

SUMMARY
Hepatitis C virus (HCV) infects chronically 3% of the world population and the current therapy against this pathogen is only partially effective. With the aim of developing novel antiviral strategies against HCV, we screened a d,l-α-peptide library using an unbiased methodology based on a cell culture infection system for HCV. We found a family of highly active amphiphilic eight-residue cyclic d,l-α-peptides that specifically blocked entry of all tested HCV genotypes into target cells at a post-binding step without affecting infection by other enveloped RNA viruses. Structure-activity relationship (SAR) studies indicate that antiviral activity was dependent on cyclic d,l-α-peptide self-assembly processes and that, although they possesses a net neutral charge, they display a characteristic charge distribution. Our results indicate that supramolecular amphiphilic peptide structures constitute a novel class of highly selective HCV entry inhibitors.
INTRODUCTION
The hepatitis C virus (HCV) is an enveloped member of the Flaviviridae family with a 9.6 kb positive stranded RNA genome that causes acute and chronic hepatitis and hepatocellular carcinoma (Hoofnagle, 2002; Liang et al., 2000). More than 170 million people are chronically infected with this blood-borne pathogen worldwide (Alter, 1997) making HCV a major worldwide killer and the most common cause of liver transplantation in the United States (Alter, 1999). Despite the fact that new therapies based on combinations of interferon with direct-acting antivirals (DAAs) have improved the proportion of responding patients, the concern for rapid emergence of resistant variants fuels the search for new antivirals targeting different aspects of the viral lifecycle that could be used in combination therapies (Gelman and Glenn, 2010).
HCV infection is initiated by binding of the viral particle to cellular receptors at the surface of the target cell (reviewed in (Burlone and Budkowska, 2009)). Subsequently, internalization of the bound virus particles through receptor-mediated endocytosis (Blanchard et al., 2006) results in the fusion of viral and cellular membranes triggered by endosome acidification (Tscherne et al., 2006), delivery of the viral genome to the cytosol, and primary translation of the incoming HCV genome by a process that is dependent on the cellular autophagy machinery (Dreux et al., 2009). The HCV genome encodes a single polyprotein that is co- and post-translationally cleaved by cellular and viral proteases into structural (core, E1, E2 and p7) and non-structural (NS2, NS3, NS4A, NS4B, NS5A and NS5B) proteins (Bartenschlager and Lohmann, 2000) that mediate viral genome replication and progeny virus assembly and secretion (reviewed in (Moradpour et al., 2007; Murray et al., 2008) by a process that exploits the cellular lipoprotein biosynthetic machinery (Chang et al., 2007; Gastaminza et al., 2008; Huang et al., 2007; Jiang and Luo, 2009; Kapadia and Chisari, 2005; Su et al., 2002; Ye et al., 2003).
Until recently, the lack of cell culture and small animal infection systems limited analysis of HCV infection to surrogate models, e.g. autonomously self-replicating viral RNAs (replicons) (Blight et al., 2000; Lohmann et al., 1999) and pseudotyped virus particles (Bartosch et al., 2003) that reproduced only selected aspects of the virus life-cycle (Hao et al., 2007; Kim et al., 2007). With the advent of cell culture models of HCV infection in human hepatoma-derived cell lines based on the genotype 2a JFH-1 strain of HCV (Lindenbach et al., 2005; Wakita et al., 2005; Zhong et al., 2005), the entire viral life cycle became accessible in cell culture, thus providing the opportunity to study the impact of known and novel antiviral compounds on all aspects of the viral life cycle. We recently developed a miniaturized colorimetric system that permits efficient analysis of HCV infection, replication, assembly, secretion, and spread in cell culture (Gastaminza et al., 2010). We adapted this methodology to screen a library of 144 pre-selected non-toxic, amphipathic, membrane-active cyclic d,l-α-peptides (Fernandez-Lopez et al., 2001) for antiviral activity against the parental JFH-1 HCV strain.
Self-assembling peptide nanotubes are a versatile class of supramolecular structures with expanding utility in materials and biological settings. Cyclic peptides made up of an even number of alternating d- and l-α-amino acid residues adopt (or sample) flat ring-shaped conformations in which all amide backbone functionalities reside approximately perpendicular to the plane of the ring structure (Ghadiri et al., 1993; Ghadiri et al., 1995; Hartgerink et al., 1996; Kobayashi et al., 1995). In this conformation, because of the spatial disposition and juxtaposition of amide hydrogen-bond donor and acceptor sites on either side of the ring structure, the peptide subunits can stack under conditions that favor hydrogen bonding, such as lipid membranes, to form uniformly-shaped and contiguously hydrogen-bonded β-sheet like tubular ensembles (Clark et al., 1998; Ghadiri et al., 1994; Granja and Ghadiri, 1994; Kim et al., 1998). We have shown that appropriately designed amphiphilic cyclic d,l-α-peptides are able to selectively target and self assemble in bacterial membranes and exert antibacterial activity by increasing the membrane permeability (Dartois et al., 2005; Fernandez-Lopez et al., 2001; Fletcher et al., 2007). Furthermore, using adenovirus as a model non-enveloped virus, we previously identified antiviral cyclic d,l-α-peptides that specifically prevented the development of low pH in endocytic vesicles, thereby arresting the escape of virions from the endosome and abrogating adenovirus infection without adversely affecting cell viability (Horne et al., 2005). Due to their abiotic backbone structures, these peptides are both chemically and proteolytically stable and can be targeted to operate in a variety of settings and environments. In the present work, we report the discovery of a family of novel HCV-specific antiviral molecules. We demonstrate that these peptides operate by preventing viral entry and require a cyclic configuration and the ability to self-assemble as nanotubes to exert their antiviral activity.
RESULTS
Identification of a family of HCV inhibitors
Over the past few years we have amassed a collection of >1,200 eight-residue cyclic d,l-α-peptides in pure forms that were derived from various libraries designed to probe the sequence-space for effective antimicrobial agents (Dartois et al., 2005; Fernandez-Lopez et al., 2001; Horne et al., 2005; Motiei et al., 2009; Wilcoxen, 2002). From this peptide depository, which represents a small sampling of a rather diverse cyclic d,l-α-peptide sequence-space, a panel of 144 sequences known to be nontoxic in HeLa cells (LD50 > 100 µM) was screened for antiviral activity against HCV (Table S1) in a cell-based ELISA assay that integrates the number of infected cells and the amount of viral antigen per cell in a 96-well format (Gastaminza et al., 2010). This screen yielded nine cyclic amphiphilic octapeptides (Table 1) that were characterized by a neutral charge, the presence of either Ser or positively charged residues adjacent to the hydrophobic region of the molecule, and predominantly Glu, Asp, or Gln residues at the remaining positions. All the peptides identified by the counterscreen displayed an EC50 between 8 to 16 µM and the virtual absence of toxicity (≥100 µM in Huh-7 cells).
