

Abstract
The atrioventricular node controls cardiac impulse conduction and generates pacemaker activity in case of failure of the sino-atrial node. Understanding the mechanisms of atrioventricular automaticity is important for managing human pathologies of heart rate and conduction. However, the physiology of atrioventricular automaticity is still poorly understood. We have investigated the role of three key ion channel-mediated pacemaker mechanisms namely, Cav1.3, Cav3.1 and HCN channels in automaticity of atrioventricular node cells (AVNCs). We studied atrioventricular conduction and pacemaking of AVNCs in wild-type mice and mice lacking Cav3.1 (Cav3.1−/−), Cav1.3 (Cav1.3−/−), channels or both (Cav1.3−/−/Cav3.1−/−). The role of HCN channels in the modulation of atrioventricular cells pacemaking was studied by conditional expression of dominant-negative HCN4 channels lacking cAMP sensitivity. Inactivation of Cav3.1 channels impaired AVNCs pacemaker activity by favoring sporadic block of automaticity leading to cellular arrhythmia. Furthermore, Cav3.1 channels were critical for AVNCs to reach high pacemaking rates under isoproterenol. Unexpectedly, Cav1.3 channels were required for spontaneous automaticity, because Cav1.3−/− and Cav1.3−/−/Cav3.1−/− AVNCs were completely silent under physiological conditions. Abolition of the cAMP sensitivity of HCN channels reduced automaticity under basal conditions, but maximal rates of AVNCs could be restored to that of control mice by isoproterenol. In conclusion, while Cav1.3 channels are required for automaticity, Cav3.1 channels are important for maximal pacing rates of mouse AVNCs. HCN channels are important for basal AVNCs automaticity but do not appear to be determinant for β-adrenergic regulation.
Key words: genetically-engineered mice, pacemaker activity, atrioventricular node, congenital heart block, sino-atrial node dysfunction, ion channels, Cav1.3 channels, Cav3.1 channels, HCN channels, electrophysiology, conduction, heart rate
Introduction
The atrioventricular node (AVN) sets the proper delay between atrial and ventricular activation, and can drive the heartbeat in case of failure of the sino-atrial node (SAN).1 The AVN is also an important pharmacological target for controlling ventricular rate upon atrial arrhythmias. One of the most fascinating features of AVN tissue is the coexistence of a highly regulated conduction function and automaticity. Interestingly, SAN and AVN dysfunction are often found in association in human pathologies of heart rhythm. For instance, sick sinus syndrome caused by genetic mutations in the Scn5A gene is characterized by SAN bradycardia and dysfunction of atrioventricular (AV) conduction.2,3 In congenital heart block (CHB) SAN bradycardia is associated with different degrees of AV conduction dysfunction.4 It has been proposed that CHB is caused by circulating maternal autoantibodies against L-type Cav1.3 and possibly T-type Cav3.1 channels in plasma of affected children.5,6 In the recently described “sinus node dysfunction and deafness syndrome” (SNDD), SAN bradycardia is associated to atrioventricular blocks and rhythm dissociation.7 SNDD is caused by insertion of a glycine residue into a highly conserved alternatively spliced region of the Cav1.3 pore-forming subunit, a condition that produces non-conducting Cav1.3 channels.7 Specific phenotypes of genetically-modified mouse strains in which altered genes coding for ion channels involved in cardiac automaticity have been described (reviewed in ref. 8), among them, mice lacking Cav1.3 channels,9,10 Cav3.1 channels,11 or Scn5A haplo-insufficient mice (Scn5A+/−),12 with reduced cardiac TTX-resistant Na+ current (INa,r) and mice lacking Ca2+ activated SK2 K+ channels13 show dysfunction in both SAN pacemaker activity and impulse conduction through the AVN. Association between SAN and AVN dysfunction in mouse models indicates that genetically-modified strains are a promising approach towards understanding the bases of AVN automaticity and AV conduction, but we have limited information of the functional role of ion channel isoforms in spontaneously active mouse AVN cells (AVNCs). Better knowledge of AVN automaticity is thus important for understanding human pathologies of heart rhythm and also for developing new therapies which aim to stimulate AVN pacemaking with concomitantly improved ventricular rate control. Since genetic loss of Cav1.3 and Cav3.1 channels affects both SAN automaticity and AV conduction,9–11 the primary objective of the present study was to investigate the functional roles of these channels in AVNCs pacemaking. Furthermore, because cAMP-dependent regulation of hyperpolarization-activated HCN (f-) channels is considered to play a key role in autonomic control of SAN pacemaking14 and maximal heart rates,15 we aimed to compare the effects of genetic modification of Cav1.3, Cav3.1 and HCN channels on the β-adrenergic regulation on AVNC automaticity. Interestingly, we found that these channels differentially impact AVNC automaticity. Furthermore, inactivation of Cav1.3 channels prevents spontaneous pacemaking of AVNCs, a finding that can explain the pathophysiologic bases of SNDD. We also report that combined loss of Cav1.3 and Cav3.1 channels induces SAN and AVN dysfunction in mice similar to human CHB. Our work establishes the role of Cav1.3, Cav3.1 and HCN channels in AVNCs pacemaking and gives insights into the pathogenesis of dysfunction of human AV conduction such as CHB and SNDD.
Results
Atrioventricular dysfunction in mice lacking Cav channels.
We first evaluated heart rate and AV conduction dysfunction in mice lacking Cav1.3, Cav3.1 channels or both in telemetric recordings in freely moving animals. Cav3.1−/− mice showed regular heartbeats with moderately reduced heart rates and longer PR intervals than wild-type (WT) mice, consistently to what has been reported previously in animals with a different genetic background (Fig. 1A and B and Sup. Table S1).11 Cav1.3−/− mice showed irregular heart rate with intermittent AV blocks. PR intervals of Cav1.3−/− mice were longer than that of Cav3.1−/− mice (Fig. 1A and B). To determine if concomitant loss of Cav1.3 and Cav3.1 channels resulted in an additive effect on AV conduction we generated Cav1.3−/−/Cav3.1−/− mice by crossing Cav1.3−/− with Cav3.1−/− mice. Cav1.3−/−/Cav3.1−/− mice showed severely delayed AV conduction, with longer PR intervals compared to single mutants for each channel (Fig. 1A and B). Complete AV block and rhythm dissociation were also observed (Fig. 1A). Furthermore, more frequent episodes of high degree AV block were observed in Cav1.3−/−/Cav3.1−/− mice compared to each single mutant strain. These episodes were characterized by the presence of multiple isolated P waves that were not followed by a QRS complex. Thus, the averaged number of isolated P waves was higher in Cav1.3−/−/Cav3.1−/− mice than in Cav1.3−/− mice (Fig. 1C and Sup. Table S1). The mean heart rate was increasingly slower in Cav3.1−/−, Cav1.3−/− and Cav1.3−/−/Cav3.1−/− mice. These observations showed that combined loss of Cav1.3 and Cav3.1 channels produced a higher impact on heart rate and AV conduction than the Cav1.3−/− mutation alone.
