

SUMMARY
The mycobacterial biotin protein ligase (MtBPL) globally regulates lipid metabolism in Mtb through the posttranslational biotinylation of acyl coenzyme A carboxylases involved in lipid biosynthesis that catalyze the first step in fatty acid biosynthesis and pyruvate coenzyme A carboxylase, a gluconeogenic enzyme vital for lipid catabolism. Here we describe the design, development and evaluation of a rationally designed bisubstrate inhibitor of MtBPL. This inhibitor displays potent sub-nanomolar enzyme inhibition and antitubercular activity against multi- and extensively drug resistant Mtb strains. We show that the inhibitor decreases in vivo protein biotinylation of key enzymes involved in fatty acid biosynthesis and that the anti-bacterial activity is MtBPL-dependent. Additionally, the gene encoding BPL was found to be essential in M. smegmatis. Finally, the X-ray co-crystal structure of inhibitor bound MtBPL was solved providing detailed insight for further structure-activity analysis. Collectively, these data suggest that MtBPL is a promising target for further antitubercular therapeutic development.
INTRODUCTION
Tuberculosis (TB) caused by the bacterium Mycobacterium tuberculosis (Mtb) continues to burden human health on a global scale. Despite an impressive amount of effort directed at eradicating tuberculosis, TB is still the second leading cause of mortality among infectious diseases superseded only by HIV. The emergence of multidrug-resistant (MDR) and extensively drug-resitant (XDR) TB strains coupled with the high incidence of HIV-TB co-infection and the lack of any new approved chemotherapeutic agents for TB in over four decades, demands the development of new antitubercular agents, ideally with novel mechanisms of action, to combat this devastating pathogen.
Fatty acid biosynthesis is a validated pathway for antitubercular drug discovery and the first line agent isoniazid inhibits the type II fatty acid synthase (FAS-II) in Mtb (Slayden et al., 2000; Timm et al., 2003). Acyl coenzyme A carboxylases (ACCs) catalyze the first committed step in fatty acid biosynthesis through synthesis of the monomeric malonyl-CoA building blocks. However, unlike most bacteria that encode for a single ACC complex, Mtb encodes for multiple ACCs responsible for the biosynthesis of malonyl coenzyme A (CoA), (methyl)malonyl CoA, and (C22–C24) malonyl CoA for construction of the structurally diverse lipids in Mtb including simple linear fatty acids, methyl-branched lipids and mycolic acids (Gago et al., 2006; Kurth et al., 2009; Portevin et al., 2005). In order for the ACCs to become functionally active, the biotin-carboxylase carrier protein (BCCP) domains of these proteins must be posttranslationally modified to their active holo biotinylated form by a biotin protein ligase (BPL) encoded by birA in Mtb (MtBPL) (Purushothaman et al., 2008). The biotin serves as the cofactor that mediates carboxyl group transfer onto the acetyl CoA substrates. In addition to fatty acid biosynthesis, fatty acid degradation is also an essential process and mycobacteria are thought to be primarily lipolytic in vivo deriving their energy via fatty acid catabolism (Timm et al., 2003). MtBPL may also play a key role in fatty acid degradation, as this enzyme is also responsible for biotinylation of the BCCP domain of pyruvate carboxylase, which can channel oxaloacetate to the phosphoenolpyruvate carboxykinase, a gluconeogenic enzyme that is essential for growth on fatty acids and critical for mycobacterial pathogenesis (Marrero et al., 2010). MtBPL thus globally regulates lipid metabolism and represents an attractive target for development of new antitubercular agents.
There are two classes of biotin protein ligases (BPLs): the monofunctional enzymes represented by the BPL from Pyrococcus horikoshii (PhBPL), that solely catalyze posttranslational biotinylation and the bifunctional enzymes represented by the prototypical BPL from Escherichia coli (EcBPL) that also act as transcriptional repressors (Brown et al., 2004; Chapman-Smith and Cronan, 1999). Recent biochemical characterization of the BPL from Mtb (MtBPL) has revealed it to be a monofunctional enzyme that exists as a monomer in both the unliganded apo-and holo-forms as well as in the biotinyl adenylate (Bio-AMP, 2) liganded form (Purushothaman et al., 2008). BPLs catalyze the transfer of biotin onto a conserved lysine residue of the biotin-carboxylase carrier protein (BCCP) domain of ACCs. This reaction is accomplished in two steps: in the first-half reaction, BPLs bind biotin (1, Figure 1A) and ATP and catalyze the nucleophilic attack of the biotin carboxylate onto the α-phosphate of ATP to yield the acyl-adenylate Bio-AMP 2 (Figure 1A) and pyrophosphate. In the second-half reaction, BPLs transfer biotin from Bio-AMP 2 onto a conserved lysine residue within the BCCP domain 3 to provide biotinylated-BCCP 4 (Figure 1B). The bisubstrate inhibitor 5 (Figure 1C) has been described, which mimics the intermediate acyl-adenylate Bio-AMP 2 by replacement of the labile acylphosphate linkage in Bio-AMP 2 with the bioisosteric acylsulfamate moiety (Brown and Beckett, 2005; Brown et al., 2004). Interestingly, the bisubstrate inhibitor 5 was found to bind 1.4-fold more weakly to EcPBL than the single substrate biotin, suggesting the acylsulfamate was a poor isostere for the native acylphosphate linkage.
Figure 1.

Enzyme reaction catalyzed by BPL. A. First half-reaction to form biotin adenylate. B. Second half-reaction involving transfer of biotin to acetyl CoA carboxylases. C. Bisubstrate inhibitors of BPL and degradation products of 5. See also Figure S1.
Here we describe the design, synthesis, biochemical, structural, and biological evaluation of a bisubstrate inhibitor of MtBPL that we term Bio-AMS (6, Figure 1C) that binds more than 1700-fold more tightly than biotin and possesses potent and selective antimicrobial activity against MDR- and XDR-TB strains of Mtb. We used M. smegmatis to confirm that the BPL-encoding gene, birA, is essential as well as to assess susceptibility of MtBPL to inhibition in vivo. Support for the designed mechanism of action is provided using a combination of proteomic profiling and genetic studies. Finally, we describe the detailed molecular interactions present between MtBPL and Bio-AMS that establish the molecular basis for potent inhibition and establish a framework for future structure-aided drug design.
RESULTS
Inhibitor Design and Synthesis
Bisubstrate inhibitors that mimic acyl-adenylate reaction intermediates typically bind much more tightly than the individual substrates owing to the large number of interactions between the inhibitor and both substrate binding sites (Ferreras et al., 2005; Somu et al., 2006). The reported lack of potency of 5 towards EcBPL is surprising (Brown and Beckett, 2005), so we synthesized this compound as described (Supplemental Experimental Procedures). We immediately observed that it is chemically unstable and decomposes through cyclonucleoside formation to afford N-biotinylsulfamic acid 7 (Figure 1C) and 3,5′-cyclo-5′-deoxyadenosine 8 (experimental details and complete characterization are found in the Supplemental Information). We hypothesized that a more stable compound would result if the 5′-oxygen atom in sulfamate 5 were replaced with a nitrogen atom of sulfamide 6 as a result of the decreased nucleofugality of the sulfamide (Broeckaert et al., 2008). Compound 6 (Bio-AMS) was synthesized, and as predicted, it did not undergo cyclonucleoside formation. Due to its improved stability, Bio-AMS represents an excellent probe to chemically validate MtBPL as a target for antitubercular therapeutic development.