Table 1. Peptide sequences of hits from primary library screening and peptide 9 derivatives used in the SAR studies.
Primary hits and derivatives of peptide 9 were generated as described in the text and assayed for their antiviral activity (EC50) and toxicity (LD50) as shown. EC50 values were determined by colorimetric evaluation of HCV infection in the presence of increasing peptide concentrations (0.75–50 µM). LD50 values were determined by monitoring parallel cultures for cell viability using the XTT assay (See Experimental Procedures). Values are shown as average and mean error of a minimum of three experiments. Net charge was estimated by the charge of the individual amino acids. Amino acids shown in lower case represent D-configuration. Z = 2,3-Diaminopropionic acid; Ac, acetyl group; Me, N- methylation; nd, not determined. See also Tables S1 and S2.
| Primary Hits: | ||||
|---|---|---|---|---|
| Name | Sequence | EC50 (µM) | LD50 (µM) | Net charge |
| 1 | 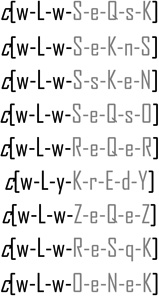 |
3.3 ± 0.5 | 65 | 0 |
| 2 | 7.7 ± 3.8 | >100 | 0 | |
| 3 | 7.9 ± 2.1 | >100 | 0 | |
| 4 | 10.3 ± 3.4 | >100 | 0 | |
| 5 | 15.5 ± 2.8 | >100 | 0 | |
| 6 | 8.9 ± 1.6 | >100 | 0 | |
| 7 | 6.9 ± 3.4 | 80 | 0 | |
| 8 | 3.4 ± 0.4 | 90 | 1 | |
| 9 | 11 ± 2.8 | >100 | 0 | |
| SAR (Peptide 9 derivatives): | ||||
| Name | Sequence | EC50 (µM) | LD50 (µM) | Net charge |
| 10 | 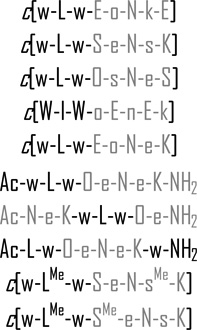 |
>50 | >100 | 0 |
| 11 | 6.5 ± 0.8 | >100 | 0 | |
| 12 | 7.8 ± 0.3 | >100 | 0 | |
| 13 | 5.5 ± 1.2 | 45 | 0 | |
| 14 | 18.4 ± 3.6 | >100 | 0 | |
| 15 | >50 | nd | 0 | |
| 16 | >50 | nd | 0 | |
| 17 | >50 | nd | 0 | |
| 18 | >50 | nd | 0 | |
| 19 | >50 | nd | 0 | |
To ascertain the mode of antiviral action of this peptide family, we treated the target cells with the peptides at different times relative to the time of inoculation with the virus (Figure 1). This analysis showed that the peptides displayed an antiviral effect when the peptide was present during the initial inoculation period (five hours) or throughout the experiment, but not if they were pre-incubated with the cells for five hours before virus inoculation or if they were added to the cells five hours after inoculation, i.e. after infection was complete. These results suggested that the peptides interfered with an early step of the infection, such as steps involved in viral entry. Another possibility was that the peptides reduced the infectivity of the viral particles by directly interacting with and/or destabilizing them, as was recently described for a linear amphiphilic anti-HCV peptide (Cheng et al., 2008). To test that hypothesis, we incubated the virus with the peptides (20 µM) for 5 hours at 37 °C, after which viral infectivity was determined by dilution of the virus-peptide mixture to a final peptide concentration at least 50-fold below the EC50. As shown in Figure S1, the peptides did not notably reduce the infectivity or the amount of viral RNA in the inoculum, suggesting that the peptides are not virocidal. This finding is particularly important since the antiviral peptides are capable of interacting with lipid membranes, as measured by dye release assays in unilamellar vesicles (Table S2). This property, however, did not correlate with the antiviral activity of the peptides, indicating that liposome lysis capacity is not sufficient for the observed anti-HCV activity. Collectively, these results imply that the peptides target an early step in the HCV infection cycle and likely share a common mode of action.
Figure 1. Time of addition studies reveal inhibition of early steps of HCV infection with d,l-α-peptides.

Peptides (20 µM) were added to target cells 5 h prior to inoculation (Cell pre-treatment), during the 5 hour (37 °C) inoculation period (Co-treatment), after virus infection (Treatment post-infection), or were present at all times (Present all-time). Infection efficiency was measured by colorimetry 72 hours postinfection as described in Materials and Methods. Results are shown as average and mean error of two independent infections. See also Figure S1.
Structural features required for antiviral activity (SAR analysis)
To probe the features required for antiviral activity, we modified the amino acid sequence of a representative member of the amphiphilic cyclic d,l-α-peptide family (peptide 9) in a manner that would preserve its overall neutral charge. To test whether one or more acidic residues can be tolerated at positions next to the hydrophobic domain, compounds 10 and 14 were synthesized and tested for antiviral activity. Reversing the amino acid positions of Lys/Orn and Glu pairs to place one or two negatively charged Glu residues at the hydrophobic interface in cyclic peptides 10 and 14, led to either complete or significant loss of antiviral activity, respectively (Table 1). Interestingly, despite lack of antiviral activity of peptide 10, this compound was found to be membranolytic in liposome dye release assays (Table S2), reinforcing the notion that membrane activity is not sufficient for antiviral activity. The above data thus suggest that, in addition to the amphiphilic net neutral charge structure, the amino acid sequence is also strong determinant of antiviral activity.
In order to probe the influence of Lys/Orn and Glu pairs on antiviral activity, we synthesized net neutral peptides 11 and 12 in which a Lys/Orn and a Glu residue were simultaneously replaced with Ser residues. These derivatives displayed slightly increased potency (EC50 of 6.5 µM for 11 and 7.5 µM for 12; Table 1) in line with the level of activities observed for similar sequences (peptides 1, 2, and 3; Table 1), suggesting that placing a Ser residue at the hydrophobic/hydrophilic interface can have a positive influence on the antiviral activity.
