

Abstract
Expression and purification to homogeneity of the apolipoprotein B mRNA editing subunit, APOBEC1, has allowed the demonstration that this apoenzyme has considerable residual enzymatic activity on a minimal apoB mRNA substrate, even in the absence of any auxiliary factors. Assay of this activity as a function of various experimental conditions has led to substantial optimization of assay conditions through the use of incomplete factorial and response surface experiments. Surprisingly, the apoenzyme is thermostable, and has a temperature optimum near 45°C. We have used these optimized conditions, to assess steady-state kinetic parameters for APOBEC1 mRNA editing activity with and without the auxiliary factor, ACF. An important effect of the auxiliary factor is to broaden the temperature range of APOBEC1 activity, lowering the optimal temperature and enabling it to function optimally at lower temperatures. A model consistent with this observation is that at lower temperatures ACF promotes a conformational transition in the RNA substrate that occurs spontaneously at higher temperature. Notably, the substantial RNA editing activity of APOBEC1 alone may be responsible for the “hyperediting” observed upon overexpression of APOBEC1 in transgenic mice.
Keywords: RNA editing, apoB, APOBEC1, ACF, kinetics
INTRODUCTION
Two forms of apolipoprotein B (apoB) circulate in the blood. In humans, apoB100 is made in the liver and transports endogenously synthesized cholesterol and triglyceride in the circulation (Knott et al. 1986; Yang et al. 1986). In the intestine, apoB48 mediates the absorption and delivery of dietary lipid. ApoB100 and apoB48 are products of the same gene. An early posttranscriptional pre-mRNA RNA editing event deaminates cytidine6666 to uridine in apoB100 mRNA (Chen et al. 1987; Powell et al. 1987; Scott 1995), and this converts glutamine codon 2153 (CAA) to an in-frame stop codon (UAA) and thereby generates apoB48 (Chester et al. 2000). As APOBEC1 activity determines the relative proportions of the two forms of apoB, the basis for regulating it is of considerable interest. This work describes new aspects of APOBEC1 activity obtained from homogeneous enzyme.
The catalytic subunit of the apoB RNA-editing enzyme, APOBEC1, is a 27-kDa member of the cytidine deaminase family of enzymes (Navaratnam et al. 1993a; Teng et al. 1993; Betts et al. 1994; Bhattacharya et al. 1994), and also includes several thermostable cytidine deaminases (Woo et al. 2002). The crystal structure of Escherichia coli cytidine deaminase (ECCDA) has been determined (Betts et al. 1994). Molecular modeling and extensive mutagenesis have demonstrated that APOBEC1 shares quaternary and tertiary structural features with ECCDA (Navaratnam et al. 1998). Both enzymes form homodimers with composite active sites constructed with contributions from each monomer. Significant gaps are present in the APOBEC1 sequence, compared to ECCDA. The combined mass of the gaps (10 kDa) matches that for the minimal RNA substrate. The size of these gaps, together with their location in ECCDA, suggests how APOBEC1 has evolved from related cytidine deaminases to accommodate its RNA substrate (Navaratnam et al. 1998).
ApoB mRNA editing is a highly sequence-specific process dependent upon the primary, secondary, and perhaps even tertiary structure of the RNA substrate (Davies et al. 1989; Bostrom et al. 1990; Chen et al. 1990; Shah et al. 1991; Driscoll et al. 1993; Hersberger and Innerarity 1998). RNA binding (Anant et al. 1995; Navaratnam et al. 1995) is mediated through residues both in and around the APOBEC1 active site, including two opposed phenylalanine residues at sites which in ECCDA (residues 107 and 123) are 10–14 Å away from the active site, and which in the homology model face into a cleft created by one of the gaps (Navaratnam et al. 1998). These and other features presumably recognize unique features of the RNA substrate.
The apoB mRNA sequence around the editing site (C6666) is AU-rich and highly conserved from marsupials to humans (Richardson et al. 1998; Hersberger et al. 1999). An 11-nt sequence downstream of the editing site, designated the “mooring” sequence, is essential for editing (Backus and Smith 1991; Shah et al. 1991). The RNA forms a stem-loop at the apoB mRNA editing site to which APOBEC1 and its auxiliary editing proteins have also been shown to bind (Richardson et al. 1998; Anant and Davidson 2000).
APOBEC1 participates with additional protein factors to form a multicomponent editing holoenzyme (Navaratnam et al. 1993b; Teng et al. 1993). ACF (APOBEC1 complementation factor) and its splice variant ASP (APOBEC1 stimulating protein) have been recently identified and shown to complement APOBEC1 in the editing of apoB mRNA (Lellek et al. 2000; Mehta et al. 2000). Although ACF has been demonstrated to bind to the U-rich “mooring” sequence of apoB mRNA (Navaratnam et al. 1993b; Mehta et al. 2000), the functional role of this binding has not been detailed. Deletion mutagenesis of ACF indicated that ACF binds to APOBEC1 through its amino terminal region, and this region is also involved in binding the RNA substrate (Blanc et al. 2001; Mehta and Driscoll 2002).
