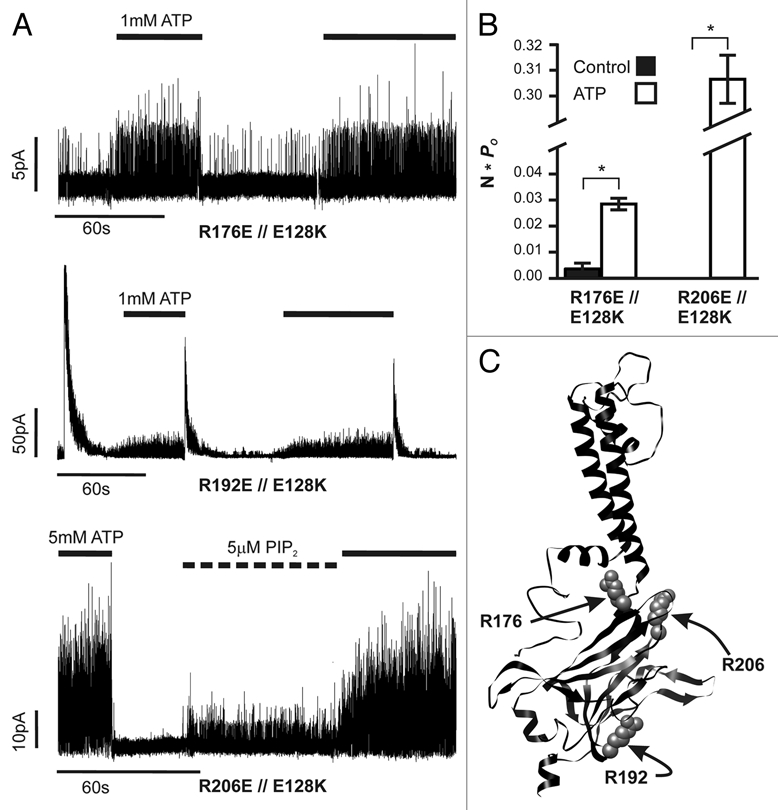
ATP-activation observed with co-expression of E128K and Kir6.2 mutations that diminish PIP2 response. (A) Representative inside-out patch voltage-clamp records from ATP-activation pairs identified in the E128-interacting screen. Recordings are all from transiently transfected COSm6 cells and control solution is Kint/EDTA. Application of 1 or 5 mM ATP or 5 µM PIP2 is indicated by bars above each trace. (B) Average currents in the absence and presence of ATP were quantified as N*Po values for R176E//E128K (0 vs. 1 mM ATP) and R206E//E128K (0 vs. 5 mM ATP); no activity was seen during control conditions (i.e., no ATP) in any R206E//E128K patch. Error bars represent SEM for each condition; number of patches tested is given in Figure 1B. (C) Placement of ATP-activation mutations in Kir6.2. The three residues mutated are shown using space-filling atoms on a ribbon homology model of Kir6.2.
In the presence of Mg2+, ATP stimulates channel activity by interacting with the nucleotide binding domains of SUR1.24 To exclude the possibility that the low level of stimulation by ATP is due to incomplete chelation of Mg2+ by EDTA, we further examined the effect of ATP on channels formed by one of the Kir6.2 mutants, R176E, and a SUR1 harboring both the E128K mutation and another mutation located in the second NBF, G1479R, that abolishes MgATP stimulation.25 The Kir6.2 R176E//SUR1 E128K-G1479R triple mutant channel was still more active in the presence of 1 mM ATP than in Kint/EDTA (data not shown), indicating that the ATP-activation effect observed is not due to MgATP stimulation via SUR1. We next considered the possibility that ATP might activate the mutant channels through a mechanism independent of the ATP-binding site in Kir6.2 responsible for channel inhibition. To test this, we introduced a mutation at another Kir6.2 site, R50, which has been shown to contribute to ATP binding.26 The R50E mutation renders the channel less sensitive to ATP inhibition. In mutant channels formed by E128K SUR1 and doubly mutant Kir6.2 containing R50E and R176E or R50E and R206E, no stimulatory or inhibitory effects on the low spontaneous channel activity were observed with exposure to 1 mM ATP (data not shown). The results suggest that the stimulatory effect of ATP seen in the Kir6.2 R176E or R192E or R206E//E128K SUR1 mutant channels involves the ATP binding site that normally leads to inhibition of channel activity.