By virtue of their flat ring structure and special backbone amide bond disposition, cyclic d,l-α-peptides have a propensity to undergo intermolecular hydrogen bond-directed self-assembly into nanotubular assemblies. Previous studies in the design and discovery of antimicrobial cyclic d,l-α-peptides have strongly correlated peptide self-assembly and antimicrobial activity (Fernandez-Lopez et al., 2001). Similarly, in this study we examined whether the cyclic structure and supramolecular assembly are also required for the antiviral activity of the anti-HCV peptides. Accordingly, acetylated linear versions of prototypic peptide 9 were synthesized and tested against HCV. These linearized sequences (peptides 15, 16, and 17; Table 1) failed to block HCV infection, establishing that a cyclic structure is required for potency. To establish whether peptide self-assembly is also required for the observed antiviral activity, backbone N-methylated analogs of peptide 11 were synthesized (compounds 18 and 19). Specific backbone N-alkylation of cyclic d,l-α-peptides has been shown to limit peptide self-assembly to only dimeric structures (Bong et al., 2001; Ghadiri et al., 1994; Ghadiri et al., 1993). As shown in Table 1, backbone N-alkylation resulted in complete loss of activity. Another line of support for the supramolecular nature of the active species is that compound 13, the enantiomeric version of 9, was also active in blocking HCV infections. Overall, the above studies support the notion that a mechanistic requirement for these cyclic d,l-α-peptides to reduce HCV infection is their self-assembly into supramolecular species.
Having identified the major structural determinants of this group of antiviral molecules, we next set out to gain insights into the mode of action by defining at which step of the viral lifecycle these molecules impede HCV infection. To accomplish this goal, we used the most potent and least toxic peptides from the above studies (peptides 9, 11, and 12) in the following studies.
Mechanism of action
The time-of-addition experiments suggested that these peptides target early steps in the viral lifecycle. To test this hypothesis, we performed single-step infection experiments in which Huh-7 cells were inoculated with a high virus dose (m.o.i. of five) for five hours in the presence of the indicated peptides (20 µM), after which the cells were extensively washed and incubated with fresh complete medium for an additional 24 hours. As shown in Figure 2A, the peptides inhibited intracellular and extracellular virus particle production in this experiment, confirming that they indeed interfere at an early step in HCV infection.
Figure 2. d,l-α-peptides inhibit infection in a post-binding step preceding viral RNA replication.

(A) Single cycle infection in the presence of antiviral peptides. Huh-7 cells were inoculated at multiplicity of 5 (m.o.i 5) with HCV for 5 h at 37 °C in the presence of vehicle (DMSO) or the designated peptide (20 µM). After the inoculation period, the cells were washed once with PBS and further incubated in complete medium without peptides. Infectivity was determined in total cell extracts and supernatants 24 h post-inoculation. Intracellular infectivity (FFU/ml) (Left). Extracellular infectivity (FFU/ml) (Right). Data are shown as average and mean error of duplicate infections. *Peptide 4 not shown, tested under same conditions displayed inconsistent results.
(B) d,l-α-peptides do not display anti-HCV activity in the replicon model. Huh-7 cells harboring a full-length JFH-1 replicon were treated with cyclic peptide (20 µM), the protease inhibitor BILN2061 (1 µM), or the polymerase inhibitor 2’-C-methyl-cytidine (1 µM) for 48 hours at 37 °C. Intracellular HCV RNA levels were determined by RT-qPCR and normalized with GAPDH mRNA levels. Results are shown as average and mean error of triplicate experiments.
(C) d,l-α-peptides do not interfere with infectious particle binding but target a post-binding step of the viral lifecycle. Huh-7 cells were inoculated with increasing amounts of infectious HCV for 1 h at 4 °C in the presence of a constant amount of peptide (20 µM). Susceptibility of binding or post-binding events to inhibition was studied by adding the peptide during inoculation or after virus particle binding to target cells (see Materials and Methods). Infection efficiency was calculated by determining the infectivity titer in each condition and comparing it with that obtained in the presence of vehicle (DMSO).
To determine if peptide 9 and its derivatives inhibit HCV RNA replication, we incubated Huh-7 cells harboring a full-length JFH-1 replicon with the peptides (20µM) or DMSO and measured the intracellular HCV RNA content 48 hours later. Parallel cultures were incubated with the HCV protease inhibitor BILN2061 (1 µM) (Lamarre et al., 2003) or polymerase inhibitor 2’-C-methylcytidine (2-CMC) (1 µM) (Carroll and Olsen, 2006) as a control. As shown in Figure 2B, BILN2061 and 2-CMC efficiently reduced HCV RNA levels while peptide 9 and its derivatives did not. These results suggest that the peptides target events upstream of replication, probably at the level of viral entry. These findings also confirm the observation that the peptides do not show any antiviral activity when added after the virus had entered the cell in the time-of-addition experiment (Figure 1).
To determine if the peptides inhibit a binding or post-binding step in viral entry, we temporally separated these two events by inoculating the target cells at 4 °C to allow virus particle binding while minimizing internalization (Koutsoudakis et al., 2006; Tscherne et al., 2006). Huh-7 cells were inoculated for 1 hour at 4 °C in the presence or absence of peptides 9, 11, and 12 (20 µM) after which the cells were washed twice with ice-cold PBS and replenished with warm medium to promote viral internalization (Tscherne et al., 2006). Cultures were then incubated for 72 hours at 37 °C and the viral titer was determined as described in Zhong et al. (Zhong et al., 2005). As shown in Figure 2C, the inhibitory peptides did not alter the binding of the virus to the target cell, reinforcing the notion that they are not virocidal, as shown in Figure S1, and suggesting that they inhibit an event downstream of binding. To test this hypothesis directly, Huh-7 cells were inoculated at low temperature after which unbound virus was removed by extensive washing with cold PBS and bound virus internalization was triggered by addition of warm medium containing the antiviral peptides (20 µM). After a 5 h incubation at 37°C, medium containing the peptides was removed and the cells were replenished with complete medium lacking the peptides after extensive washing with PBS. Cultures were incubated for additional 72 hours after which infection efficiency was evaluated by immunofluorescence. Treatment of Huh-7 cells with the antiviral peptides during viral internalization resulted in profound inhibition of viral infection (Figure 2C), suggesting that the antiviral peptides inhibit a step in viral entry downstream of cell binding.
To further confirm these findings, we studied the ability of increasing concentrations of peptides 9–12 to inhibit entry of HCV-pseudotyped lentiviral particles (HCVpp) into Huh-7 cells (Hsu et al., 2003). As shown in Figure 3A, HCV-pseudotyped lentiviral particle entry was efficiently inhibited in a dose-dependent manner by peptides 11 and 12 but not 10, confirming that these molecules inhibit HCV entry. Interestingly, lentiviral particles bearing VSV glycoprotein (VSVpp) were not susceptible to inhibition by the same peptide concentrations, (Figure 3B) suggesting that the peptides display selective antiviral activity against HCV in a process mediated by the viral glycoprotein complex.
Figure 3. HCV-pseudotyped lentiviral particles are susceptible to the antiviral action of d,l-α-peptides.

(A) Serial dilutions of the peptides were mixed with constant amounts of HCV-pseudotypes and used to inoculate Huh-7 cells.
(B) HCV and VSV-G pseudotype infections were carried out in parallel in the presence of peptide (20 µM) to assess the specificity of inhibition. Infection efficiency in the presence of the peptides was estimated by reporter gene expression (luciferase) and compared to that obtained in the control (DMSO). Relative luciferase activity values are shown as average and mean error of triplicate infections.