ACF shares significant sequence homology to the RNA binding protein GRY-RBP (Blanc et al. 2001). Both proteins have three nonidentical RRM motifs. Whereas ACF complements the activity of APOBEC1, GRY-RBP inhibits editing, suggesting that the activity of APOBEC1 may be regulated by the coordinated action of these two proteins.
Although previous work suggests that APOBEC1 requires at least some of the auxiliary proteins for full specificity on apoB mRNA substrates, this conclusion has rested heavily on studies with impure catalytic subunit, performed under conditions that have not been optimized, and which may intensify the requirement for such additional components. In this study, we describe the purification of APOBEC1 as a glutathione S-transferase (GST) fusion protein. When we first tested the purified enzyme, a small amount of editing was observed in the absence of auxiliary factors. Therefore, we decided to optimize this activity with the hope of addressing questions related to the role of auxiliary factors using steady-state kinetic analysis.
We show here that the purified fusion protein possesses the same biochemical characteristics as endogenous APO-BEC1 but, unusually, that under appropriate conditions, purified APOBEC1 subunit has the ability to edit the apoB mRNA substrate without the addition of any auxiliary factors. Moreover, identification and optimization of the appropriate assay conditions, carried out using incomplete factorial and response-surface methods (Yin and Carter 1996), respectively, have led to an 18-fold increase in the specific activity of the APOBEC1 subunit. Optimized assay conditions further have enabled us to determine steady-state Michaelis-Menten parameters in the presence and absence of the ACF auxiliary factor, and hence, to identify key features of its role in the editing reaction.
Interestingly, we observe that without ACF, APOBEC1 catalysis is remarkably thermostable with optimal RNA editing at 45°C. ACF stimulates editing less at high temperatures. Rather, it appears to modulate the temperature dependence of catalytic activity, enabling comparable editing activity at physiological temperatures, both by increasing Vmax and by lowering Km at physiological temperatures. These observations suggest that the auxiliary factor acts by stabilizing at ambient temperatures an altered form of the RNA substrate that forms spontaneously at higher temperatures.
RESULTS
Expression, purification, and biochemical characterization the GST-APOBEC1 fusion protein
APOBEC1 as an N-terminal GST fusion protein was cloned into a baculovirus expression vector, allowing high-level expression in Sf9 cells using the strong viral polyhedrin promoter. The yield of purified GST-APOBEC1 was in the region of 3–5 mg from 1 liter of culture. A single band on SDS-PAGE, stained either with silver or Coomassie blue, with an apparent mass of 46 kDa characterized the purified GST-APOBEC1 (Fig. 1A▶). This molecular weight is less than the calculated molecular mass of 52 kDa; however, it is consistent with our previous studies using GST-APOBEC1 expressed in E. coli (Navaratnam et al. 1995). The reason for this anomalous migration is unknown.
FIGURE 1.

RNA editing activity of GST-APOBEC1. (A) GST-APOBEC1 and GST-ACF purified from Sf9 and E. coli cells, respectively, using glutathione sepharose 4B and size exclusion, analyzed by SDS-PAGE and stained with Coomassie blue (top). Silver-stained (1) and overloaded Coomassie blue-stained (2) SDS-PAGE gel of purified GST-APOBEC1 (bottom). (B) Primer extension analysis showing the editing of synthetic apoB RNA by GST-APOBEC1 and GST-ACF, in the in vitro conversion assay. Increasing amounts of GST-APOBEC1 (0.25, 0.5, and 0.75 μg) in the absence of GST-ACF and increasing amounts of GST-ACF (0.25, 0.5, 0.75, and 1.0 μg) along with GST-APOBEC1 (0.25 μg) were incubated with apoB RNA. The negative control was performed in the absence of both GST-APOBEC1 and GST-ACF. UAA denotes the edited codon and CAA shows the unedited codon.
To verify whether creation of an N-terminal GST APOBEC1 fusion protein has any altered its biochemical characteristics, its ability to bind zinc, and bind and edit apoB mRNA was evaluated. RNA–protein interactions were assessed using UV cross-linking experiments and zinc binding by the denaturation and subsequent renaturation of the protein in the presence of 65Zn. As expected, GST-APOBEC1 was able to bind apoB and AU-rich RNA substrates; it also bound zinc, as previously observed with GST-APOBEC1 expressed in E. coli (Navaratnam et al. 1995; data not shown).
APOBEC1 alone has site-specific, cofactor-independent C-to-U editing activity
Previous work established an in vitro editing assay, in which the amount of cytidine that is converted to uridine is measured by primer extension analysis (Driscoll et al. 1989) and used to estimate apoB mRNA editing. Under those conditions, which were also used in the present experiments, this assay is linear both with time and enzyme concentration. Crude and partially purified preparations of APOBEC1 had previously been shown to edit apoB mRNA only in the presence of auxiliary factors. Surprisingly, the fractions of pure GST-APOBEC1 also edited apoB mRNA in the absence of ACF (Fig. 1B▶).