Biochemical Evaluation of Bio-AMS
We next evaluated Bio-AMS in a kinetic continuous coupled assay with 250 nM MtBPL to determine the concentration-response curve for inhibition of MtBPL at fixed saturating substrate concentrations. As shown in Figure 2A, the half-maximum inhibitor concentration (IC50 = 135 ± 9 nM) is approximately equal to half of the enzyme concentration, a characteristic inherent to tight-binding inhibitors (Copeland, 2005). Attempts to determine the true inhibition constant (Ki) were complicated by the tight-binding behavior and the bisubstrate nature of inhibition, which precluded assessment by traditional steady-state kinetic methods (Yu et al., 2006).
Figure 2.

In vitro analysis and mechanism of action studies of Bio-AMS inhibitor. A. Dose-response of MtBPL with inhibitor Bio-AMS. vi and v0 are velocities of inhibitor and DMSO treated samples, respectively. B. Competitive ITC trace of a solution of Bio-AMS (200 μM) in 10 mM Tris pH 7.5, 200 mM KCl, and 2.5 mM MgCl2 titrated into a solution of MtBPL (20 μM) and biotin (150 μM) in the same buffer. C. Western blot analysis of biotinylated protein levels in lysates from Mtb H37Rv cells treated with varying concentrations of Bio-AMS. Lane M: biotinylated molecular weight marker; Lane 1: DMSO-treated MTB cells; Lane 2: Mtb cells treated with 0.1 μM Bio-AMS; Lane 3: Mtb cells treated with 1.0 μM Bio-AMS. The dashed box corresponds to the section of the gel that was excised for trypsin digestion and LC-MS/MS analysis. D. Normalized band intensities of bands in lanes 1–3 from C. See also Figure S2 and Table S2.
We employed isothermal titration calorimetry (ITC) to investigate the binding affinity of Bio-AMS for MtBPL. Accurate determinations of the binding constants using ITC are typically limited to a range of 103–108 M−1 (Sigurskjold, 2000). A more reliable approach for determining binding constants greater than 108 M−1 is to use displacement ITC in which a high affinity inhibitor is titrated into a solution of protein that is bound with a weaker ligand (Leavitt and Freire, 2001; Velazquez-Campoy et al., 2001). Displacement ITC experiments were performed (Figure 2B) using biotin as the competitive ligand to obtain values for KA and n (Methods). The binding enthalpy was determined by stoichiometric titration of MtBPL with Bio-AMS at a high c value (Methods). The thermodynamic parameters are listed in Table 1. The ITC studies provided an experimental KD of 0.530 ± 0.13 nM with an n value of 1.12 ± 0.02 indicating one substrate binding site per MtBPL monomer. Bio-AMS binds approximately 1700-fold more tightly to MtBPL than biotin (Table 1) (Purushothaman et al., 2008). The binding is solely enthalpically driven (ΔH = −15.4 ± 0.1 kcal/mol) with an unfavorable entropic component (−TΔS = 3.0 ± 0.0 kcal/mol). The large observed enthalpic component is commonly observed among tight-binding bisubstrate inhibitors and can be attributed to numerous hydrogen bonds and electrostatic interactions between inhibitor and protein (Neres et al., 2008; Sikora et al., 2010).
Table 1.
Thermodynamic parameters of Bio-AMS binding to MtBPL.
| n | KD, nM | ΔH, kcal mol−1 | ΔG, kcal mol−1 | −TΔS, kcal mol−1 | |
|---|---|---|---|---|---|
| Biotina | 0.98 | 940 | −11.1 | −8.1 | 3.0 |
| Bio-AMS | 1.12 ± 0.02 | 0.530 ± 0.134 | −15.4 ± 0.1 | −12.4 ± 0.1 | 3.0 ± 0.0 |
Thermodynamic values for biotin were taken from reference (Purushothaman et al., 2008)
Antitubercular Activity, Antimicrobial Specificity, and Therapeutic Index
The antitubercular activity of Bio-AMS was evaluated against Mtb H37Rv, the common laboratory virulent strain. The minimum inhibitory concentration that inhibited greater than 99% of cell growth (MIC99) is 0.31–0.78 μM (Table 2). Based on the promising antitubercular activity, we then tested Bio-AMS against ten phenotypically characterized multidrug- and extensively drug-resistant TB strains. The MIC99 values range from 0.16–0.625 μM (Table 2) demonstrating that Bio-AMS is equally effective against susceptible and MDR- and XDR-TB strains. We also tested the ability of Bio-AMS to inhibit cell growth in other bacterial and fungal strains since BPLs are present in most organisms. Remarkably, Bio-AMS was inactive against a panel of Gram-negative bacteria (A. baumannii, E. coli, K. pneumoniae, P. aeruginosa,), Gram-positive bacteria (E. faecalis and S. aureus) and fungi (C. neoformans, C. albicans) (Table S1). We speculate the antimicrobial specificity results from limited intracellular accumulation due to differences in cellular penetration and efflux, rather than biochemical selectivity. Since mammals also contain a biotin protein ligase (holocarboxylase synthetase (Campeau and Gravel, 2001)), we evaluated the cytotoxicity of Bio-AMS against two mammalian cell lines, Vero cells and DU145 human prostate cells. DU145 cells were insensitive to Bio-AMS, while Bio-AMS displayed very low cell cytotoxicity to Vero cells inhibiting 50% growth (CC50) at 58 μM providing a therapeutic index (CC50/MIC99) of 74–187 based on the MIC against Mtb H37Rv.
Table 2.
Biological activity and selectivity of Bio-AMS.
| Strain/Cell Line | Resistance Phenotype | Classificationd | MIC99 (μM) | EC50 (μM) |
|---|---|---|---|---|
| M. tuberculosis H37Rv | wt | – | 0.31 – 0.78 | – |
| M. tuberculosis XDR-1 | HRESPOCTh | XDR | 0.625 | – |
| M. tuberculosis XDR-2 | HREPKOTh | XDR | 0.625 | – |
| M. tuberculosis MDR-1 | HRESPO | MDR | 0.16 | – |
| M. tuberculosis MDR-2 | HREKO | MDR | 0.16 | – |
| M. tuberculosis MDR-3 | HRESP | MDR | 0.625 | – |
| M. tuberculosis MDR-4 | HRCPTh | MDR | 0.625 | – |
| M. tuberculosis MDR-5 | HRERb | MDR | 0.625 | – |
| M. tuberculosis MDR-6 | HREZSKPTh | MDR | 0.625 | – |
| M. tuberculosis MDR-7 | HRERb | MDR | 0.625 | – |
| M. tuberculosis MDR-8 | HREZRbTh | MDR | 0.625 | – |
| A. baumannii | - | Gram-negative | > 400 | – |
| E. coli | - | Gram-negative | > 400 | – |
| K. pneumoniae | - | Gram-negative | > 400 | – |
| P. aeruginosa | - | Gram-negative | > 400 | – |
| E.faecalis | - | Gram-positive | > 400 | – |
| S. aureus | MRSA | Gram-positive | > 400 | – |
| C. albicans | - | Fungal | > 400 | – |
| C. neoformans | - | Fungal | > 400 | – |
| DU145 | n.a.c | Mammalian | – | >100 |
| Vero | n.a. | Mammalian | – | 58b |
Letter codes represent drugs which that strain is resistant to. H = isoniazid; R = rifampicin; E = ethambutol; S = streptomycin; P = para-aminosalicylic acid; O = ofloxacin; Th = thiacetazone; C = capreomycin; K = kanamycin; Rb = rifabutin; Z = pyrazinamide.