Antiviral Spectrum and Specificity
To determine the antiviral spectrum of these molecules against different HCV genotypes, we tested their efficacy against particles bearing structural proteins from JFH-1 and J6CF (2a), H77 (1a) and Con1 (1b). Serial dilutions of infectious HCV particles corresponding to the different genotypes were used to inoculate Huh-7 cells in the presence of 20 µM of the most potent peptides (compounds 11 and 12), as well as in the presence of peptide 10 (Figure 4), which was shown to have no effect on JFH-1 virus infection (Table 1). Remarkably, compounds 11 and 12 strongly inhibited HCV infection by particles bearing structural proteins from genotypes 1a, 1b and 2a, as shown in Figure 4, suggesting that they are active against all the tested HCV genotypes.
Figure 4. d,l-α-peptides inhibit all tested HCV genotypes.

Supernatants containing infectious HCV particles from genotypes 1a, 1b, or 2a were serially diluted in complete medium containing the indicated antiviral peptides (20 µM) and used to inoculate naive Huh-7 cells. Seventy-two hours post-inoculation, cells were fixed and processed for immunofluorescence to determine viral infectivity titer. Data are shown as average and mean error of duplicate infections.
To determine if the antiviral effect of the peptides is limited to HCV, we asked if compounds 10–12 (20 µM) could inhibit the entry of influenza A (Flu A), VSV, or LCMV. Interestingly, in contrast to HCV, none of these viruses was susceptible to the antiviral action of the selected peptides (Figure 5), suggesting that they have a selective effect against HCV.
Figure 5. d,l-α-peptides are specific for HCV and do not interfere with infection by other enveloped RNA viruses.

Dilutions of the selected antiviral peptides (20 µM) or the endosome acidification inhibitor chloroquine (20µM) were mixed with supernatants containing infectious HCV, influenza virus, VSV and LCMV. Relative infectivity titers where determined as described in Materials and Methods and shown in each case as average and mean error of duplicate experiments. Values are expressed as percentage relative to the control.
Efficient control of viral spread
Having shown that these peptides interfere with viral entry in a single infection cycle (Figure 2A), we sought to determine if they could also restrict viral spread during multiple rounds of infection. Thus, we infected Huh-7 cells with infectious virus at low multiplicity (m.o.i. 0.01 ffu per cell) in the presence of the most active antiviral peptides (compounds 11 and 12; 20 µM). Four days post-inoculation, we observed that, as expected, only a small fraction of the DMSO-treated cells were infected and expressed viral envelope protein (E2) as revealed by immunofluorescence microscopy (Figure 6A, DMSO day 4; red cells). However, the virus was capable of efficiently spreading to almost all the DMSO-treated cells, as shown by the large proportion of HCV-antigen positive cells observed at day 12 postinfection (Figure 6A, DMSO day 12; red cells). However, the virus infected and spread ineficciently or did not spread at all in cells treated with peptide 11 or peptide 12 respectively, as shown by the virtual absence of HCV-positive cells at day 12 postinfection in the peptide-treated cells (Figure 6A; peptide 11 and 12) and the greatly reduced accumulation of viral RNA in the cultures over time (Figure 6B). These results suggest that these antiviral peptides can efficiently control viral spread in cell culture even if they are only replenished every four days.
Figure 6. Efficient control of viral spread by selected d,l-α-peptides.

Huh-7 cells were infected at low multiplicity (m.o.i of 0.01) and incubated for the designated periods of time in the presence of 20 µM of the antiviral peptides. Cells were replenished with fresh medium containing the peptides at days 4 and 8 postinfection.
(A) HCV-infected cells are revealed by immunofluorescence microscopy with antibodies against the viral protein E2and are shown in red (E2). Cell nuclei of infected and uninfected cells were stained with Hoescht dye and are shown in blue (nuclei).
(B) Intracellular HCV RNA accumulation was measured by RT-qPCR at the indicated time points. Results are shown as average and mean error of triplicate samples.
DISCUSSION
In this study, we discovered a family of net-charge neutral peptides that inhibit HCV infection at a postbinding step during viral entry by screening a small library of amphiphilic eight-residue cyclic d,l-α-peptides. Chemical and structural modifications to a representative member of the family, and analysis of the antiviral activity of the resulting derivatives, revealed some of the key requirements for the observed anti-HCV activity. A characteristic pattern of acidic and basic amino acids in the peptide sequence, a cyclic backbone configuration, and the ability for the peptide to undergo self-assembly were all necessary for anti-HCV activity. Together, these results strongly suggest a supramolecular nanotubular nature of the active species (Table 1).
Time-of-addition experiments suggested that early events in the viral lifecycle are targeted by the peptides (Figure 1). The lack of inhibitory effect on HCV replicons (Figure 2B) suggested that an event prior to RNA replication step was inhibited by the peptides, while cold-inoculation studies (Figure 2C) suggested that the peptides inhibit a post-binding step during viral entry into the target cell. Furthermore, these compounds showed comparable potency (EC50) against HCV-pseudotyped lentiviral particles (Figure 3A), further suggesting that viral entry is targeted by the peptides. In contrast, VSV-pseudotyped lentiviral particle entry was not inhibited by the peptides (Figure 3B), suggesting that they are targeting a mechanism that is unique to HCV and is probably mediated by the envelope glycoproteins. This was confirmed by the lack of susceptibility of other enveloped RNA viruses (Influenza, VSV, and LCMV) to inhibition by the peptides (Figure 5).
The amphiphilic nature of these peptides and their known ability to interact with and self-assemble in synthetic lipid membranes (Table S2) suggest that their site of action is most likely either the viral envelope or cellular membranes (Fernandez-Lopez et al., 2001) (Horne et al., 2005). We have shown that the intrinsic infectivity of the particles is not altered after incubation with the antiviral peptides, so we therefore hypothesize that they exert their antiviral activity by interacting with specialized cellular membranes. It is also important to note that although membrane activity seems to be required, it is not sufficient for anti-HCV activity, since we have shown that peptide 10, a related membrane-active peptide (Table S2), does not inhibit HCV entry. Therefore, in addition to the apparent requirement for amphiphilic cyclic d,l-α-peptide structure that enables membrane interaction and potential self-assembly into self-assembling nanotubular species, the observed level of sequence specificity seems to be the key for HCV-selective activity and the low apparent toxicity. The cellular receptors that mediate HCV entry have been shown to localize in specific areas of the plasma membrane (Voisset et al., 2007). Therefore, it is tempting to speculate that the differential composition of these plasma membrane domains through which HCV penetrates the cells forms the basis of the specificity of the peptides. Further studies will be required to determine the exact biochemical and biophysical mechanisms underlying inhibition of HCV infection by amphiphilic cyclic d,l-α-peptide structures. Remarkably, the polymeric amphipatic nature of these inhibitory peptides evokes the amphipatic DNA polymers shown to inhibit HCV infection at a similar post-binding entry step (Matsumura et al., 2009), or the previously described small molecule LJ001 that specifically intercalates into a broad-spectrum of viral membranes, inhibiting entry of the virus at a step after virus binding but before virus–cell fusion (Wolf et al., 2010). Whether the molecular mechanism underlying these inhibitions is similar remains to be determined.