Figure 1.

(A) Telemetric surface ECGs of freely moving WT, Cav3.1−/−, Cav1.3−/− and Cav1.3−/−/Cav3.1−/− mice showed additive effect of Cav gene inactivation on AV conduction dysfunction. Solid bars indicate PR interval duration in different genotypes. Dotted bars indicate RR intervals. Arrows indicate isolated P waves. Note that in Cav1.3−/−/Cav3.1−/− mice, some P waves elicited a QRS complex and some isolated P waves fall into the T wave of the QRS complex. PR intervals were progressively longer in Cav3.1−/−, Cav1.3−/− and Cav1.3−/−/Cav3.1−/− mice. (B) Stars indicate statistical significance compared to WT (one-way ANOVA). (C) Isolated P waves were not found in WT and Cav3.1−/− mice. The number of two or three consecutive isolated P waves measured was higher in Cav1.3−/−/Cav3.1−/− than in Cav1.3−/− mice, indicating that AV conduction dysfunction in a Cav1.3−/− genetic background was worsened by additional loss of Cav3.1 channels. Stars indicate statistical significance compared to WT (Kruskal-Wallis).
Cav1.3 channels are required for mouse AVNC automaticity.
Previous studies have shown that Cav1.3-mediated ICa,L is important for pacemaking of SANCs.10,16 The dependence of WT AVNC automaticity from ICa,L (see the accompanying paper in ref. 17) and the dysfunction of AV conduction observed in Cav1.3−/− mice suggest that Cav1.3 channels are functionally important in AVNCs. We thus speculated that Cav1.3 channels could play a role in automaticity of AVNCs and tested the contribution of Cav1.3-mediated ICa,L to AVNC pacemaking using Cav1.3−/− mice. Inactivation of Cav1.3-mediated ICa,L had severe effects on AVNC automaticity. All tested Cav1.3−/− AVNCs did not show spontaneous pacemaking, but displayed a depolarized membrane potential of −32 ± 4 mV (Fig. 2A and n = 11). This membrane potential did not significantly differ from that observed in WT cells treated with isradipine (p > 0.05).17 Also similar to isradipine treated WT AVNCs, Cav1.3−/− AVNCs displayed low amplitude oscillations of membrane potential (Fig. 2A). The membrane voltage oscillations recorded in isradipine treated AVNCs were thus unlikely to be caused by unblocked Cav1.3 channels.17
Figure 2.
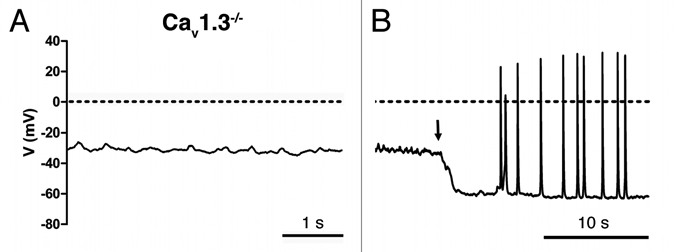
(A) Cav1.3−/− AVNCs display positive membrane potential and no basal pacemaker activity. (B) Tonic hyperpolarizing current injection (black arrow) induced spontaneous AP firing in Cav1.3−/− AVNCs.
Both submaximal (0.02 µM) and saturating (0.1 µM) concentrations of isoproterenol (ISO) could not restore spontaneous pacemaking of Cav1.3−/− AVNCs (data not shown).
To test the possibility that the lack of automaticity in Cav1.3−/− AVNCs was due to failure to generate the AP upstroke, we hyperpolarized Cav1.3−/− AVNCs by injection of a constant negative current (−25 ± 2 pA, n = 11) until the cell fired APs spontaneously (Fig. 2D). Out of eleven cells tested, seven Cav1.3−/− AVNCs showed spontaneous APs upon current injection, while four cells (36% of total) remained silent. Because Cav1.3−/− cells could fire APs only upon tonic negative current injection, we will refer to their activity as “initiated automaticity” rather than “pacemaker activity”. Cav1.3−/− AVNCs exhibited initiated automaticity from a MDP of −60 ± 2 mV n = 7, a value that did not differ from the MDP of WT AVNCs (see Table 1 of the accompanying paper ref. 17). However, initiated automaticity of negative current-injected Cav1.3−/− AVNCs was slower than pacemaker activity of WT cells (Sup. Table S2). Furthermore, while pacemaking of WT AVNCs was regular and presented linear diastolic depolarization, initiated automaticity of negative current-injected Cav1.3−/− AVNCs was intermittent and did not present a depolarizing diastolic phase (Fig. 2B). Instead rather than a diastolic depolarization phase, Cav1.3−/− AVNCs presented subthreshold oscillations of the membrane potential similar to those seen after isradipine block of WT APs.17 These randomly reached the threshold for AP discharge (Fig. 2B). The AP amplitude was significantly lower in injected Cav1.3−/− AVNCs than in WT cells suggesting that Cav1.3 channels not only determine the diastolic depolarization but also contribute to the AP itself. The AP duration of negative current-injected Cav1.3−/− AVNCs tended to be longer than that of WT cells, but such a difference did not reach statistical significance (Sup. Table S2). Low residual (11% of the WT density) ICa was recorded in AVNCs of Cav1.3−/− mice (−2 ± 0.3 pA/pF n = 4 and −0.21 ± 0.03 pA/pF n = 6 in WT and Cav1.3−/− AVNCs, respectively). Most likely, Cav1.2 channels activating at higher voltages than Cav1.3 channels underlie this residual ICa,L (Fig. 3). These observations indicate that Cav1.3 channels are required for pacemaking of mouse AVNCs and constitute the predominant L-type channel isoform in these cells.
Figure 3.