Vero cells were still 25% viable at 500 μM,
Not applicable
XDR = extensively drug-resistant; MDR = multidrug-resistant
The following ATCC strains were used: A. baumannii, 19606; E. coli, 25922; K. pneumoniae, 13883; P. aeruginosa, 27853; E. faecalis, VRE, 51299; S. aureus, MRSE, 43300; C. albicans, 10231; C. neoformans, 66031.
Mechanism of Action
To demonstrate that Bio-AMS inhibits protein biotinylation in vivo, a culture of Mtb H37Rv was treated with Bio-AMS (0.1 μM or 1.0 μM) along with a DMSO control. After treatment for 26 h, cells were harvested and lysed. Protein concentrations were determined and lysates were separated by denaturing gel electrophoresis and imaged via western blot (Figure 2C). The biotinylated protein pattern of the DMSO treated cells (lane 1) shows two biotinylated proteins above 100 kDa, one of which is likely pyruvate carboxylase. Additionally, the proteins between 55–80 kDa are most likely the acyl CoA carboxylases (accA1–A3). Lastly, there is one unidentified biotinylated protein that is approximately 25 kDa. When Mtb was incubated with 0.1 μM (~1/3–1/8 × MIC99, lane 2), a minimal decrease in band intensity (protein biotinylation) was seen. However, when 1.0 μM Bio-AMS (~1.5–3 × MIC99, lane 3) was incubated with Mtb, a clear decrease in protein biotinylation of all bands were observed. Band intensities in each lane were calculated and normalized to the amount of protein loaded into each well to provide normalized biotinylation levels (Figure 2D). The biotinylated proteins highlighted by the dashed box in Figure 2C were confirmed to be the acetyl/propionyl CoA carboxylases by mass spectrometry (Supplementary Figure S2 and Table S2). Since cells were treated with Bio-AMS for only 26 h before being harvested, the slow doubling time of Mtb (24 h in GAST medium) likely prevented growth arrest in the presence of inhibitor, but still allowed for a measurable decrease in protein biotinylation. These results clearly demonstrate that Bio-AMS decreases global biotinylation levels of proteins involved in fatty acid biosynthesis.
Validation of the essentiality of birA in M. smegmatis and susceptibility to inhibition by Bio-AMS
For genetic studies we first turned to M. smegmatis, a fast-growing mycobacterium whose genome encodes homologs for each of the three ACC alpha subunits and the six ACC beta subunits of Mtb and that, like the genome of Mtb, contains a single BPL-encoding gene (MsbirA). After repeated attempts to delete MsbirA in wild-type (wt) M. smegmatis had failed, we constructed a merodiploid strain, which contained a second MsbirA integrated into the attachment site of the phage L5. This allowed us to delete the wt copy of MsbirA resulting in M. smegmatis ΔMsbirAMsbirA-attL5 (Supplementary Figure S3 and S4), and to subsequently demonstrate that M. smegmatis is unable to form colonies on agar plates without MsbirA (Supplementary Figure S3). We next used M. smegmatis ΔMsbirA MsbirA-attL5 to show that the Mtb birA gene, MtbirA, can replace MsbirA and constructed three M. smegmatis strains, MtBPL-H, MtBPL-M, and MtBPL-L, which instead of MsBPL express high, intermediate, or low levels of MtBPL, respectively (Figure 3A). These M. smegmatis strains all grew normally in liquid media (Supplementary Figure S5A), but their susceptibility to Bio-AMS increased with decreasing expression of MtBPL (Figure 3B). By contrast, these strains showed identical susceptibilities to isoniazid (INH) and ethambutol (EMB) (Supplementary Figure S5B & C). Next, we generated two Mtb strains, MtbMtBPL-H and MtBPL-M, which overexpressed MtBPL to different degrees (Figure 3C). Overexpression of MtBPL did not affect growth (not shown) or susceptibility of Mtb to INH (Supplementary Figure S5D) but did decrease susceptibility to Bio-AMS (Figure 3D). Taken together, these experiments demonstrated (i) that MsbirA is essential for growth of M. smegmatis, (ii) that MtbirA can replace MsbirA, and (iii) that the BPL expression level specifically determines susceptibility of M. smegmatis and Mtb to Bio-AMS.
Figure 3.

Impact of MtBPL expression on drug susceptibility of M. smegmatis and Mtb In A and C 10 μg of total protein extracts of the indicated strains were used to measure MtBPL expression by western blotting. Enolase (lower panels) served as the loading control. B and D show susceptibility of M. smegmatis (MtBPL-H (black), MtBPL-M (blue) and MtBPL-L (red)) and Mtb (MtBPL-H (black), MtBPL-M (blue) and wt (red)) to BioAMS. Normalized growth was calculated as OD580 at the indicated drug concentration divided by OD580 without drug. Error bars represent SEMs of 4 to 8 measurements. See also Figures S3–S5.
Co-crystal Structure
To identify important molecular interactions critical to the potency of Bio-AMS, we solved the co-crystal structure with MtBPL to 1.7-Å resolution (Supplementary Figure S6 and Table S3 for data collection and refinement statistics). Compared to the apo structures of MtBPL (Gupta et al., 2010; Ma and Wilmanns, 2007), the complex structure reveals that two loops (residues 65–76 and 162–169) undergo a disorder-to-order transition upon binding of Bio-AMS and form extensive, specific contacts (Figure 4), consistent with the displacement ITC results. The biotin moiety and adenine nucleobase are recognized by a common constellation of hydrogen bonds observed in all liganded biotin-protein ligase crystal structures to date (Bagautdinov et al., 2005; Tron et al., 2009; Wood et al., 2006). The adenine nucleobase is specifically recognized via hydrogen bonds with the side chain of Asn158 and backbone of Ala75. Consistent with Bio-AMS being competitive with biotin, the buried urea moiety of biotin is hydrogen bonded to the backbone carbonyl and amide of Arg67, the hydroxyl of Ser38, the side-chain amide of Gln63, and the hydroxyl of Thr39. Analogous to the isosteric 5′-biotinyl-AMP structures reported from Pyrococcus hiorikoshii (Bagautdinov et al., 2005), Trp74 packs between the adenine ring and the alkyl tail of biotin. In contrast to all other reported biotin-protein ligase structures, MtBPL possesses a unique interaction between the sidechain amide of Gln81 and both the indole nitrogen of Trp74 and carbonyl of Ala75. Likely unique to MtBPL, Asp167 interacts with the 2′-hydroxyl of the ribose; the only other reported interaction between a ribose hydroxyl and a BPL is seen with Gln221 on a separate portion of the ordered loop with the EcBPL (Wood et al., 2006). With the acylsulfamide linkage, there are extensive interactions between the guanidinium groups of Arg69 and Arg72 and the sidechain of Asn130 with the sulfamide oxygens. Because of the low pKa of the central nitrogen of the acylsulfamide linker, it is likely in the deprotonated amide form; the crystal structure supports this with a hydrogen bond distance of 3.0 Å with good geometry to the backbone N-H of Arg69. The terminal amine of Lys138 – thought to be critical for adenylation reaction – interacts with and stabilizes the amide oxygen of the acylsulfamide linker, one sulfamide oxygen, the carboxylate of Asp131 and a water molecule hydrogen bonded to the distal nitrogen of the sulfamide linkage.