Finally, compounds that efficiently inhibit viral spread, like the peptides presented in this study, can complement the antiviral activity of other compounds, as part of a combination therapy. Such combination therapies could lessen the likelihood of the emergence of resistant variants by reducing their spread to other cells.
SIGNIFICANCE
The Hepatitis C virus is comprised of multiple genotypes, and significant heterogeneity exists even within genotype subgroups. Moreover, emergence of drug-resistant quasispecies is a major issue, and combinatorial drug therapy targeting different aspects of the viral lifecycle is required for sustained efficacy. Therefore, there is an urgent need for new classes of anti-HCV drugs. In this study we report a new class of supramolecular agents specifically targeting HCV entry at a post-binding step. Our results suggest that these antiviral peptides can efficiently control viral spread in cell culture, making them potential candidates for controlling spread of the infection in vivo including restricting the spread of emerging drug-resistant virus variants in combination therapy. It is noteworthy that, based on our previous studies, membrane-selective self-assembling cyclic d,l–α-peptides can be derived from a vast sequence space, so there is considerable room for further structure-activity optimizations. Moreover, as a class of bioactive supramolecular species, cyclic d,l–α-peptides are in general stable to proteolysis and have proven in vivo utility and antimicrobial efficacy. Therefore, it seems likely that the basic findings described here could catalyze the development of practical therapeutic applications.
EXPERIMENTAL PROCEDURES
Peptide Chemistry
Synthesis of the cyclic peptides
Linear peptides were synthesized using standard Fmoc solid phase synthesis protocols, manually or with an Advanced Chemtech Peptide Synthesizer Model 348 Ω. A typical synthesis was performed on 0.07 mmol scale using 0.8–1.0 mmol/g 2-chlorotrityl resin for preparation of Fmoc-aa-(OAll)-chlorotrityl resin. Chain elongations were achieved using DIC and HOBt in NMP with coupling reaction times ranging from 4 to 12 h either manually or 65 minutes when using the peptide synthesizer. Fmoc deprotection was achieved using 20% piperidine in NMP (2 × 15 min.). The allyl deprotection step was carried out manually after swelling of the resin in dry DCM for 20 min. To the resin was added a degassed solution of 0.5 eq Pd(PPh3)4 in DCM followed by 10 eq. phenylsilane. After shaking under nitrogen for 2 h, the resin was washed sequentially with DMF, DCM, and DMF. Resin was treated again for 2 h with 0.5 eq Pd(PPh3)4 DCM and phenylsilane under nitrogen and washed with a solution of 1% sodium dimethylthiocarbamate in DMF and 1% DIEA in DMF. After the final Fmoc deprotection, subsequent cyclization was achieved using HATU as coupling agent in the presence of DIEA in NMP. Peptides were cleaved from the resin with concomitant side chain deprotection by agitation in a solution of 95:2.5:2.5 trifluroacetic acid (TFA)/H2O/triethylsilane (TES) for two hours. The crude peptide was precipitated with ether, centrifuged, and washed twice with ether. After air-drying, peptides were purified by preparative RP-HPLC.
Virological Procedures
Viruses and cells
Huh-7 cells were cultured as previously described (Zhong et al., 2005). JFH-1 virus was generated by transfection and viral stocks were produced by infection of Huh-7 cells at a multiplicity of infection of 0.01 as described (Zhong et al., 2005). At day 8–10 post-inoculation, supernatants were collected and the infectivity titer was determined as described below. High titer stocks of D183 virus (Zhong et al., 2006) were prepared by infection of a Huh-7.5.1 cells at low multiplicity (m.o.i. of 0.01) as described (Zhong et al., 2005). Generation of recombinant chimeric HCV viruses J6CF-, H77, and Con1-JFH-1) chimeric HCV genomes described (Pietschmann et al., 2006) was previously described.
Influenza A virus (A/WSN/33) was kindly provided by Dr. Adolfo García-Sastre (Mount Sinai School of Medicine-New York, NY). Lymphocytic choriomeningitis virus (LCMV/Armstrong) was kindly provided by Dr. Juan Carlos De La Torre (The Scripps Research Institute). Vesicular stomatitis virus (Indiana) was acquired from ATCC.
Viral infectivity in the presence of antiviral peptides
HCV
Infectivity titers were determined on Huh-7 cells by end point dilution and immunofluorescence as previously described (Zhong et al., 2005). Typically, 25 µl of each sample were serially diluted five-fold in D-MEM 10%FCS and 100 µl were used to inoculate Huh-7 cells. Infection was examined 72 hours post inoculation by immunofluorescence using a 1:1000 dilution of a recombinant monoclonal human IgG anti-E2 (C1 antibody, a gift from Dr. Dennis Burton-The Scripps Research Institute) with the appropriate secondary Alexa555-conjugated antibodies. For analysis of antiviral activity, an HCV focus reduction assay was performed as described (Cheng et al., 2008). Briefly, the peptides were reconstituted in 100% DMSO at a final concentration of 10 mM and stored at −20°C. A virus-peptide mixture containing 50–75 FFU and 20 µM of peptide was prepared in complete medium (D-MEM+10%FCS) and used to inoculate Huh-7 cells plated (104 cells per well in a 96-well plate, seeded the day before). The cells were incubated at 37 °C for 3 days after which the cells were immunostained as described above. The number of HCV-positive foci was counted by fluorescence microscopy and expressed as percentage of the number of control foci in cells incubated with a virus-DMSO mixture. Infectivity of intracellular virions was determined by four cycles of freezing-thawing of infected cell suspensions as previously described (Gastaminza et al., 2006).
LCMV
Different dilutions of LCMV were used to inoculate Huh-7 cells for 1 hour at 37°C. The cells where washed and replenished with complete medium. Infectivity was measured 20 hours post-infection by immunofluorescence as described above for HCV using a specific antibody against LCMV NP (NP 1.1.3, a gift from Dr. Juan Carlos De La Torre- The Scripps Research Institute). LCMV “focus reduction assay” was performed identically to that of HCV using a specific antibody against LCMV NP (see above), except for a different incubation period post-inoculation of 20 hours.