Discussion
In this study, we present the finding that ATP activates recombinant KATP channels composed of mutant SUR1 and Kir6.2 with diminished PIP2 sensitivity. How can ATP increase channel openings when the KATP channel has evolved to be inhibited by ATP? To explain these ATP-activation pairs, we propose a mechanism that involves open state instability and ATP-induced conformational changes (Fig. 3). In this scheme, the E128K-induced destabilization of PIP2 interactions in combination with an additional Kir6.2 mutation that diminishes channel response to PIP2 leads to dramatic channel instability such that it cannot maintain open conformations. ATP binding to Kir6.2 induces a con-formational change that is conducive to channel opening when ATP unbinds. The increased activity observed in the ATP-activation double mutants exposed to high concentrations of ATP is not activation by ATP per se, but rather brief openings of the channel following temporary unbinding of ATP. That the E128K mutation in SUR1 causes the channel to become less sensitive to ATP inhibition—as a result of functional uncoupling between SUR1 and Kir6.2—helps unmask the temporary opening of the channel during the brief transitions between ATP-bound and ATP-unbound states. The ATP-induced conformational change referred to here is analogous to the previously reported effect of ATP on several mutations that cause spontaneous channel inactivation, including E128W in SUR1 and R192E, R301E and R314E in Kir6.2.14,23 In the inactivation mutants, exposure to high concentrations of ATP followed by subsequent washout to remove the inhibitory effect of ATP recovers channels from inactivation and allows channels to open briefly before they inactivate again (this inactivation phenomenon can be seen in the R192E//E128K mutant following removal of ATP, Fig. 2A, middle). We have proposed that ATP recovers those mutants from inactivation by causing a conformational change that allows channels to interact with PIP2 once ATP is washed out. Unlike the inactivation mutants in which the critical PIP2-interacting residues remain intact, however, the ATP-activation mutants only exhibit a very small increase in open probability because of the greatly diminished ability of the Kir6.2 mutant (R176E, R206E, and also R192E when it is in the inactivated state) to interact with PIP2.
Figure 3.
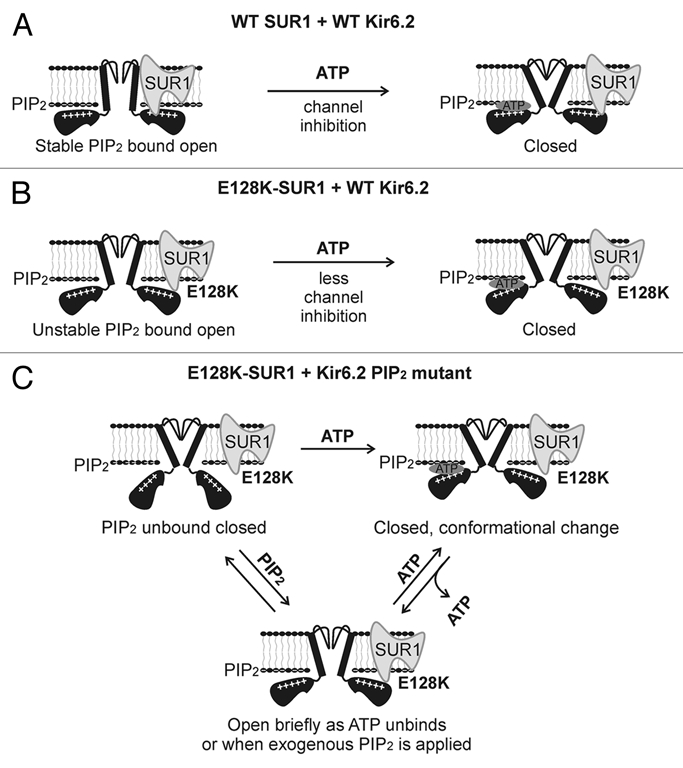
Proposed mechanism by which ATP stimulates the activity of channels formed by E128K-SUR1 and R176E-, R192E- or R206E-Kir6.2. (A) In WT channels SUR1 and Kir6.2 are functionally coupled. This leads to stable interactions between Kir6.2 and PIP2 and efficient ATP inhibition of channel activity. (B) In E128K-SUR1 + WT Kir6.2 channels, functional coupling between SUR1 and Kir6.2 is disrupted causing unstable Kir6.2-PIP2 interactions thereby reduced open probability. The uncoupling between SUR1 and Kir6.2 also renders the channel less sensitive to ATP inhibition. (C) In channels formed by E128K-SUR1 and Kir6.2 PIP2 mutants, most channels are unable to interact with PIP2 resulting in no channel activity in Kint/EDTA. ATP binding to Kir6.2 causes a conformational change that resets the channel from closed to a “ready to conduct” state such that when ATP unbinds, the channel opens briefly before relapsing into PIP2-unbound closed state or before ATP rebinds again to close the channel. High concentrations of exogenous PIP2 can also force the channel to open briefly. Note schemas do not represent quantitative kinetic or structural information.