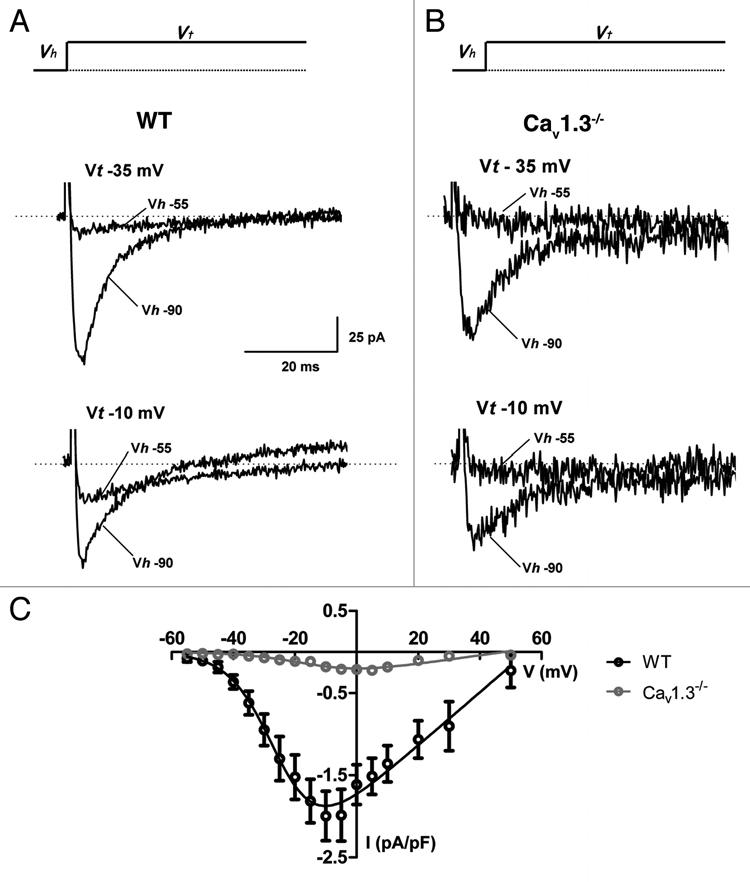
AVNCs of Cav1.3−/− mice display drastically reduced ICa,L. (A) Sample traces of ICa,L in WT AVNCs at the two different test potentials (Vt) indicated (−35 and −10 mV). ICa,L was activated from a holding potential (Vh) of −55 mV which inactivates ICa,T. (B) In contrast, very few ICa,L is recorded in Cav3.1−/− AVNCs in the same condition. ICa,T is activated from a HP of −90 mV in (A and B). (C) IV-curve of ICa,L in WT (open black circles and line) and residual ICa,L Cav1.3−/− mice (open red circles and line).
Cav3.1−/− channels are important for AVNCs automaticity and determine maximal pacemaking rates in AVNCs.
We reported in a previous study, that inactivation of Cav3.1 channels abolished ICa,T in both SANCs and AVNCs.11 The importance of Cav3.1-mediated ICa,T in AVNCs automaticity is unknown. We thus investigated the impact of Cav3.1 channel inactivation in AVNC automaticity. Furthermore, because combined inhibition of Cav1.3 and Cav3.1 channel activity in CHB cause AV dysfunction, which is similar to that observed in Cav1.3−/−/Cav3.1−/− mice (Fig. 1), we studied pacemaker activity in Cav1.3−/−/Cav3.1−/− AVNCs.
Contrary to Cav1.3−/− cells, Cav3.1−/− AVNCs had spontaneous pacemaking (Fig. 4). However, Cav3.1−/− cells showed irregular and slower pacemaking than WT AVNCs (Fig. 4A and B). Similarly to Cav1.3−/− cells (Fig. 4C), Cav1.3−/−/Cav3.1−/− AVNCs were quiescent at rest and injection of negative current (−23 ± 3 pA, n = 6) was necessary to initiate automaticity (Fig. 4D). Cav3.1 channel deficiency reduced AVNC pacemaking frequency by 70% compared to that of WT cells. Initiated automaticity of negative current-injected Cav1.3−/−/Cav3.1−/− AVNCs was even slower than that of firing of negative current-injected Cav1.3−/− or pacemaker activity of Cav3.1−/− AVNCs. The firing rate of Cav1.3−/−/Cav3.1−/− AVNCs was 86% slower than pacemaking of WT cells (Fig. 4D and E and Sup. Table S2). This observation indicates that Cav3.1-mediated ICa,T does not only participate in the pacemaking of WT AVNCs, but contributes also to initiated automaticity of negative current-injected Cav1.3−/− AVNCs.
Figure 4.
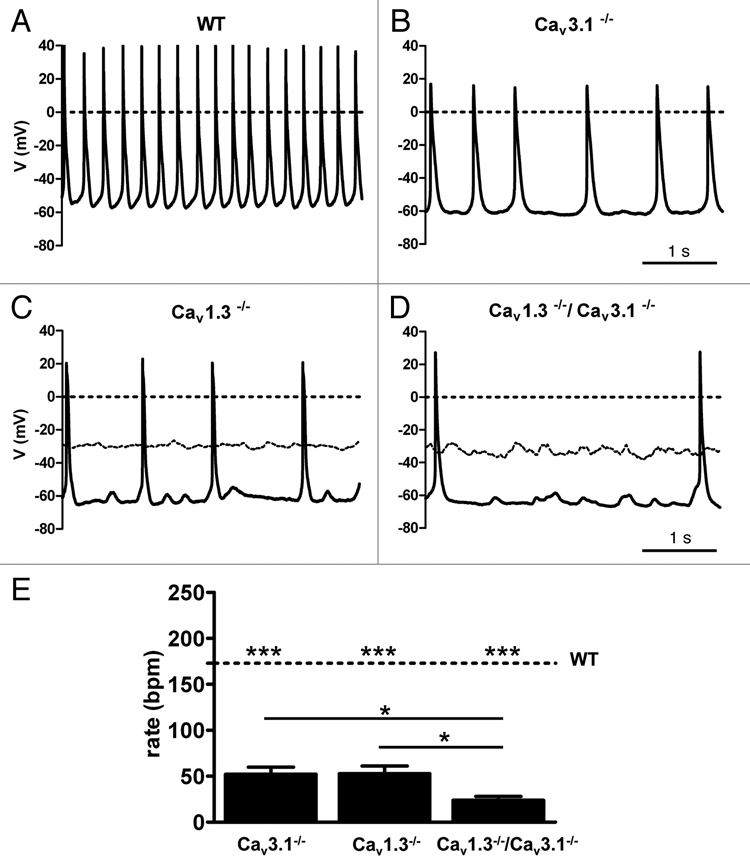
Pacemaker activity of AVNCs from WT (A), Cav3.1−/− (B), Cav1.3−/− (C) and Cav1.3−/−/Cav3.1−/− (D) mice. Cells from Cav1.3−/− and Cav1.3−/−/Cav3.1−/− mice were injected with tonic negative hyperpolarizing current (see text). Dashed lines indicate the spontaneous membrane potential before starting current injection. Inactivation of Cav3.1 channels significantly slowed AVNCs pacemaking and produced erratic beating rate. Only sporadic actions potentials could be observed in negative-current injected Cav1.3−/− and Cav1.3−/−/Cav3.1−/− AVNCs. (E) Histogram showing comparison between averaged beating rates of Cav1.3, Cav3.1−/− and Cav1.3−/−/Cav3.1−/− AVNCs. The dotted line indicates the averaged beating rate of WT AVNCs. Stars indicate statistical significance (one-way ANOVA).