Figure 4.
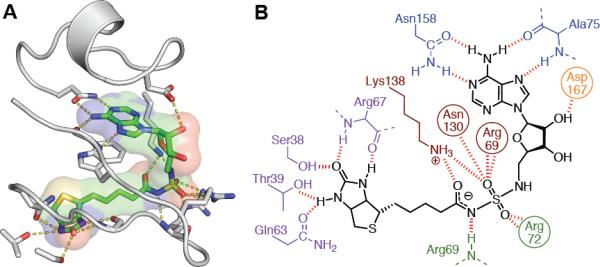
Structure of Bio-AMS with MtBPL. A. The MtBPL binding site showing the loops undergoing the order-to-disorder transition (grey) upon binding of Bio-AMS (green carbons). Specific hydrogen bonds are shown in yellow dashes. Asn130 is omitted for clarity. B. Schematic view of polar interactions between biotin (purple), acylsulfamide linker (red/green), ribose (orange) and adenosine (blue) portions of Bio-AMS and MtBPL. See also Figure S6 and Table S3.
DISCUSSON
Bio-AMS is considered a bisubstrate inhibitor since it interacts with both substrate (biotin and ATP) binding pockets. Bisubstrate inhibitors (A−B) can realize substantial enhancement in binding energy compared to the sum of the Gibbs binding energies of the respective fragments (A + B) because of a smaller entropy barrier to binding of A−B compared to A + B (Jencks, 1981). Thus, the report that 5′-O-[N-(biotinyl)sulfamoyl]adenosine 5 bound more weakly than the single substrate biotin was incongruous, particularly in light of several recent reports demonstrating related bisubstrate inhibitors bound with high affinity (Brown and Beckett, 2005; Ferreras et al., 2005; Somu et al., 2006)). Competitive displacement isothermal titration calorimetry revealed the true potency of Bio-AMS providing a KD of 0.530 nM that is driven exclusively by a large favorable enthalpy of −15.4 kcal/mol. The high affinity and favorable thermodynamic parameters exhibited in the interaction of MtBPL with Bio-AMS correlates well with the detailed molecular interactions observed (Figure 4). The large number of hydrogen bonds and the electrostatic interactions with Lys138 and the backbone of Arg69 result in the large, favorable enthalpy. The conformational restriction of the ligand within the binding pocket, combined with the disorder-to-order transition observed for the two loops of MtBPL covering Bio-AMS, is likely compensated for by the increased hydrophobic interactions with the alkyl tail of biotin and Trp74 to net only a small unfavorable entropy.
Another important finding from our work was the demonstration that 5 slowly decomposes through cyclonucleoside formation, which has precedent in related 5′-activated nucleosides (Kristinsson et al., 1995; Liang et al., 2008; Yanachkov and Wright, 1994). Replacement of the 5′-oxygen atom in 5 with a nitrogen atom afforded the more stabile acylsulfamide linkage of Bio-AMS. Although the acylsulfamate linkage has historically been used as a bioisostere of the acylphosphate to generate adenylate inhibitors, caution is warranted due to its potential lack of stability. Several strategies exist to improve the stability including replacement of the 5′-oxygen with a nitrogen atom as shown herein or a carbon atom. Alternate strategies to prevent the undesired cyclonucleoside formation include installation of a bulky group at the C-2 position of the adenine ring to hinder the required syn conformation about the glycosidic linkage or deletion of the N-3 nitrogen atom of adenine to prevent nucleophilic attack. As demonstrated in this work, the replacement of the 5′ oxygen with a nitrogen provides the required stability while maintaining isostere integrity.
The observation that Bio-AMS exhibited potent whole-cell activity toward Mtb was unexpected. Indeed related acyladenylate bisubstrate inhibitors developed for the functionally related aminoacyl tRNA synthetases have typically been devoid of whole cell activity owing to the limited membrane permeability of these highly polar nucleoside analogs (Hurdle et al., 2005; Kim et al., 2003; Pohlmann and Brotz-Oesterhelt, 2004). Mycobacteria are considered to have the most complex and fortified cellular wall of any bacterial species, thus the potent and selective antitubercular activity was intriguing. However, the ability of these bisubstrate adenylate inhibitors to cross the cell wall of Mtb is not unprecedented, as our lab and others have shown these inhibitors possess potent whole cell anti-tubercular activity (Ferreras et al., 2005; Somu et al., 2006). We hypothesize that the observed selectivity is likely due to selective accumulation facilitated by one of the ~30 ATP binding cassette (ABC) transporters present in Mtb (Braibant et al., 2000) that are responsible for assimilation of nucleosides and cofactors.
Biotinylation, catalyzed by the mycobacterial biotin protein ligase MtBPL, is a posttranslation modification that represents a key nodal point in mycobacterial fatty acid metabolism and controls flux through both fatty acid biosynthetic and catabolic pathways. We designed and synthesized the bisubstrate inhibitor Bio-AMS, which was shown by isothermal titration calorimetry to bind MtBPL with a sub-nanomalor dissociation constant. Despite its highly polar structure, Bio-AMS also displayed impressive whole cell activity against Mtb H37Rv as well as a panel of MDR- and XDR Mtb strains. The selective antitubercular activity of this compound is intriguing and may be due to selective mycobacterial accumulation. Further biochemical and cell based accumulation studies will be required to delineate the origin of the remarkable selective antitubercular activity. Nevertheless, our genetic experiments with M. smegmatis demonstrated that MsbirA is an essential gene and also confirmed that the anti-bacterial activity of Bio-AMS is BPL-dependent. Proteomic analysis demonstrated that Bio-AMS also inhibits protein biotinylation in Mtb. The detailed molecular interactions visualized in the co-crystal structure of MtBPL with Bio-AMS pinpoint key opportunities to engineer selectivity and further augment binding affinity. Thus, the work presented here validates MtBPL as an attractive target for the development of novel antitubercular agents. Beyond their role as potential antitubercular agents, Bio-AMS may also find utility as tool compound for chemical biology studies since biotinylation is widely used for site-specific protein labeling and other biotechnological applications.
SIGNIFICANCE
With the rapid emergence of drug resistant strains of Mycobacterium tuberculosis comes the need to identify new drug targets and develop small molecule inhibitors for these enzymes. Here, we synthesized a potent bisubstrate inhibitor for studying a promising enzyme target from Mtb. In designing this inhibitor, we have identified an important decomposition mechanism among bisubstrate adenylation inhibitors, indicating that careful attention needs to be given to engineering stability into these small molecules. The co-crystal structure of the inhibitor-bound BPL enzyme provides key insight into the structural differences between BPL from Mtb and other organisms, which builds the foundation for future medicinal chemistry efforts. Additionally, these studies represent the first to use genetic knockdowns to determine the mechanism of action of a new small molecule probe in mycobacteria. Lastly, our genetic knockout results demonstrate that BPL is an essential enzyme, further validating this enzyme as a promising candidate target for antibiotic development.
METHODS
See Supplementary Information for full synthetic procedures, cloning and expression of MtBPL, antimicrobial and cytotoxicity assays, and biotinylated protein identification.