In conclusion, the abnormal ATP-stimulation gating behavior we observed in the combined SUR1 E128K and Kir6.2 mutants that have severely impaired ability to interact with PIP2 has provided a glimpse of a gating step associated with ATP binding. Together with previous studies of mutations that cause channel inactivation, a picture is emerging that ATP binding to the cytoplasmic domain of Kir6.2, which normally leads to channel closure, can switch the mutant channels from a non-conducting state to a ready-to-conduct state such that when ATP unbinds, the channel opens. Alternatively, the transition between a non-conducting to a ready-to-conduct state may occur when ATP unbinds. Our study reveals a dynamic nature of ligand response in the KATP channel complex. It demonstrates that the response of KATP channels to intracellular ATP is not a fixed property but is a function of the relationships between the channel subunits, ATP and PIP2.
Methods
Electrophysiology.
As described elsewhere in reference 14, rat Kir6.2 cDNA and hamster SUR1 cDNA constructs were in pCDNAI/Amp and pECE plasmids, respectively. Inside-out voltage-clamp recordings were collected using an Axon ID amplifier and pClamp9 acquisition software (Axon Instruments, Inc.). Non-heparinized Kimble glass (Thermo Fisher Scientific) pipettes were pulled to a resistance of ∼1.5–2.0 MΩ on a horizontal puller (Sutter Instruments Co.). Recordings were performed at room temperature in symmetrical Kint/EDTA solutions (140 mM KCl, 10 mM K-HEPES, 1 mM K-EGTA, 1 mM K-EDTA, pH 7.3), with differing concentrations of ATP (as the potassium salt, Sigma-Aldrich) added to the bath solution and with a membrane potential of –50 mV. Inward currents are shown as upward deflections. Cells expressing E128K mutants were treated overnight with 300 µM tolbutamide to augment expression, which was washed out for 2 hours prior to recording.
Data analysis.
Data are presented as means ± standard error of the mean (SEM), unless otherwise noted. Statistical analysis was performed using independent two-population, two-tailed Student's t test, with p < 0.05 considered statistically significant. N*Po analysis (where N is the number of individual channels detected and Po is the probability of an open channel) in Figure 2B was performed using the pCLAMP9 automated channel event detector. Conditions within a record, i.e., control versus ATP, were analyzed independently of each other with regard to both N and Po.
Acknowledgments
We thank Jeremy Bushman for assistance in preparing Figure 2C and Paul Tewson for technical assistance. This work was supported by grants from NIDDK to Emily B. Pratt (F30DK081305) and to Show-Ling Shyng (DK066485 and DK066485S1).
References
- 1.Clement JPt, Kunjilwar K, Gonzalez G, Schwanstecher M, Panten U, Aguilar-Bryan L, Bryan J. Association and stoichiometry of KATP channel subunits. Neuron. 1997;18:827–838. doi: 10.1016/s0896-6273(00)80321-9. [DOI] [PubMed] [Google Scholar]
- 2.Inagaki N, Gonoi T, Seino S. Subunit stoichiometry of the pancreatic beta-cell ATP-sensitive K+ channel. FEBS Lett. 1997;409:232–236. doi: 10.1016/s0014-5793(97)00488-2. [DOI] [PubMed] [Google Scholar]
- 3.Shyng S, Ferrigni T, Nichols CG. Regulation of KATP channel activity by diazoxide and MgADP. Distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. J Gen Physiol. 1997;110:643–654. doi: 10.1085/jgp.110.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JPt, Boyd AE, 3rd, Gonzalez G, et al. Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–426. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- 5.Conti LR, Radeke CM, Shyng SL, Vandenberg CA. Transmembrane topology of the sulfonylurea receptor SUR1. J Biol Chem. 2001;276:41270–41278. doi: 10.1074/jbc.M106555200. [DOI] [PubMed] [Google Scholar]
- 6.Tusnady GE, Bakos E, Varadi A, Sarkadi B. Membrane topology distinguishes a subfamily of the ATP-binding cassette (ABC) transporters. FEBS Lett. 1997;402:1–3. doi: 10.1016/s0014-5793(96)01478-0. [DOI] [PubMed] [Google Scholar]
- 7.Baukrowitz T, Schulte U, Oliver D, Herlitze S, Krauter T, Tucker SJ, et al. PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science. 1998;282:1141–1144. doi: 10.1126/science.282.5391.1141. [DOI] [PubMed] [Google Scholar]