Because SAN and AVN activity are tightly controlled by β-adrenergic regulation, we also investigated how the loss of Cav3.1 and Cav1.3 channels affected the regulation of AVNC automaticity by ISO. In Cav3.1−/− AVNCs, 0.1 µM ISO significantly increased pacemaker activity, although maximal frequency was still lower than pacemaking of WT AVNCs under the same conditions (Fig. 5A and B and Sup. Table S3). Similarly, ISO stimulated initiated automaticity of negative current-injected Cav1.3−/− AVNCs but did not restore ISO-stimulated AP frequency measured in WT cells (Fig. 5A and C and Sup. Table S4). In both Cav3.1−/− and negative current-injected Cav1.3−/− AVNCs, the relative increase in pacemaking of Cav3.1−/− and initiated automaticity of negative current-injected AVNCs by ISO was similar to that of WT AVNCs (Fig. 5D and E). ISO did not normalize cellular arrhythmia of both Cav3.1−/− and negative current-injected Cav1.3−/− AVNCs (Fig. 5B).
Figure 5.

β-adrenergic modulation of firing in AVNCs from WT (A), Cav3.1−/− (B) and Cav1.3−/− mice (C). AVNCs were challenged with a saturating dose of ISO (0.1 µM). ISO cannot compensate for cellular arrhythmia in Cav3.1−/− cells (B). (D) Histogram showing maximum firing rates of WT, Cav3.1−/− and Cav1.3−/− AVNCs under ISO. Stars indicate statistical significance (two-way ANOVA). (E) The relative sensitivity of pacemaker activity to ISO was similar in WT, spontaneously pacemaking Cav3.1−/− and negative current-injected Cav1.3−/− AVNCs. The diagram shows that the relative increase in firing rate is constant in these genotypes. Significance was assessed with the one-way ANOVA test.
cAMP regulation of HCN channels is important for basal AVNCs automaticity, but is not required to reach maximal pacemaking rates.
In SANCs, HCN channels are considered to play a key role in both the generation and regulation of pacemaker activity, primarily because of the direct cAMP-dependent regulation of the channel open probability, which modulates If availability at voltages spanning that of the diastolic depolarization.18 Because initiated automaticity of negative current-injected Cav1.3−/− and pacemaking of Cav3.1−/− AVNCs are still responsive to ISO (Fig. 5D and E) we investigated the importance of cAMP-dependent regulation of HCN-channels in AVNC pacemaking. For this purpose, we used the mutant mouse strain hHCN4-573X (see Materials and Methods). This strain expresses a dominant-negative cAMP-insensitive, HA-tagged human HCN4 channel construct in a heart-specific and inducible way.15 Mutant mice expressing hHCN4-573X showed a negative shift of the If activation curve, which induces a functional loss of If activity at physiological diastolic voltages.15
AVNCs from control mice (Fig. 6A and right part) expressed HCN4 (see Sup. Expanded Materials and Methods and Fig. 1 in the accompanying paper in ref. 17). No specific anti-HA staining was observed in these cells (Fig. 6A and left part). In contrast, anti-HA staining was consistently found along membrane borders of mutant cells, showing that hHCN4-573X channels were targeted to the cell membrane (Fig. 6B). In perforated-patch voltage-clamp experiments, expression of hHCN4-573X in mutant AVNCs did not affect peak If density (Fig. 6C–F). Application of 0.1 µM ISO enhanced If in control cells (Fig. 6D), but failed to stimulate If in mutant AVNCs throughout the voltage range tested (Fig. 6F). Under current-clamp conditions, hHCN4-573X expressing AVNCs displayed irregular pacemaking (Fig. 7A). The AP amplitude of hHCN4-573X expressing cells was lower that that of control cells (Sup. Table S4). The membrane potential of hHCN4-573X expressing AVNCs was 9 mV more positive than that of control AVNCs. (Sup. Table S4). Application of a submaximal dose of ISO (0.02 µM) induced a small increase in pacemaking of control and mutant AVNCs (Fig. 7B). No significant rate difference between hHCN4-573X expressing cells and control AVNCs was observed at 0.1 µm ISO. At this ISO concentration, the AP amplitude and the MDP of hHCN4573X expressing cells were indistinguishable from that of control AVNCs (Sup. Table S4).
Figure 6.