Influenza
Increasing dilutions of influenza A virus (A/WSN/33) were used to inoculate Vero cells for 1hour at 37 °C. Cells were washed and replenished with complete medium (D-MEM+10%FCS). Infectivity was measured 72 hours post-infection by endpoint dilution. Influenza infectivity reduction assays were performed by endpoint dilution titration in the presence of 20 µM peptide. The inoculum was removed 2 hours postinoculation and non-internalized virus was inactivated by washing the cells with an acid PBS buffer (pH 5.2). The cells were washed again with PBS and replenished with DMEM +10% FCS and incubated for 72 hours. Infectivity titer was determined as described above.
In all of the foregoing experiments, chloroquine (25 µM) was included as positive control since it has been shown to interfere with endosomal acidification, a process required for internalization of HCV, Influenza and LCMV (reviewed in (Smith and Helenius, 2004).
Peptide library screening
The methodology employed to screen the peptide library was adapted from that described in (Gastaminza et al., 2010) and (Gastaminza et al., 2011). The modified procedure involves using the parental JFH-1 strain (Kato et al., 2001) instead of the cell culture-adapted D183 virus (Zhong et al., 2006) and the Huh-7 cell line instead of the hyperpermissive cell clone Huh-7.5.1.c2 cells (Pedersen et al., 2007). Peptide stock solutions (10 mM) were diluted to the desired final concentration of 20 µM in D-MEM (10%FCS) growth medium containing 500 focus-forming units FFU of JFH-1 per well. This mixture was used to inoculate 104 Huh-7 cells/well in a 96-well format. The cells were incubated at 37 °C for 3 days after which the cells were fixed with 4% paraformaldehyde. Infection efficiency was evaluated by colorimetry in a cell-based ELISA-type assay using a recombinant monoclonal antibody against the viral envelope as described in (Gastaminza et al., 2010).
Discrimination of false positives was carried out by determining the cell biomass in the control and experimental wells by staining with crystal as described (Bernhardt et al., 1992), and discarding compounds that reduce the biomass of the control wells by more than 25%.
Determination of potency (EC50) and toxicity (LD50)
Serial dilutions of the compound were prepared in 9% sucrose solutions using a Tecan 550 Genesis Pipetting Robot. These solutions (20 µl) were mixed with 80 µl of a virus dilution in complete medium containing 500 FFU leading to final compound concentrations of 50, 40, 35, 30, 25, 20, 15, 10, 5, 2.5, 1 and 0.1 µM. The mixture was transferred into a 96-well plate containing 104 Huh-7 cells plated the day prior to the experiment The cultures were then incubated for a period of 72 hours at 37 °C, after which the cells are fixed and processed for colorimetry as described above. OD650 values were transformed using a standard curve as described above and the HCV infection values relative to the vehicle control were used to calculate EC50 values by interpolation of the 50% in the linear portion of the inhibition curve. LD50 values were determined by evaluating cell viability using the XTT-formazan method at different times post-treatment, 24 and 120 hours (Roehm et al., 1991).
Time-of-addition assay
Peptides were added at a final concentration of 20 µM under the following conditions: 1) pre-inoculation: peptide was added to cells for 5 hours at 37° C followed by washing once with PBS medium before virus infection; 2) co-inoculation: peptide was added to cells together with virus for 5 hours at which time the cells were washed as above and replenished with complete media without peptide; 3) post-inoculation: cells were infected for 5 hours at which point the virus was removed, the cells were washed once with PBS and the peptide was added and left on the cells for the duration of the experiment without washing. These conditions were compared with the infection where the peptides where present at all times. Cells were fixed and processed for colorimetric analysis seventy-two hours post infection as described above.
HCV pseudotype particle infectivity inhibition assay
HCV (H77 genotype 1a strain) E1/E2-pseudotyped (or VSV-G pseudotyped as a control) lentiviral particles bearing the luciferase reporter gene were generated as described (Flint et al., 2004). For the inhibition assays, equal amounts of infectious particles were mixed with serial dilutions of the cyclic peptides or the vehicle (DMSO). The mixture was used to inoculate Huh-7 cells that were incubated in complete medium for another 72 hour period after which infection efficiency was evaluated by measuring reporter gene (luciferase) activity in total cell extracts (Hsu et al., 2003). Relative luciferase activity values were obtained using untreated cells as control (100%) after background (uninfected cells) substraction.
Inhibition of viral entry: binding and post-binding events
In order to discriminate inhibition of particle binding and post-binding events, we incubated pre-chilled target cells (Huh-7) for 1 hour on ice in the presence of the inhibitors (20 µM). After this adsorption period, cells were washed twice with cold PBS and replenished with warm D-MEM 10% FCS and incubated for 72 hours at 37 °C before infection was revealed by immunostaining.
Parallel cultures inoculated with virus on ice in the absence of peptides (1 h), were washed twice with cold PBS and replenished with D-MEM 10%FCS containing candidate peptides (20 µM). Cultures were incubated for 5 hours at 37 °C period to allow virus internalization after which the cells were washed twice with PBS and incubated in D-MEM 10% FCS for extra 72 hours. Infection efficiency was revealed by immunostaining as described above.
Highlights.
-
-
A miniaturized colorimetric screening identified a family of net-charge neutral compounds with antiviral activity against HCV.
-
-
A family of highly active amphiphilic eight-residue cyclic d,l-α-peptides specifically blocks entry of all tested HCV genotypes into target cells at a post-binding step.
-
-
Amphiphilic cyclic d,l-α-peptides efficiently control viral spread in cell culture at low micromolar concentrations without measurable toxicity.
-
-
Membrane active cyclic d,l-α-peptides that self-assemble into supramolecular nanotubular structures constitute a novel class of highly selective HCV entry inhibitors.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Dr. Takaji Wakita (National Institute of Infectious Diseases, Tokyo, Japan) for kindly providing the infectious JFH-1 molecular clone and replicon constructs; Dr. Jane McKeating (University of Birmingham, Birmingham, UK) for providing the vectors necessary for HCVpp production and Dr. Zhong from Gilead Sciences (Foster City, CA) for providing the 2’-m-C-cytosine. We thank Dr. Adolfo García-Sastre (Mount Sinai School of Medicine, New York, NY) for Influenza A virus (A/WSN/33), Dr. Juan Carlos De La Torre (The Scripps Research Institute, La Jolla, CA) for providing Lymphocytic choriomeningitis virus (LCMV/ Armstrong). We thank Drs. Marlene Dreux, Urtzi Garaigorta, and Stefan Wieland, for their expert advice and useful discussions. We are grateful to Brian Boyd, Christina Whitten-Bauer, and Josan Chung for excellent technical assistance. This work was supported by NIH grants R01GM52190 (MRG) and R01AI079043 (FVC). This is manuscript number 19611 from the Scripps Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions: A.M., P.G., F.V.C., and M.R.G. designed research, analyzed the data and wrote the paper; A.M., P.G., M.L., and G.C. performed research.