Bio-AMS Synthesis
Bio-AMS was synthesized in two steps starting from 5′-amino-5′-N-tert-butoxycarbonyl-5′-deoxy-2′,′-O-isopropylidene-5′-N-(sulfamoyl)adenosine (S5 (Lu et al., 2008), Supplementary Scheme S3) in 55% overall yield. Biotin was coupled to S5 (Lu et al., 2008) followed by global TFA deprotection to yield crude Bio-AMS (6). The title compound was purified by reverse phase HPLC. Detailed experimental procedures are provided in the Supplementary Information. 1H NMR (600 MHz, DMSO-d6) δ 0.96 (t, J = 4.8 Hz, 18H, 2 eq. TEA), 1.22–1.33 (m, 2H), 1.39–1.50 (m, 3H), 1.57–1.63 (m, 1H), 2.08 (t, J = 7.2 Hz, 2H), 2.49 (p, J = 1.8 Hz, 9H, 2 eq. TEA), 2.56 (d, J = 5.4 Hz, 1H), 2.79 (dd, J = 12.6, 4.8 Hz, 1H), 3.05 (ddd, J = 8.4, 6.1, 4.7 Hz, 1H), 3.09 (dd, J = 13.2, 4.2 Hz, 2H), 4.03 (dd, J = 7.2, 4.2 Hz, 1H), 4.09–4.11 (m, 2H), 4.26 (dd, J = 7.8, 5.4 Hz, 1H), 4.68 (q, J = 4.8 Hz, 1H), 5.25 (s, 1H) 5.43 (d, J = 6.0 Hz, 1H), 5.82 (d, J = 6.6 Hz, 1H), 6.34 (s, 1H), 6.52 (s, 1H), 7.38 (s, 2H), 8.18 (s, 1H), 8.29 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 11.7, 25.7, 28.5, 28.7, 37.3, 40.5, 45.8, 46.2, 55.8, 59.6, 61.5, 71.8, 73.1, 84.1, 88.5, 119.9, 140.7, 149.4, 153.0, 156.7, 163.2, 173.2; HRMS (ESI–) calcd for C20H28N9O7S2 [M – H]− 570.1559, found 570.1523 (error 6.3 ppm).
Kinetic Inhibition Assays
Enzyme assays to determine the IC50 value for Bio-AMS were run under initial velocity conditions using our recently described coupled adenylation–hydroxamate formation assay employing saturating concentrations of both ATP and biotin (Wilson and Aldrich, 2010). Varying concentrations of inhibitor Bio-AMS (20 mM DMSO stock), providing a final DMSO concentration of 1% (v/v) or DMSO only (control) were preincubated with a master mix [250 nM MtBPL, 50 mM Tris (pH 8.0), 5.0 mM MgCl2, 0.5 mM DTT, 2.5 mM ATP, 0.1 unit of nucleoside phosphorylase, 0.04 unit of pyrophosphatase, 0.2 mM 7-methylthioguanosine (MesG), 150 mM hydroxylamine (pH 7.0)] for 10 min at 25 °C. The master mix (95 μL) was then added to 5.0 μL of a biotin solution to provide a final biotin concentration of 250 μM. Reactions (100 μL) were run in 96-well half-area UVStar plates (Greiner), and the formation of 7-methyl-6-thioguanine was measured at 360 nm (ε360 = 11,000 mol−1 cm−1) at 25 °C on a Molecular Devices SpectraMax M5e. The initial velocity data were fit by nonlinear regression analysis to the sigmoidal Hill equation (eq 1)
| (1) |
using GraphPad prism version 4.0 to obtain the IC50 value, where vi/v0 is the initial fractional velocity, h is the Hill slope, I is the logarithm of the inhibitor concentration, and A and B are the top and bottom of the curve, respectively.
Isothermal Titration Calorimetry
ITC experiments were conducted on a Microcal VPITC microcalorimeter (Northhampton, MA, USA). All titration experiments were performed at 20 °C in ITC buffer (10 mM Tris (pH 7.5), 200 mM KCl, and 2.5 mM MgCl2). MtBPL was exchanged into ITC buffer using an Amicon Ultra concentrator (Millipore, Billerica, MA, USA), and final enzyme concentrations were determined using an ε280 of 35540 M−1 cm−1. In individual titrations, ligands were injected into solutions of the enzyme. The quantity c = KAMt(0), in which Mt(0) is the initial enzyme concentration is crucial for successful ITC experiments (Wiseman et al., 1989). Experiments for the determination of ΔH (the change in binding enthalpy in kilocalories per mole) were performed with a c value in the range of 1.2–2.4×104, while displacement ITC experiments were performed with a c value in the range of 206–415. Ligand and protein concentrations for the determination of ΔH were 16 μM MtBPL and 160 μM Bio-AMS. The KA (the association constant in M−1) and n (the number of binding sites per monomer) values were determined by ITC displacement experiments with 150 μM biotin added to both the enzyme and ligand solutions. Enzyme concentrations for displacement experiments were increased to 42 μM for titration with 420 μM Bio-AMS. All titrations were performed in duplicate using a stirring speed of 307 rpm and a 300 s interval between 10 μL injections. The first injection was omitted from data fitting. Titrations were run past the enzyme saturation point to correct for heats of dilution. The data for the stoichiometric titration of Bio-AMS in MtBPL provided ΔH values. The experimental data for the competitive displacement ITC experiments were fitted to a theoretical titration curve using the Origin software package (version 7.0) to provide KAapp and n values (Velazquez-Campoy and Freire, 2006). The KA value for Bio-AMS was obtained from the KAapp value using eq. 2:
| (2) |
where [B] ([biotin]) equals 150 μM and KAB (association constant for biotin) = 1.08 × 106. The thermodynamic parameters (ΔG and −TΔS) were calculated using equation 3:
| (3) |
where ΔG, ΔH, and ΔS are the changes in free energy, enthalpy, and entropy of binding, respectively, R = 1.98 cal mol−1 K−1 and T is the absolute temperature. The affinity of Bio-AMS for MtBPL is provided as the dissociation constant (KD = 1/KA). Direct and competitive ITC experiments were performed in two independent experiments and analyzed independently. The thermodynamic values were then averaged. ITC experiments for determining the binding affinity of biotin (KAB in eq. 2) for MtBPL were performed as described above. Ligand and enzyme concentrations were 195 μM and 19.5 μM, respectively. The data were then fit using the single-site binding model provided in Origin 7.0 to determine KAB.
Antitubercular Assays
For determination of minimum inhibitory concentrations (MICs) against Mtb strains, broth microdilution assays were determined in quadruplicate in GAST media supplemented with Fe3+ (De Voss et al., 2000) using Bio-AMS from a DMSO stock solution or with control wells treated with an equivalent volume of DMSO or with isoniazid as positive control. Cells were grown to an OD650 of 0.2, diluted 1000-fold in the GAST/Fe medium and 50 μL added per well of a 96-well plate containing 50 μL of GAST/Fe medium with compound in a two-fold dilution series across the wells giving an initial cell density of 104 cells/assay. Plates were incubated at 37 °C and growth was monitored at 7 and 14 days. The MIC was recorded as the lowest concentration of compound that resulted in complete inhibition of growth. MICs were determined against the laboratory strain Mtb H37Rv as well as a panel of drug resistant clinical isolates of Mtb (Jeon et al., 2008).