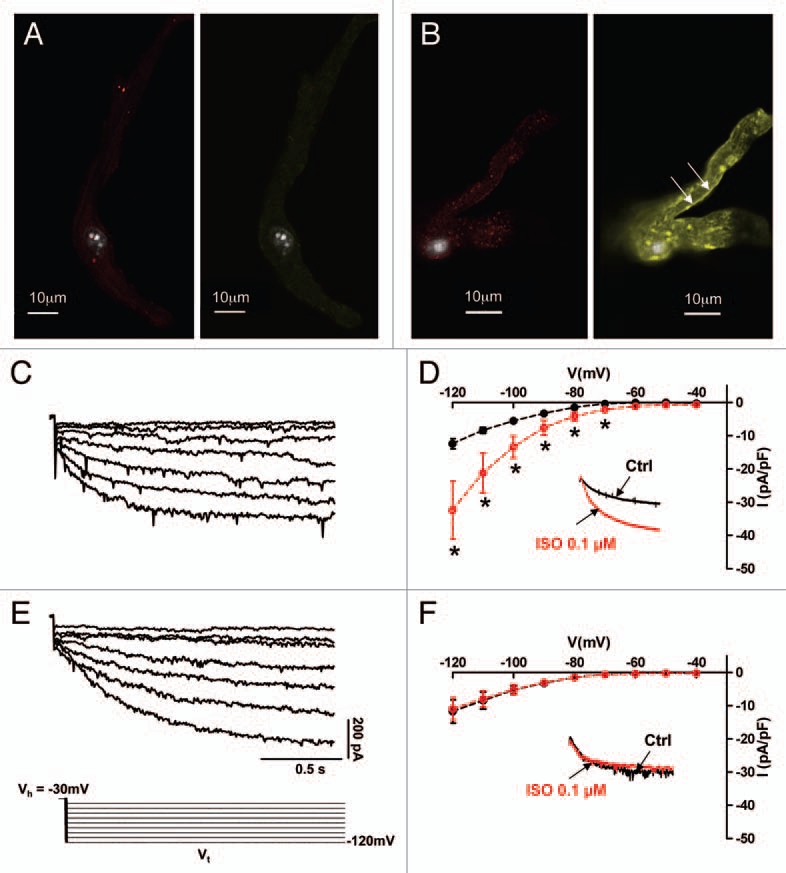
Conditional heart-specific expression of hHCN4-573X transgene suppressed regulation of HCN-mediated If by ISO in AVNCs. Staining of cells with an anti-HA antibody evidences expression of hHCN4-573X in AVNCs of mutant mice (A and B). AVNCs from control mice (A) showed only anti HCN4 staining (left part, red) because of expression of endogenous HCN4 channels. No significant anti-HA immunoractivity was observed in control AVNCs. One of three representative experiments is illustrated. In contrast, AVNCs from mutant mice (B), had stronger anti-HCN4 immunoreactivity (red) than control AVNCs and membrane bound anti-HA staining (arrows). If sample traces (C) and isochronal I–V curves (D) in control solution (black symbols) and 0.1 µM ISO (red symbols) showed normal responsiveness of If to ISO in AVNCs from control mice. In contrast, mutant AVNCs showed that hHCN4-573X expression slowed If activation kinetics (E) and abolished If sensitivity to ISO (F). Stars indicate statistical significance (Student's t-test). The voltage-clamp protocol of (C and E) is shown. Insets in parts (D and F) show the effect of 0.1 µM ISO in sample traces recorded at −100 mV.
Figure 7.
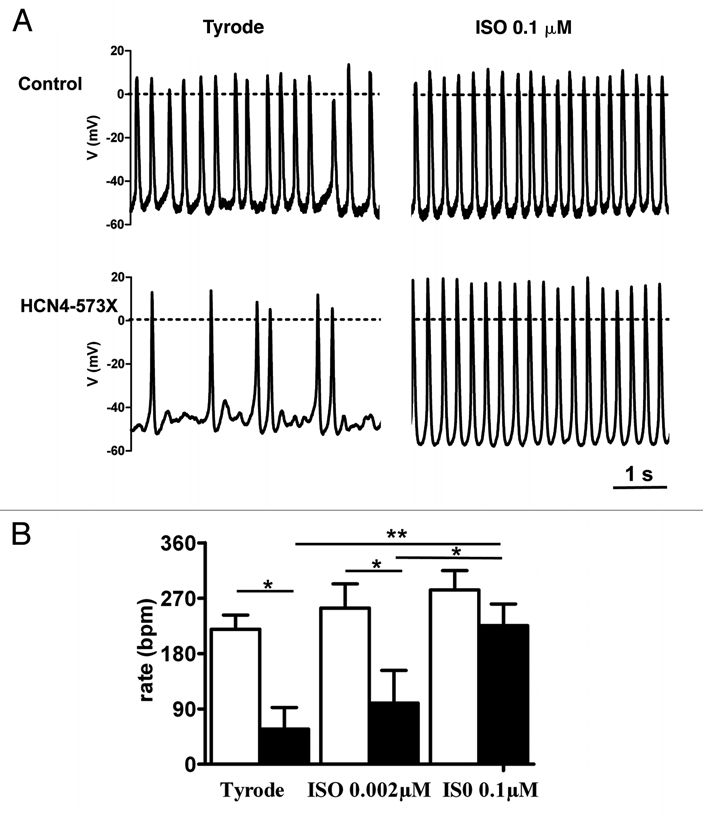
(A) Pacemaker activity in AVNCs from transgenic control mice (upper line) and mice expressing hHCN4-573X (bottom line). Control cells showed regular pacemaker activity in both control conditions and under stimulation by saturating ISO concentration, while AVNCs expressing hHCN4-573X channels showed slow and irregular pacemaking at rest with sporadic AP discharge. ISO induced regular pacemaking in mutant AVNCs. (B) Histogram showing averaged beating rates of control and mutant HCN4-573X AVNCs in basal conditions (Tyrode), after application of a submaximal (0.002 µM) and saturating (0.1 µM) dose of ISO. Pacemaking of HCN4-573X cells reached that of control cells at saturating concentrations of ISO (0.1 µM). Stars indicate statistical significance (two-way ANOVA).
Discussion
This is the first study investigating the functional role of three key ion channel genes in pacemaker activity of mouse AVNCs. Four major new findings are presented. First, Cav1.3 channels constitute the predominant ICa,L channel isoform and are necessary for spontaneous automaticity of mouse AVNCs. In this respect, our results on Cav1.3−/− AVNCs give an important insight on the pathophysiologic mechanism of AV dysfunction in human SNDD. Second, we show that the loss of Cav3.1 channels restricts both basal and maximal spontaneous pacemaking rates of AVNCs. Third, we report that combined inactivation of Cav1.3 and Cav3.1 channels produces additive effects on both in vivo AV conduction and AVNCs firing and yields a phenotype similar to human CHB. Forth, we found that cAMP-dependent regulation of HCN channels appears to be important for AVNC automaticity under basal conditions, but is not strictly required for regulation of pacemaking and for reaching maximal pacing rates, especially under β-adrenergic receptor activation. Our study thus provides a comprehensive description of the pacemaker activity of murine AVNCs.
Cav1.3 channels are required for AVNC pacemaking.