- 8.Shyng SL, Nichols CG. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science. 1998;282:1138–1141. doi: 10.1126/science.282.5391.1138. [DOI] [PubMed] [Google Scholar]
- 9.Babenko AP, Gonzalez G, Aguilar-Bryan L, Bryan J. Sulfonylurea receptors set the maximal open probability, ATP sensitivity and plasma membrane density of KATP channels. FEBS Lett. 1999;445:131–136. doi: 10.1016/s0014-5793(99)00102-7. [DOI] [PubMed] [Google Scholar]
- 10.Enkvetchakul D, Loussouarn G, Makhina E, Shyng SL, Nichols CG. The kinetic and physical basis of KATP channel gating: toward a unified molecular understanding. Biophys J. 2000;78:2334–2348. doi: 10.1016/S0006-3495(00)76779-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- 12.Babenko AP, Bryan J. SUR domains that associate with and gate KATP pores define a novel gatekeeper. J Biol Chem. 2003;26:26. doi: 10.1074/jbc.C300363200. [DOI] [PubMed] [Google Scholar]
- 13.Chan KW, Zhang H, Logothetis DE. N-terminal transmembrane domain of the SUR controls trafficking and gating of Kir6 channel subunits. EMBO J. 2003;22:3833–3843. doi: 10.1093/emboj/cdg376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pratt EB, Tewson P, Bruederle CE, Skach WR, Shyng SL. N-terminal transmembrane domain of SUR1 controls gating of Kir6.2 by modulating channel sensitivity to PIP2. J Gen Physiol. 137:299–314. doi: 10.1085/jgp.201010557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pratt EB, Yan FF, Gay JW, Stanley CA, Shyng SL. Sulfonylurea receptor 1 mutations that cause opposite insulin secretion defects with chemical chaperone exposure. J Biol Chem. 2009;284:7951–7959. doi: 10.1074/jbc.M807012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Antcliff JF, Haider S, Proks P, Sansom MS, Ashcroft FM. Functional analysis of a structural model of the ATP-binding site of the KATP channel Kir6.2 subunit. EMBO J. 2005;24:229–239. doi: 10.1038/sj.emboj.7600487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schulze D, Krauter T, Fritzenschaft H, Soom M, Baukrowitz T. Phosphatidylinositol-4,5-bisphosphate (PIP2) modulation of ATP and pH sensitivity in Kir channels. A tale of an active and a silent PIP2 site in the N terminus. J Biol Chem. 2003;278:10500–10505. doi: 10.1074/jbc.M208413200. [DOI] [PubMed] [Google Scholar]
- 18.Cukras CA, Jeliazkova I, Nichols CG. The role of NH2-terminal positive charges in the activity of inward rectifier KATP channels. J Gen Physiol. 2002;120:437–446. doi: 10.1085/jgp.20028621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan Z, Makielski JC. Anionic phospholipids activate ATP-sensitive potassium channels. J Biol Chem. 1997;272:5388–5395. doi: 10.1074/jbc.272.9.5388. [DOI] [PubMed] [Google Scholar]
- 20.Shyng SL, Cukras CA, Harwood J, Nichols CG. Structural determinants of PIP2 regulation of inward rectifier KATP channels. J Gen Physiol. 2000;116:599–608. doi: 10.1085/jgp.116.5.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haider S, Tarasov AI, Craig TJ, Sansom MS, Ashcroft FM. Identification of the PIP2-binding site on Kir6.2 by molecular modelling and functional analysis. EMBO J. 2007;26:3749–3759. doi: 10.1038/sj.emboj.7601809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 23.Lin YW, Jia T, Weinsoft AM, Shyng SL. Stabilization of the activity of ATP-sensitive potassium channels by ion pairs formed between adjacent Kir6.2 subunits. J Gen Physiol. 2003;122:225–237. doi: 10.1085/jgp.200308822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gribble FM, Tucker SJ, Haug T, Ashcroft FM. MgATP activates the beta cell KATP channel by interaction with its SUR1 subunit. Proc Natl Acad Sci USA. 1998;95:7185–7190. doi: 10.1073/pnas.95.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nichols CG, Shyng SL, Nestorowicz A, Glaser B, Clement JPt, Gonzalez G, et al. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science. 1996;272:1785–1787. doi: 10.1126/science.272.5269.1785. [DOI] [PubMed] [Google Scholar]
- 26.Tucker SJ, Gribble FM, Proks P, Trapp S, Ryder TJ, Haug T, et al. Molecular determinants of KATP channel inhibition by ATP. EMBO J. 1998;17:3290–3296. doi: 10.1093/emboj/17.12.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]