Biotinylated Proteome Assay
A 30 mL culture of Mtb H37Rv was grown at 37 °C to an OD650 of 0.194, and was split into 3 × 10 mL. The 10 mL cultures were treated with either DMSO, 0.1 μM, or 1.0 μM Bio-AMS for 26 h at 37 °C. After this time, the OD650 values for the DMSO and 1.0 μM treated cells were 0.616 and 0.600, respectively. The cells were harvested and the cell pellets were washed once with PBS (10 mM Sodium Phosphate, 150 mM NaCl, pH 7.8) and frozen at −80 °C. The cells were subjected to bead beating in PBS containing protease inhibitor cocktail (Roche). After removing unlysed cells and cell wall debris by centrifuging, the total protein concentrations were determined for the DMSO, 0.1 μM, and 1.0 μM Bio-AMS treated cells and were found to be 2.0, 2.2, and 2.2 mg/mL, respectively. An equal volume of 2× SDS reducing gel electrophoresis loading buffer was added to the samples, which were subsequently heated for 10 min at 80 °C. A 5.0 μL aliquot of each sample was separated by denaturing gel electrophoresis (4–15% Tris-HCl Ready Gel, BioRad) and transferred to a PVDF membrane (iBlot System, Invitrogen). The membrane was blocked with Tris-buffered saline containing 0.05% Tween-20 (TBST) containing 1.0% BSA for 30 min at 25 °C. The membrane was incubated with an AlexaFluor 647 fluorescent-streptavidin conjugate (Invitrogen) at a dilution of 1:1000 in TBST for 1 h at 25 °C, after which the membrane was washed 3 × 10 mL TBST. Biotinylated proteins within the membrane were visualized by fluorescence scanning using an FMBIO III fluorescent scanner (MaraiBio). Band intensities were calculated using the Image Analysis software version 3.0 provided with the scanner. Band intensities were normalized for protein concentration to provide the normalized % band intensity values in Figure 2D.
Essentiality of birA
For mutant construction, Mycobacterium smegmatis mc2155 was grown at 37 °C on Difco 7H11 agar (BD) or in Difco Middlebrook 7H9 broth (BD) with 0.2% v/v glycerol and 0.05% v/v Tween 80. Antibiotics were added, where appropriate, at 50 μg/ml (hygromycin B, Calbiochem), 20 μg/ml (streptomycin, Sigma), 25 μg/ml (kanamycin A, Sigma), or 25 μg/ml (zeocin, Invitrogen). Preparation of competent cells, electroporations, and the preparation of genomic DNAs were performed as described (Ehrt et al., 2005). Plasmids were constructed using standard procedures (details are available upon request). Polyclonal anti-MtBPL antibodies were generated using a commercial immunization service (Covance).
Initially we attempted to delete MsbirA from the chromosome of M. smegmatis and cloned a knockout plasmid, pKO-MsbirA (derived from pSM270 (Manganelli et al., 2001)) in which ~800 bp DNA fragments containing the upstream and downstream region of MsbirA were separated by a hygromycin resistance gene. Integration of pKO-MsbirA into the M. smegmatis chromosome by a single crossover conferred resistance to hygromycin (hygR) and streptomycin (strepR) as well as sensitivity to sucrose (sucS). A second crossover that would have deleted MsbirA would have resulted in a strain that is hygR, streptomycin sensitive and sucrose resistance. We analyzed 150 sucrose resistant descendants of two different single crossover strains, but were unable to identify a single MsbirA deletion mutant among them. This strongly suggested that MsbirA is essential for growth of M. smegmatis.
We next constructed a birA merodiploid (Supplemental Figure S3, step 1) by transforming M. smegmatis with a kanamycin resistance (kanR) conferring MsbirA expression plasmid that integrated into the attachment site of the mycobacteriophage L5 (attL5). Deletion of the wt copy of MsbirA in the merodiploid (Supplemental Figure S3, step 2) with pKO-MsbirA proved to be straightforward and was confirmed by southern blotting (Supplemental Figure S4). We refer to the resulting strain as ΔMsbirA MsbirA-attL5 to indicate that the only functional copy of MsbirA is located in the attL5-site. Plasmids in the attL5 site can be efficiently replaced by site-specific recombination (Pashley and Parish, 2003), which allowed us to determine if M. smegmatis is able to grow after deletion of MsbirA. Replacement of the kanR MsbirA expression plasmid with a zeocin resistance (zeoR) conferring plasmid that contained MsbirA was efficient (Supplemental Figure S3, step 4). In contrast, no zeoR colonies were obtained in several independent transformations of ΔMsbirA MsbirA-attL5 with a zeoR plasmid that did not contain MsbirA (Supplemental Figure S3, step 3). This demonstrated that deletion of MsbirA prevents growth of M. smegmatis on agar plates.
To generate M. smegmatis mutants that expressed MtBPL instead of MsBPL we transformed ΔbirA MsbirA-attL5 with zeoR plasmids containing MtbirA. Mutants in which MsbirA-attL5 was replaced by MtbirA-attL5 were identified by confirming sensitivity to kanamycin (which is a result of deleting the MsbirA plasmid from the attL5) and in immunoblots performed with a polyclonal antibody that was generated against MtBPL and did not recognize MsBPL. Plasmids that contained different promoters upstream of MtbirA were used to generate strains that constitutively expressed different levels of MtBPL (Figure 3A). Transformation of plasmids in which (i) the native upstream region of MtbirA, (ii) the promoter Psmyc (Kaps et al., 2001), or (iii) Ptb38 were used to transcribe MtbirA yielded Msm MtBPL-L, Msm MtBPL-M, and Msm MtBPL-M, respectively. Promoter Ptb38 is contained within a DNA fragment that has been identified in a promoter trap screen (Ehrt et al., 2005). This promoter has strong activity in M. smegmatis and slightly lower activity in Mtb (unpublished). Plasmids containing MtbirA transcribed by Ptb38 and Psmyc were transformed in Mtb H37Rv to generate Msm MtBPL-M, and Msm MtBPL-M, respectively. The Enolase was used as loading control and detected using a polyclonal antibody provided by Dr. Sabine Ehrt. All M. smegmatis mutants in which MsbirA was replaced by MtbirA grew similar to wt M. smegmatis (Supplemental Figure S5). Overexpression of MtBPL also did not affect growth of Mtb (not shown). The activity of Bio-AMS against the M. smegmatis strains was assessed as described for Mtb with two exceptions: growth was analyzed after 2 to 4 days instead of 10 to 14 days and no iron supplement was added to the GAST medium.
Crystallization
BirA was incubated with a slight molar excess of Bio-AMS for 30 min at 37 °C prior to crystallization. Crystals were grown in 1–2 weeks at 20 °C by micro-seeding with apo crystals in hanging drops using the vapor-diffusion method. The protein solution contained 4.6 mg/mL BirA, 10 mM HEPES pH 7.5, 50 mM NaCl, 1 mM DTT, 1.05% DMSO and 200 μM Bio-AMS. Equal volumes were mixed of protein solution and mother liquor containing 20% MPEG 2000, 50 mM trimethylamine N-oxide, and 100 mM Tris pH 8.5. Crystals were cryo-protected in a solution containing mother liquor supplemented with 20% PEG 400 and flash frozen in liquid nitrogen.