The loss of Cav1.3 channels induces AV conduction dysfunction in mice (Fig. 1), suggesting that these channels play an important role in AVN physiology. In this study, we show that Cav1.3 channels play an essential role in AVNCs pacemaking. Several lines of evidence support this. First, under normal conditions (no injection of constant hyperpolarizing current), Cav1.3−/− AVNCs did not display spontaneous automaticity and, even in the presence of saturating doses of ISO, pacemaking could not be restored. Second, even in negative current-injected Cav1.3−/− AVNCs, we recorded low pacing frequencies and irregular AP generation. Interestingly, the diastolic depolarization was absent and the membrane voltage of Cav1.3−/− AVNCs during the diastolic phase showed mainly subthreshold membrane potential oscillations. This observation indicates that Cav1.3 channels are a critical determinant in the generation of the diastolic depolarization in AVNCs. Third, while initiated firing of negative current-injected Cav1.3−/− AVNCs did accelerate under ISO, maximal rates were still much lower than those of WT counterparts (Fig. 4). We did not directly investigate the reason for the positive resting membrane potential in Cav1.3−/− AVNCs. A likely hypothesis is that the loss of crosstalk between Cav1.3 channels and SK2 K+ channels13 in Cav1.3−/− AVNCs induces depolarization of the membrane resting potential. The observation that Cav1.3−/− AVNCs lacked spontaneous automaticity when isolated in vitro may not imply unexcitability in vivo. Indeed, we showed that Cav1.3−/− AVNCs are still able to fire INa-dependent action potentials upon membrane hyperpolarization (Fig. 2). It is possible that in the intact AVN, Cav1.3−/− myocytes are also sufficiently hyperpolarized to enable INa-dependent action potentials to be triggered by SAN impulse. In vivo, the both the SAN and AVN are subject of the hyperpolarizing load imposed by the electrical coupling with the right atrium. This phenomenon explains, for instance, why IKr blockers completely stop pacemaker activity of isolated SANCs, but not in rabbit SAN-atrial preparations19 or in intact mouse hearts.20 Hyperpolarizing load by the right atrium could also explain why in another Cav1.3−/− mouse line, Zhang and co-workers21 measured residual AVN automaticity in intact atrial preparations containing the AVN. Finally, it is worth noting that the AVN is a highly heterogeneous structure in which different cell types, possibly serving different conduction pathways can co-exist. Cell-cell interactions between automatic and non-automatic AVNCs, together with the alternative use of different conduction pathways could also explain why Cav1.3−/− mice do not show chronic complete AV block. However, the importance of Cav1.3 channels in AV conduction is stressed by the presence of dissociated rhythms in SNDD patients.7
Co-expression of both Cav1.3- and Cav1.2-mediated ICa,L has been reported in SANCs of two different Cav1.3−/− mouse strains.10,16 We estimated in a previous study that in mouse SANCs Cav1.3-mediated ICa,L accounts for about 70% of the total ICa,L.16 In mouse AVNCs functional expression of Cav1.3 channels appears to be predominant, since inactivation of Cav1.3 channels reduced AVNC ICa,L by about 90%. The very low residual ICa,L density in Cav1.3−/− AVNCs prevented us to directly measure the half activation and kinetics of Cav1.2-mediated ICa,L.
The prominent role of INa in AVNC AP firing (see Fig. 3 of the accompanying paper in ref. 17) suggests that this current is a major contributor of APs in Cav1.3−/− cells. It is possible that, especially under ISO stimulation, residual Cav1.2-mediated ICa,L can contribute to the AP upstroke. We thus propose that the absence of basal pacemaker activity in Cav1.3−/− cells is not due to incapability of generating the AP upstroke, but rather to insufficient Cav1.3-mediated ICa,L in the diastolic depolarization range. In this respect, we performed pilot numerical simulations that suggest that the difference in density and activation of Cav1.3-mediated ICa,L could contribute to the dependence of AVNCs pacemaking from INa (Sup. Fig. S2). Thus, the phenotype of Cav1.3−/− AVNCs differed from that of Cav3.1−/− SANCs, which paced at lower rates than WT cells but retained residual automaticity.16 Zhang and co-workers21 recorded ICa,L using a distinct Cav1.3−/− mouse line and did not find differences in current densities between WT and Cav1.3−/− SANCs10 or AVNCs.21 These authors attributed the lack of change in ICa,L densities to compensation by Cav1.2 channels in Cav1.3−/− SANCs10 and AVNCs.21 Discrepancies between results by Zhang et al. and ours may be due to the different genetic background of our Cav1.3−/− line and/or to the different strategy of genetic targeting of the Cav1.3 gene.
Role of Cav3.1 channels in AVNCs pacemaking.
The loss of Cav3.1-mediated ICa,T had strong consequences on AVNC automaticity. Indeed, pacemaking of Cav3.1−/− AVNCs was intermittent and slower than that of WT cells. Stimulation by ISO augmented the pacemaking rate of Cav3.1−/− AVNCs, but maximal rates measured under these conditions were lower than that of WT cells. Interestingly, negative current-injected AVNCs from Cav1.3−/−/Cav3.1−/− mice had slower firing rate than negative current-injected Cav1.3−/− cells, indicating that Cav3.1 channels can contribute also to initiated automaticity of negative currentinjected Cav1.3−/− AVNCs. The phenotype of Cav3.1−/− cells demonstrates that Cav3.1-mediated ICa,T plays an important role in automaticity of AVNCs, by contributing to basal and maximal (under ISO) pacemaking rates. The mechanism of cellular dysrhythmia in Cav3.1−/− AVNCs is not known. ISO was only partially effective in increasing pacemaking rates and did not normalize cellular arrhythmia.
HCN channels in AVNCs pacemaking.
HCN channels are considered to play a key role in both the generation and regulation of cardiac pacemaker activity.14 Because current-injected Cav1.3−/−/Cav3.1−/− AVNCs are still responsive to ISO, we evaluated the role of cAMP-dependent regulation of HCN channels in AVNCs automaticity. To avoid embryonic lethality due to constitutive loss of HCN4 protein22 we employed our new mouse model, which conditionally expresses the HCN4-573X transgene abolishing cAMP sensitivity of HCN channels in a dominant negative way.15 A striking finding is that the loss of cAMP dependent regulation of HCN channels induced slow and irregular pacemaker activity in mouse AVNCs. However, ISO application raised regular pacemaking of mutant AVNCs and maximal rates of control and mutant AVNCs were comparable. Our results thus indicate that HCN channels are important for basal excitability of mouse AVNCs, but are not required for acceleration of pacemaker activity under β-adrenergic receptor stimulation by ISO and for reaching maximal pacemaking rates. This result is in agreement with a recent study that showed no effect of the f-channel blocker ZD-7288 on ISO-induced acceleration of pacemaker activity in isolated canine AVN preparations.23 The preserved β-adrenergic regulation of AVNCs pacemaking observed in Cav1.3−/−, Cav3.1−/− and HCN4-573X expressing AVNCs is intriguing. One possibility is that other mechanisms such as RYR-dependent Ca2+ release24 or CaMKII,25 mechanisms that are operant in SANCs can mediate regulation of AVNCs pacemaking in mutant AVNCs. Also, we cannot exclude that residual Cav1.2-mediated ICa,L can compensate, in part, for the lack of Cav1.3 channels in Cav1.3−/− AVNCs.