REFERENCES
- Alter MJ. The epidemiology of acute and chronic hepatitis C. Clinical Liver Disease. 1997;1:559–568. vi–vii. doi: 10.1016/s1089-3261(05)70321-4. [DOI] [PubMed] [Google Scholar]
- Alter MJ. Hepatitis C virus infection in the United States. J Hepatol. 1999;31 Suppl 1:88–91. doi: 10.1016/s0168-8278(99)80381-x. [DOI] [PubMed] [Google Scholar]
- Bartenschlager R, Lohmann V. Replication of hepatitis C virus. J Gen Virol. 2000;81(Pt 7):1631–1648. doi: 10.1099/0022-1317-81-7-1631. [DOI] [PubMed] [Google Scholar]
- Bartosch B, Dubuisson J, Cosset FL. Infectious hepatitis C virus pseudo-particles containing functional E1–E2 envelope protein complexes. J Exp Med. 2003;197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt G, Reile H, Birnbock H, Spruss T, Schonenberger H. Standardized kinetic microassay to quantify differential chemosensitivity on the basis of proliferative activity. J Cancer Res Clin Oncol. 1992;118:35–43. doi: 10.1007/BF01192309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard E, Belouzard S, Goueslain L, Wakita T, Dubuisson J, Wychowski C, Rouille Y. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J Virol. 2006;80:6964–6972. doi: 10.1128/JVI.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blight KJ, Kolykhalov AA, Rice CM. Efficient initiation of HCV RNA replication in cell culture. Science. 2000;290:1972–1975. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- Bong DT, Clark TD, Granja JR, Ghadiri MR. Self-Assembling Organic Nanotubes. Angew Chem Int Ed Engl. 2001;40:988–1011. [PubMed] [Google Scholar]
- Burlone ME, Budkowska A. Hepatitis C virus cell entry: role of lipoproteins and cellular receptors. J Gen Virol. 2009;90:1055–1070. doi: 10.1099/vir.0.008300-0. [DOI] [PubMed] [Google Scholar]
- Carroll SS, Olsen DB. Nucleoside analog inhibitors of hepatitis C virus replication. Infect Disord Drug Targets. 2006;6:17–29. doi: 10.2174/187152606776056698. [DOI] [PubMed] [Google Scholar]
- Chang KS, Jiang J, Cai Z, Luo G. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J Virol. 2007;81:13783–13793. doi: 10.1128/JVI.01091-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G, Montero A, Gastaminza P, Whitten-Bauer C, Wieland SF, Isogawa M, Fredericksen B, Selvarajah S, Gallay PA, Ghadiri MR, et al. A virocidal amphipathic {alpha}-helical peptide that inhibits hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A. 2008;105:3088–3093. doi: 10.1073/pnas.0712380105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark TD, Buehler LK, Ghadiri MR. Self-Assembling Cyclic β3-Peptide Nanotubes as Artificial Transmembrane Ion Channels. Journal of the American Chemical Society. 1998;120:651–656. [Google Scholar]
- Dartois V, Sanchez-Quesada J, Cabezas E, Chi E, Dubbelde C, Dunn C, Granja J, Gritzen C, Weinberger D, Ghadiri MR, et al. Systemic antibacterial activity of novel synthetic cyclic peptides. Antimicrob Agents Chemother. 2005;49:3302–3310. doi: 10.1128/AAC.49.8.3302-3310.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreux M, Gastaminza P, Wieland SF, Chisari FV. The autophagy machinery is required to initiate hepatitis C virus replication. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14046–14051. doi: 10.1073/pnas.0907344106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Lopez S, Kim HS, Choi EC, Delgado M, Granja JR, Khasanov A, Kraehenbuehl K, Long G, Weinberger DA, Wilcoxen KM, et al. Antibacterial agents based on the cyclic D,L-alpha-peptide architecture. Nature. 2001;412:452–455. doi: 10.1038/35086601. [DOI] [PubMed] [Google Scholar]
- Fletcher JT, Finlay JA, Callow ME, Callow JA, Ghadiri MR. A Combinatorial Approach to the Discovery of Biocidal Six-Residue Cyclic d,l-α-Peptides Against the Bacteria Methicillin-Resistant Staphylococcus aureus (MRSA) and E. coli and the Biofouling Algae Ulva linza and Navicula perminuta. Chemistry – A European Journal. 2007;13:4008–4013. doi: 10.1002/chem.200((......)). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint M, Logvinoff C, Rice CM, McKeating JA. Characterization of infectious retroviral pseudotype particles bearing hepatitis C virus glycoproteins. J Virol. 2004;78:6875–6882. doi: 10.1128/JVI.78.13.6875-6882.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastaminza P, Cheng G, Wieland S, Zhong J, Liao W, Chisari FV. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J Virol. 2008;82:2120–2129. doi: 10.1128/JVI.02053-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastaminza P, Kapadia SB, Chisari FV. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J Virol. 2006;80:11074–11081. doi: 10.1128/JVI.01150-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastaminza P, Pitram SM, Dreux M, Krasnova LB, Whitten-Bauer C, Dong J, Chung J, Fokin VV, Sharpless KB, Chisari FV. Antiviral Stilbene 1,2-Diamines Prevent Initiation Of Hepatitis C Viral RNA Replication At The Outset of Infection. J Virol. 2011 doi: 10.1128/JVI.02116-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastaminza P, Whitten-Bauer C, Chisari FV. Unbiased probing of the entire hepatitis C virus life cycle identifies clinical compounds that target multiple aspects of the infection. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:291–296. doi: 10.1073/pnas.0912966107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman MA, Glenn JS. Mixing the right hepatitis C inhibitor cocktail. Trends Mol Med. 2010 doi: 10.1016/j.molmed.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghadiri MR, Granja JR, Buehler LK. Artificial transmembrane ion channels from self-assembling peptide nanotubes. Nature. 1994;369:301–304. doi: 10.1038/369301a0. [DOI] [PubMed] [Google Scholar]
- Ghadiri MR, Granja JR, Milligan RA, McRee DE, Khazanovich N. Self-assembling organic nanotubes based on a cyclic peptide architecture. Nature. 1993;366:324–327. doi: 10.1038/366324a0. [DOI] [PubMed] [Google Scholar]
- Ghadiri MR, Kobayashi K, Granja JR, Chadha RK, McRee DE. The Structural and Thermodynamic Basis for the Formation of Self-Assembled Peptide Nanotubes. Angewandte Chemie International Edition in English. 1995;34:93–95. [Google Scholar]
- Granja JR, Ghadiri MR. Channel-Mediated Transport of Glucose across Lipid Bilayers. Journal of the American Chemical Society. 1994;116:10785–10786. [Google Scholar]
- Hao W, Herlihy KJ, Zhang NJ, Fuhrman SA, Doan C, Patick AK, Duggal R. Development of a novel dicistronic reporter-selectable hepatitis C virus replicon suitable for high-throughput inhibitor screening. Antimicrob Agents Chemother. 2007;51:95–102. doi: 10.1128/AAC.01008-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartgerink JD, Granja JR, Milligan RA, Ghadiri MR. Self-Assembling Peptide Nanotubes. Journal of the American Chemical Society. 1996;118:43–50. [Google Scholar]