Data collection and structure solution
X-ray diffraction data were collected at 100 K and 1.000Å wavelength on beamline 4.2.2 at the Advanced Light Source, Berkeley, CA. Data were integrated and scaled by d*TREK (Pflugrath, 1999). The structure was solved by molecular replacement using the previously determined apo MtBPL structure (PDB code 2CGH) and the program Phaser (McCoy et al., 2007) from within CCP4 (Project, 1994). Initial refinement and manual model rebuilding was performed using REFMAC5 (Murshudov et al., 1997) and Coot (Emsley and Cowtan, 2004), respectively. Topology and parameter files for Bio-AMS were generated using the PRODRG server (Schuttelkopf and van Aalten, 2004). Final refinement was carried out using Phenix (Adams et al., 2010). Ramachandran plots were calculated by MOLPROBITY and showed 96.4% of the residues were in the favored regions and all residues were in the allowed regions. OMIT density (Supplemental Figure S6) was calculated using the program SFCHECK (Vaguine et al., 1999). Images and figures were prepared using PyMOL (DeLano Scientific, Palo Alto, CA, USA). The atomic coordinates and structure factors were deposited with the Protein Data Bank with the accession code 3RUX.
Supplementary Material
ACKNOWLEDGEMENTS
This research was supported by a grant from the National Institutes of Health (AI091790 to D.S. and C.C.A.), the Bill and Melinda Gates foundation and the Wellcome Trust through the Grand Challenges in Global Health Initiative (to Douglas Young, Imperial College), and the Intramural Research Program of the NIAID, NIH (to C.E.B.). The authors thank Dr. Sabine Ehrt for enolase specific antiserum, Dr. Christine Salomon and Mr. Michael Donald for antimicrobial specificity testing, and Dr. David Ferguson and Mr. Adam Benoit for DU145 cell testing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagautdinov B, Kuroishi C, Sugahara M, Kunishima N. Crystal structures of biotin protein ligase from Pyrococcus horikoshii OT3 and its complexes: structural basis of biotin activation. J. Mol.Biol. 2005;353:322–233. doi: 10.1016/j.jmb.2005.08.032. [DOI] [PubMed] [Google Scholar]
- Braibant M, Gilot P, Content J. The ATP binding cassette (ABC) transport systems of Mycobacterium tuberculosis. FEMS Microbiol. Rev. 2000;24:449–467. doi: 10.1111/j.1574-6976.2000.tb00550.x. [DOI] [PubMed] [Google Scholar]
- Broeckaert L, Moens J, Roos G, De Proft F, Geerlings P. Intrinsic nucleofugality scale within the framework of density functional reactivity theory. J. Phys. Chem. A. 2008;112:12164–12171. doi: 10.1021/jp802846q. [DOI] [PubMed] [Google Scholar]
- Brown PH, Beckett D. Use of binding enthalpy to drive an allosteric transition. Biochemistry. 2005;44:3112–3121. doi: 10.1021/bi047792k. [DOI] [PubMed] [Google Scholar]
- Brown PH, Cronan JE, Grotli M, Beckett D. The biotin repressor: modulation of allostery by corepressor analogs. J. Mol. Biol. 2004;337:857–869. doi: 10.1016/j.jmb.2004.01.041. [DOI] [PubMed] [Google Scholar]
- Campeau E, Gravel RA. Expression in Escherichia coli of N- and C-terminally deleted human holocarboxylase synthetase. Influence of the N-terminus on biotinylation and identification of a minimum functional protein. J. Biol. Chem. 2001;276:12310–12316. doi: 10.1074/jbc.M009717200. [DOI] [PubMed] [Google Scholar]
- Chapman-Smith A, Cronan JE., Jr. The enzymatic biotinylation of proteins: a post-translational modification of exceptional specificity. Trends Biochem. Sci. 1999;24:359–363. doi: 10.1016/s0968-0004(99)01438-3. [DOI] [PubMed] [Google Scholar]
- Copeland RA, editor. Evaluation of enzyme inhibitors in drug discovery. John Wiley & Sons, Inc.; New Jersey: 2005. [Google Scholar]
- De Voss JJ, Rutter K, Schroeder BG, Su H, Zhu Y, Barry CE., 3rd The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. U.S.A. 2000;97:1252–1257. doi: 10.1073/pnas.97.3.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrt S, Guo XV, Hickey CM, Ryou M, Monteleone M, Riley LW, Schnappinger D. Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res. 2005;33:e21. doi: 10.1093/nar/gni013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Ferreras JA, Ryu JS, Di Lello F, Tan DS, Quadri LE. Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat. Chem. Biol. 2005;1:29–32. doi: 10.1038/nchembio706. [DOI] [PubMed] [Google Scholar]
- Gago G, Kurth D, Diacovich L, Tsai SC, Gramajo H. Biochemical and structural characterization of an essential acyl coenzyme A carboxylase from Mycobacterium tuberculosis. J. Bacteriol. 2006;188:477–486. doi: 10.1128/JB.188.2.477-486.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta V, Gupta RK, Khare G, Salunke DM, Surolia A, Tyagi AK. Structural ordering of disordered ligand-binding loops of biotin protein ligase into active conformations as a consequence of dehydration. PLoS One. 2010;5:e9222. doi: 10.1371/journal.pone.0009222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurdle JG, O'Neill AJ, Chopra I. Prospects for aminoacyl-tRNA synthetase inhibitors as new antimicrobial agents. Antimicrob. Agents Chemother. 2005;49:4821–4833. doi: 10.1128/AAC.49.12.4821-4833.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jencks WP. On the attribution and additivity of binding energies. Proc. Natl. Acad. Sci. U.S.A. 1981;78:4046–4050. doi: 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon CY, Hwang SH, Min JH, Prevots DR, Goldfeder LC, Lee H, Eum SY, Jeon DS, Kang HS, Kim JH, et al. Extensively drug-resistant tuberculosis in South Korea: risk factors and treatment outcomes among patients at a tertiary referral hospital. Clin. Infect. Dis. 2008;46:42–49. doi: 10.1086/524017. [DOI] [PubMed] [Google Scholar]
- Kaps I, Ehrt S, Seeber S, Schnappinger D, Martin C, Riley LW, Niederweis M. Energy transfer between fluorescent proteins using a co-expression system in Mycobacterium smegmatis. Gene. 2001;278:115–124. doi: 10.1016/s0378-1119(01)00712-0. [DOI] [PubMed] [Google Scholar]
- Kim S, Lee SW, Choi EC, Choi SY. Aminoacyl-tRNA synthetases and their inhibitors as a novel family of antibiotics. Appl. Microbiol. Biotechnol. 2003;61:278–288. doi: 10.1007/s00253-003-1243-5. [DOI] [PubMed] [Google Scholar]
- Kristinsson H, Nebel K, O'Sullivan AC, Pachlatko JP, Yamaguchi Y. Herbically Active Sulfamoyl Nucleosides In Synthesis and Chemistry of Agrochemicals IV. ACS Publications; Washington D.C.: 1995. [Google Scholar]
- Kurth DG, Gago GM, de la Iglesia A, Bazet Lyonnet B, Lin TW, Morbidoni HR, Tsai SC, Gramajo H. ACCase 6 is the essential acetyl-CoA carboxylase involved in fatty acid and mycolic acid biosynthesis in mycobacteria. Microbiology. 2009;155:2664–2675. doi: 10.1099/mic.0.027714-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavitt S, Freire E. Direct measurement of protein binding energetics by isothermal titration calorimetry. Curr. Opin. Struct. Biol. 2001;11:560–566. doi: 10.1016/s0959-440x(00)00248-7. [DOI] [PubMed] [Google Scholar]