Conclusions and implications.
Our work indicates that besides well known pacemaker mechanism such as HCN-channels and RYR-dependent Ca2+ release,23,24 Cav1.3 and Cav3.1 channels are important for AVNCs automaticity. A recent pharmacological study on intact AVNs by Lu et al. suggested that the relative importance of ion channels and RyR-dependent Ca2+ release in pacemaking can vary between the SAN and AVN.26 In this respect, our results show important differences in the impact of Cav1.3, Cav3.1 and HCN channels between AVNCs and SANCs. A preliminary numerical model of AVNCs automaticity based on Cav1.3, Cav3.1 and HCN channels is provided in the supplemental online section. Interestingly, it has been reported that AVNC precursors show a distinct genetic program than that of SAN.27 It is thus tempting to speculate that such a differential genetic program is reflected into differences in the underlying pacemaker mechanism.
Materials and Methods
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US national Institute of Health (NIH Publication No. 85-23, revised 1996) and European directives (86/609/CEE).
Generation of mouse lines.
Mice lacking Cav1.3 channels (Cav1.3−/−) of C57B/6J genetic background were obtained as described previously in reference 16. Mice lacking Cav3.1 channels (Cav3.1−/−) were derived from Cav3.1−/− mice on 129Sv genetic background11 that were backcrossed on C57B/6J mice for more than ten generations. Cav1.3−/−/Cav3.1−/− mice were obtained by crossing C57B/6J Cav1.3−/− with Cav3.1−/− mice. Mice expressing hHCN4-573X channels in a conditional and heart specific way were obtained as described previously in reference 15. Briefly, mice carrying an alpha myosin heavy chain (αMHC) and Tet-Off system controlled promoter transgene were used as controls (αMHC-tTA mice). Double transgenic mutant mice were obtained by crossing αMHC-tTA mice with transgenic mice carrying an engineered human HCN4 cDNA (hHCN4) that lacked the cAMP-binding domain and contained an hemagglutinin (HA) sequence tag at the N-terminus (hHCN4-573X). Breeding mice in doxycycline-free drinking water induced expression of hHCN4-573X channels.
Telemetric ECG recordings.
ECGs recording were performed in control and mutant mice as described previously in reference 11. An expanded description of ECG recording and analysis is available in the online data supplement (Sup. online methods).
Isolation of mouse AVNCs and patch-clamp recordings.
AVN and SAN tissues were obtained from female mice aged of 2–4 months as described in the accompanying paper in reference 17. All electrophysiological experiments were carried out at 36°C. The whole-cell variation of the patch-clamp technique was used to measure Ca2+ currents. All other experiments have been performed using the perforated patch technique with β-escin (50 µM).
Statistical analysis.
Results are presented as means ± standard error of the mean (SEM, number of cells). For calculating the level of significance, the Student's t-test, the one- or two-way ANOVA tests followed by Tukey's post-hoc test or the non-parametric Kruskal-Wallis test have been employed. When testing statistical differences results were considered significant with p < 0.05. In all figures *p < 0.05, **p < 0.01 and ***p < 0.001, respectively.
Acknowledgements
We thank Elodie Kupfer and the personnel of the IFR3 animal facility of Montpellier for breeding and managing mouse lines. We thank Isabelle Bidaud (CNRS UMR 5203, Montpellier France) for excellent technical assistance. We also thank Stephanie Barrere-Lemaire (CNRS UMR 5203) and Emilio Carbone (University of Turin, Italy) for helpful discussion. This work has been supported by CavNet a Research Training Network (RTN) funded through the European Union Research Programme (6FP) MRTN-CT-2006-035367, the INSERM National Program for Cardiovascular Diseases (PNRC), the Fondation de France (Cardiovasc 2008002730), the Agence Nationale pour la Recherche (ANR-06-PHISIO-004-01), the NIH R01HL087120-A2 grant, the Deutsche Forschungsgemeinschaft (DFG, IS63/1-1/2) and the Austrian Science Fund (P-20670). Pietro Mesirca and Angelo Torrente are CavNet fellows.