- Hoofnagle JH. Course and outcome of hepatitis C. Hepatology. 2002;36:S21–S29. doi: 10.1053/jhep.2002.36227. [DOI] [PubMed] [Google Scholar]
- Horne WS, Wiethoff CM, Cui C, Wilcoxen KM, Amorin M, Ghadiri MR, Nemerow GR. Antiviral cyclic D,L-alpha-peptides: targeting a general biochemical pathway in virus infections. Bioorg Med Chem. 2005;13:5145–5153. doi: 10.1016/j.bmc.2005.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, McKeating JA. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci U S A. 2003;100:7271–7276. doi: 10.1073/pnas.0832180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M, Jr., Ye J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc Natl Acad Sci U S A. 2007;104:5848–5853. doi: 10.1073/pnas.0700760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Luo G. Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J Virol. 2009;83:12680–12691. doi: 10.1128/JVI.01476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapadia SB, Chisari FV. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc Natl Acad Sci U S A. 2005;102:2561–2566. doi: 10.1073/pnas.0409834102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Furusaka A, Miyamoto M, Date T, Yasui K, Hiramoto J, Nagayama K, Tanaka T, Wakita T. Sequence analysis of hepatitis C virus isolated from a fulminant hepatitis patient. J Med Virol. 2001;64:334–339. doi: 10.1002/jmv.1055. [DOI] [PubMed] [Google Scholar]
- Kim HS, Hartgerink JD, Ghadiri MR. Oriented Self-Assembly of Cyclic Peptide Nanotubes in Lipid Membranes. Journal of the American Chemical Society. 1998;120:4417–4424. [Google Scholar]
- Kim SS, Peng LF, Lin W, Choe WH, Sakamoto N, Schreiber SL, Chung RT. A cell-based, high-throughput screen for small molecule regulators of hepatitis C virus replication. Gastroenterology. 2007;132:311–320. doi: 10.1053/j.gastro.2006.10.032. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Granja JR, Ghadiri MR. β-Sheet Peptide Architecture: Measuring the Relative Stability of Parallel vs. Antiparallel β-Sheets. Angewandte Chemie International Edition in English. 1995;34:95–98. [Google Scholar]
- Koutsoudakis G, Kaul A, Steinmann E, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J Virol. 2006;80:5308–5320. doi: 10.1128/JVI.02460-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamarre D, Anderson PC, Bailey M, Beaulieu P, Bolger G, Bonneau P, Bos M, Cameron DR, Cartier M, Cordingley MG, et al. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature. 2003;426:186–189. doi: 10.1038/nature02099. [DOI] [PubMed] [Google Scholar]
- Liang TJ, Rehermann B, Seeff LB, Hoofnagle JH. Pathogenesis, natural history, treatment, and prevention of hepatitis C. Annals of Internal Medicine. 2000;132:296–305. doi: 10.7326/0003-4819-132-4-200002150-00008. [DOI] [PubMed] [Google Scholar]
- Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- Matsumura T, Hu Z, Kato T, Dreux M, Zhang YY, Imamura M, Hiraga N, Juteau JM, Cosset FL, Chayama K, et al. Amphipathic DNA polymers inhibit hepatitis C virus infection by blocking viral entry. Gastroenterology. 2009;137:673–681. doi: 10.1053/j.gastro.2009.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- Motiei L, Rahimipour S, Thayer DA, Wong CH, Ghadiri MR. Antibacterial cyclic D,L-alpha-glycopeptides. Chem Commun (Camb) 2009:3693–3695. doi: 10.1039/b902455g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CL, Jones CT, Rice CM. Architects of assembly: roles of Flaviviridae non-structural proteins in virion morphogenesis. Nat Rev Microbiol. 2008;6:699–708. doi: 10.1038/nrmicro1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen IM, Cheng G, Wieland S, Volinia S, Croce CM, Chisari FV, David M. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature. 2007;449:919–922. doi: 10.1038/nature06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, et al. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci U S A. 2006;103:7408–7413. doi: 10.1073/pnas.0504877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roehm NW, Rodgers GH, Hatfield SM, Glasebrook AL. An improved colorimetric assay for cell proliferation and viability utilizing the tetrazolium salt XTT. J Immunol Methods. 1991;142:257–265. doi: 10.1016/0022-1759(91)90114-u. [DOI] [PubMed] [Google Scholar]
- Smith AE, Helenius A. How viruses enter animal cells. Science. 2004;304:237–242. doi: 10.1126/science.1094823. [DOI] [PubMed] [Google Scholar]
- Su AI, Pezacki JP, Wodicka L, Brideau AD, Supekova L, Thimme R, Wieland S, Bukh J, Purcell RH, Schultz PG, et al. Genomic analysis of the host response to hepatitis C virus infection. Proc Natl Acad Sci U S A. 2002;99:15669–15674. doi: 10.1073/pnas.202608199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tscherne DM, Jones CT, Evans MJ, Lindenbach BD, McKeating JA, Rice CM. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J Virol. 2006;80:1734–1741. doi: 10.1128/JVI.80.4.1734-1741.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisset C, Lavie M, Helle F, De Beeck AO, Bilheu A, Bertrand-Michel J, Terce F, Cocquerel L, Wychowski C, Vu-Dac N, et al. Ceramide enrichment of the plasma membrane induces CD81 internalization and inhibits hepatitis C virus entry. Cell Microbiol. 2007 doi: 10.1111/j.1462-5822.2007.01070.x. [DOI] [PubMed] [Google Scholar]
- Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcoxen KM. Investigations into the Biomimetic Synthesis, Structural Determination and Therapeutic Development of Antimicrobial Peptides. 2002 [Google Scholar]
- Wolf MC, Freiberg AN, Zhang T, Akyol-Ataman Z, Grock A, Hong PW, Li J, Watson NF, Fang AQ, Aguilar HC, et al. A broad-spectrum antiviral targeting entry of enveloped viruses. Proc Natl Acad Sci U S A. 2010;107:3157–3162. doi: 10.1073/pnas.0909587107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Wang C, Sumpter R, Jr., Brown MS, Goldstein JL, Gale M., Jr. Disruption of hepatitis C virus RNA replication through inhibition of host protein geranylgeranylation. Proc Natl Acad Sci U S A. 2003;100:15865–15870. doi: 10.1073/pnas.2237238100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A. 2005;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Gastaminza P, Chung J, Stamataki Z, Isogawa M, Cheng G, McKeating JA, Chisari FV. Persistent hepatitis C virus infection in vitro: coevolution of virus and host. J Virol. 2006;80:11082–11093. doi: 10.1128/JVI.01307-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.