- Liang F, Jain N, Hutchens T, Shock DD, Beard WA, Wilson SH, Chiarelli MP, Cho BP. Alpha,beta-methylene-2'-deoxynucleoside 5'-triphosphates as noncleavable substrates for DNA polymerases: isolation, characterization, and stability studies of novel 2'-deoxycyclonucleosides, 3,5'-cyclo-dG, and 2,5'-cyclo-dT. J. Med. Chem. 2008;51:6460–6470. doi: 10.1021/jm800692a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Zhang H, Tonge PJ, Tan DS. Mechanism-based inhibitors of MenE, an acyl-CoA synthetase involved in bacterial menaquinone biosynthesis. Bioorg. Med. Chem. Lett. 2008;18:5963–5966. doi: 10.1016/j.bmcl.2008.07.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Wilmanns M. PDB code 2CGH. 2007. unpublished. [Google Scholar]
- Manganelli R, Voskuil MI, Schoolnik GK, Smith I. The Mycobacterium tuberculosis ECF sigma factor sigmaE: role in global gene expression and survival in macrophages. Mol. Microbiol. 2001;41:423–437. doi: 10.1046/j.1365-2958.2001.02525.x. [DOI] [PubMed] [Google Scholar]
- Marrero J, Rhee KY, Schnappinger D, Pethe K, Ehrt S. Gluconeogenic carbon flow of tricarboxylic acid cycle intermediates is critical for Mycobacterium tuberculosis to establish and maintain infection. Proc. Natl. Acad. Sci. U.S.A. 2010;107:9819–9824. doi: 10.1073/pnas.1000715107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Neres J, Labello NP, Somu RV, Boshoff HI, Wilson DJ, Vannada J, Chen L, Barry CE, 3rd, Bennett EM, Aldrich CC. Inhibition of siderophore biosynthesis in Mycobacterium tuberculosis with nucleoside bisubstrate analogues: structure-activity relationships of the nucleobase domain of 5'-O-[N-(salicyl)sulfamoyl]adenosine. J. Med. Chem. 2008;51:5349–5370. doi: 10.1021/jm800567v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pashley CA, Parish T. Efficient switching of mycobacteriophage L5-based integrating plasmids in Mycobacterium tuberculosis. FEMS Microbiol. Lett. 2003;229:211–215. doi: 10.1016/S0378-1097(03)00823-1. [DOI] [PubMed] [Google Scholar]
- Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr. D Biol. Crystallogr. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- Pohlmann J, Brotz-Oesterhelt H. New aminoacyl-tRNA synthetase inhibitors as antibacterial agents. Curr. Drug Targets Infect. Disord. 2004;4:261–272. doi: 10.2174/1568005043340515. [DOI] [PubMed] [Google Scholar]
- Portevin D, de Sousa-D'Auria C, Montrozier H, Houssin C, Stella A, Laneelle MA, Bardou F, Guilhot C, Daffe M. The acyl-AMP ligase FadD32 and AccD4-containing acyl-CoA carboxylase are required for the synthesis of mycolic acids and essential for mycobacterial growth: identification of the carboxylation product and determination of the acyl-CoA carboxylase components. J. Biol. Chem. 2005;280:8862–8874. doi: 10.1074/jbc.M408578200. [DOI] [PubMed] [Google Scholar]
- Project CC. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Purushothaman S, Gupta G, Srivastava R, Ramu VG, Surolia A. Ligand specificity of group I biotin protein ligase of Mycobacterium tuberculosis. PLoS One. 2008;3:e2320. doi: 10.1371/journal.pone.0002320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Sigurskjold BW. Exact analysis of competition ligand binding by displacement isothermal titration calorimetry. Anal. Biochem. 2000;277:260–266. doi: 10.1006/abio.1999.4402. [DOI] [PubMed] [Google Scholar]
- Sikora AL, Wilson DJ, Aldrich CC, Blanchard JS. Kinetic and inhibition studies of dihydroxybenzoate-AMP ligase from Escherichia coli. Biochemistry. 2010;49:3648–3657. doi: 10.1021/bi100350c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slayden RA, Lee RE, Barry CE., 3rd Isoniazid affects multiple components of the type II fatty acid synthase system of Mycobacterium tuberculosis. Mol. Microbiol. 2000;38:514–525. doi: 10.1046/j.1365-2958.2000.02145.x. [DOI] [PubMed] [Google Scholar]
- Somu RV, Boshoff H, Qiao C, Bennett EM, Barry CE, 3rd, Aldrich CC. Rationally designed nucleoside antibiotics that inhibit siderophore biosynthesis of Mycobacterium tuberculosis. J. Med. Chem. 2006;49:31–34. doi: 10.1021/jm051060o. [DOI] [PubMed] [Google Scholar]
- Timm J, Post FA, Bekker LG, Walther GB, Wainwright HC, Manganelli R, Chan WT, Tsenova L, Gold B, Smith I, et al. Differential expression of iron-, carbon-, and oxygen-responsive mycobacterial genes in the lungs of chronically infected mice and tuberculosis patients. Proc. Natl. Acad. Sci. U.S.A. 2003;100:14321–14326. doi: 10.1073/pnas.2436197100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tron CM, McNae IW, Nutley M, Clarke DJ, Cooper A, Walkinshaw MD, Baxter RL, Campopiano DJ. Structural and functional studies of the biotin protein ligase from Aquifex aeolicus reveal a critical role for a conserved residue in target specificity. J. Mol. Biol. 2009;387:129–146. doi: 10.1016/j.jmb.2008.12.086. [DOI] [PubMed] [Google Scholar]
- Vaguine AA, Richelle J, Wodak SJ. SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr. D Biol. Crystallogr. 1999;55:191–205. doi: 10.1107/S0907444998006684. [DOI] [PubMed] [Google Scholar]
- Velazquez-Campoy A, Freire E. Isothermal titration calorimetry to determine association constants for high-affinity ligands. Nat. Protoc. 2006;1:186–191. doi: 10.1038/nprot.2006.28. [DOI] [PubMed] [Google Scholar]
- Velazquez-Campoy A, Kiso Y, Freire E. The binding energetics of first- and second-generation HIV-1 protease inhibitors: implications for drug design. Arch. Biochem. Biophys. 2001;390:169–175. doi: 10.1006/abbi.2001.2333. [DOI] [PubMed] [Google Scholar]
- Wilson DJ, Aldrich CC. A continuous kinetic assay for adenylation enzyme activity and inhibition. Anal. Biochem. 2010;404:56–63. doi: 10.1016/j.ab.2010.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman T, Williston S, Brandts JF, Lin LN. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- Wood ZA, Weaver LH, Brown PH, Beckett D, Matthews BW. Corepressor induced order and biotin repressor dimerization: a case for divergent followed by convergent evolution. J. Mol. Biol. 2006;357:509–523. doi: 10.1016/j.jmb.2005.12.066. [DOI] [PubMed] [Google Scholar]
- Yanachkov IB, Wright GE. Amino-Imino tautomerization of N2-(4-n-butylphenyl)-2'-deoxy-3,5'-cycloguanosine. J. Org. Chem. 1994;59:6739–6743. [Google Scholar]
- Yu M, Magalhaes ML, Cook PF, Blanchard JS. Bisubstrate inhibition: Theory and application to N-acetyltransferases. Biochemistry. 2006;45:14788–14794. doi: 10.1021/bi061621t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.