Abberviations
- AVN
atrioventricular node
- SAN
sino-atrial node
- ICa,L
L-type Ca2+ current
- ICa,T
T-type Ca2+ current
- If
hyperpolarization-activated f-current
- Cav1.3
voltage-dependent L-type Ca2+ channel 3rd isoform
- Cav3.1
voltage-dependent T-type Ca2+ channel 1st isoform
- HCN
hyperpolarization-activated cation channel
- CHB
congenital heart block
- SNDD
sino-atrial dysfunction and deafness syndrome
Supplementary Material
References
- 1.Meijler FL, Janse MJ. Morphology and electrophysiology of the mammalian atrioventricular node. Physiol Rev. 1988;68:608–647. doi: 10.1152/physrev.1988.68.2.608. [DOI] [PubMed] [Google Scholar]
- 2.Benson DW, Wang DW, Dyment M, Knilans TK, Fish FA, Strieper MJ, et al. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (Scn5A) J Clin Invest. 2003;112:1019–1028. doi: 10.1172/JCI18062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smits JP, Koopmann TT, Wilders R, Veldkamp MW, Opthof T, Bhuiyan ZA, et al. A mutation in the human cardiac sodium channel (E161K) contributes to sick sinus syndrome, conduction disease and Brugada syndrome in two families. J Mol Cell Cardiol. 2005;38:969–981. doi: 10.1016/j.yjmcc.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 4.Brucato A, Cimaz R, Catelli L, Meroni P. Anti-Ro-associated sinus bradycardia in newborns. Circulation. 2000;102:88–89. doi: 10.1161/01.cir.102.11.e88. [DOI] [PubMed] [Google Scholar]
- 5.Hu K, Qu Y, Yue Y, Boutjdir M. Functional basis of sinus bradycardia in congenital heart block. Circ Res. 2004;94:32–38. doi: 10.1161/01.RES.0000121566.01778.06. [DOI] [PubMed] [Google Scholar]
- 6.Qu Y, Baroudi G, Yue Y, Boutjdir M. Novel molecular mechanism involving alpha1D (Cav1.3) L-type calcium channel in autoimmune-associated sinus bradycardia. Circulation. 2005;111:3034–3041. doi: 10.1161/CIRCULATIONAHA.104.517326. [DOI] [PubMed] [Google Scholar]
- 7.Baig SM, Koschak A, Lieb A, Gebhart M, Dafinger C, Nurnberg G, et al. Loss of Cav1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat Neurosci. 2011;14:77–84. doi: 10.1038/nn.2694. [DOI] [PubMed] [Google Scholar]
- 8.Mangoni ME, Nargeot J. Genesis and regulation of the heart automaticity. Physiol Rev. 2008;88:919–982. doi: 10.1152/physrev.00018.2007. [DOI] [PubMed] [Google Scholar]
- 9.Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, et al. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell. 2000;102:89–97. doi: 10.1016/s0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Z, Xu Y, Song H, Rodriguez J, Tuteja D, Namkung Y, et al. Functional roles of Cav1.3 (alpha(1D)) calcium channel in sinoatrial nodes: insight gained using gene-targeted null mutant mice. Circ Res. 2002;90:981–987. doi: 10.1161/01.res.0000018003.14304.e2. [DOI] [PubMed] [Google Scholar]
- 11.Mangoni ME, Traboulsie A, Leoni AL, Couette B, Marger L, Le Quang K, et al. Bradycardia and slowing of the atrioventricular conduction in mice lacking Cav3.1/alpha1G T-type calcium channels. Circ Res. 2006;98:1422–1430. doi: 10.1161/01.RES.0000225862.14314.49. [DOI] [PubMed] [Google Scholar]
- 12.Papadatos GA, Wallerstein PM, Head CE, Ratcliff R, Brady PA, Benndorf K, et al. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc Natl Acad Sci USA. 2002;99:6210–6215. doi: 10.1073/pnas.082121299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Q, Timofeyev V, Lu L, Li N, Singapuri A, Long MK, et al. Functional roles of a Ca2+-activated K+ channel in atrioventricular nodes. Circ Res. 2008;102:465–471. doi: 10.1161/CIRCRESAHA.107.161778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DiFrancesco D. The role of the funny current in pacemaker activity. Circ Res. 106:434–446. doi: 10.1161/CIRCRESAHA.109.208041. [DOI] [PubMed] [Google Scholar]
- 15.Alig J, Marger L, Mesirca P, Ehmke H, Mangoni ME, Isbrandt D. Control of heart rate by cAMP sensitivity of HCN channels. Proc Natl Acad Sci USA. 2009;106:12189–12194. doi: 10.1073/pnas.0810332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mangoni ME, Couette B, Bourinet E, Platzer J, Reimer D, Striessnig J, et al. Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc Natl Acad Sci USA. 2003;100:5543–5548. doi: 10.1073/pnas.0935295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marger L, Mesirca P, Alig J, Torrente A, Dubel S, Engeland B, et al. Pacemaker activity and ionic currents in mouse atrioventricular node cells. Channels. 2011 doi: 10.4161/chan.5.3.15264. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DiFrancesco D. The role of the funny current in pacemaker activity. Circ Res. 2010;106:434–446. doi: 10.1161/CIRCRESAHA.109.208041. [DOI] [PubMed] [Google Scholar]
- 19.Verheijck EE, Wilders R, Bouman LN. Atrio-sinus interaction demonstrated by blockade of the rapid delayed rectifier current. Circulation. 2002;105:880–885. doi: 10.1161/hc0702.104128. [DOI] [PubMed] [Google Scholar]
- 20.Clark RB, Mangoni ME, Lueger A, Couette B, Nargeot J, Giles WR. A rapidly activating delayed rectifier K+ current regulates pacemaker activity in adult mouse sinoatrial node cells. Am J Physiol Heart Circ Physiol. 2004;286:1757–1766. doi: 10.1152/ajpheart.00753.2003. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Q, Timofeyev V, Qiu H, Lu L, Li N, Singapuri A, et al. Expression and roles of Cav1.3 (alpha1D) L-type Ca2+ channel in atrioventricular node automaticity. J Mol Cell Cardiol. 2011;50:194–202. doi: 10.1016/j.yjmcc.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stieber J, Herrmann S, Feil S, Loster J, Feil R, Biel M, et al. The hyperpolarization-activated channel HCN4 is required for the generation of pacemaker action potentials in the embryonic heart. Proc Natl Acad Sci USA. 2003;100:15235–15240. doi: 10.1073/pnas.2434235100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim D, Shinohara T, Joung B, Maruyama M, Choi EK, On YK, et al. Calcium dynamics and the mechanisms of atrioventricular junctional rhythm. J Am Coll Cardiol. 2010;56:805–812. doi: 10.1016/j.jacc.2010.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ridley JM, Cheng H, Harrison OJ, Jones SK, Smith GL, Hancox JC, et al. Spontaneous frequency of rabbit atrioventricular node myocytes depends on SR function. Cell Calcium. 2008;44:580–591. doi: 10.1016/j.ceca.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 25.Wu Y, Gao Z, Chen B, Koval OM, Singh MV, Guan X, et al. Calmodulin kinase II is required for fight or flight sinoatrial node physiology. Proc Natl Acad Sci USA. 2009;106:5972–5977. doi: 10.1073/pnas.0806422106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu J, Noble PJ, Xiao G, Abdelrahman M, Dobrzynski H, Boyett MR, et al. Role of pacemaking current in cardiac nodes: Insights from a comparative study of sinoatrial node and atrioventricular node. Prog Biophys Mol Biol. 2008;96:294–304. doi: 10.1016/j.pbiomolbio.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 27.Horsthuis T, Buermans HP, Brons JF, Verkerk AO, Bakker ML, Wakker V, et al. Gene expression profiling of the forming atrioventricular node using a novel tbx3-based node-specific transgenic reporter. Circ Res. 2009;105:61–69. doi: 10.1161/CIRCRESAHA.108.192443. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.