Optimization of mRNA editing in the absence of cofactors
The unexpected editing activity observed in the absence of auxiliary factors suggested to us that a more careful evaluation of the assay conditions would provide a useful way to identify the role played by the auxiliary factors. To assess the editing of apoB mRNA by GST-APOBEC1 alone, the conditions required for editing were first identified and then optimized, using variations in RNA substrate concentration, pH, ionic strength, and temperature to induce differences in editing rates.
Previous editing reactions were carried out at low ionic strength, pH 8, and at 30°C. Our initial search included 36 balanced trials, sampling the effects of pH values (7.0, 8.0, and 9.0), temperature (25°, 30°, and 35°C) and substrate RNA concentration (5.5, 11, and 16.5 nM), in the presence or absence of monovalent (potassium or sodium) and divalent ions (magnesium or calcium), as detailed in the incomplete factorial design (Carter and Carter 1979) in Table 1▶. Editing assays were performed in duplicate, and levels of apoB mRNA editing were quantitated for each condition (Table 1▶; Fig. 2▶). The data in Table 1▶ were analyzed first by stepwise regression methods and ultimately verified by standard regression analysis to identify the strengths and signs of correlations between editing activity (Q) and each factor examined.
TABLE 1.
Incomplete factorial experimental design matrix to screen factors influencing the editing of mRNA by GST-APOBEC1
| Sample | pH | Temperature °C | Divalent/Monovalent cation | Ionic strength mM | Divalent ion mM | RNA concentration nM | −ACF editinga % | +ACF editinga % |
|---|---|---|---|---|---|---|---|---|
| 1 | 8 | 25 | Mg/K | 90 | 18 | 11 | 2.1 ± 1.3 | 4.3 ± 1.8 |
| 2 | 7 | 35 | Ca/Na | 190 | 9.89 | 11 | 2.0 ± 0.8 | 3.2 ± 1.2 |
| 3 | 9 | 30 | Ca/K | 90 | 6 | 11 | 3.9 ± 2.0 | 13.1 ± 0 |
| 4 | 8 | 35 | Mg/Na | 140 | 12.02 | 5.5 | 5.0 ± 0.9 | 14.5 ± 5.3 |
| 5 | 9 | 25 | Mg/Na | 140 | 18 | 5.5 | 2.0 ± 0.03 | 11.7 ± 2.2 |
| 6 | 7 | 30 | Ca/Na | 190 | 10.59 | 16.5 | 1.8 ± 0.6 | 3.4 ± 0.71 |
| 7 | 7 | 25 | Mg/K | 190 | 12.54 | 16.5 | 1.4 ± 0.3 | 3.3 ± 1.36 |
| 8 | 9 | 35 | Ca/K | 90 | 6 | 16.5 | 11.6 ± 0.4 | 15.5 ± 6.0 |
| 9 | 8 | 30 | Mg/Na | 140 | 18 | 11 | 3.5 ± 0.2 | 9.0 ± 0.7 |
| 10 | 7 | 25 | Ca/Na | 140 | 6 | 5.5 | 2.2 ± 0.7 | 3.4 ± 1.2 |
| 11 | 8 | 35 | Mg/K | 140 | 6 | 5.5 | 2.5 ± 0.03 | 3.3 ± 1.1 |
| 12 | 9 | 25 | Ca/K | 190 | 18 | 11 | 1.6 ± 0.04 | 7.7 ± 0.09 |
| 13 | 9 | 35 | Mg/Na | 190 | 18 | 16.5 | 1.7 ± 0.7 | 1.7 ± 0.2 |
| 14 | 7 | 30 | Ca/K | 90 | 6 | 5.5 | 2.1 ± 0.4 | 3.5 ± 1.5 |
| 15 | 8 | 25 | Ca/Na | 90 | 12.05 | 5.5 | 6.2 ± 0.2 | 16.2 ± 5.0 |
| 16 | 8 | 30 | Mg/K | 190 | 6 | 16.5 | 2.9 ± 0.08 | 6.8 ± 1.3 |
| 17 | 7 | 35 | Mg/Na | 90 | 18 | 11 | 8.6 ± 0.4 | 19.5 ± 4.7 |
| 18 | 9 | 30 | Ca/K | 140 | 17.26 | 16.5 | 3.6 ± 0.4 | 10.5 ± 1.3 |
| 19 | 7 | 35 | Mg/K | 90 | 11.03 | 11 | 3.3 ± 0.3 | 3.3 ± 1.1 |
| 20 | 9 | 35 | Ca/Na | 140 | 15.54 | 11 | 3.3 ± 1.3 | 2.7 ± 1.0 |
| 21 | 8 | 30 | Mg/K | 90 | 18 | 11 | 1.3 ± 1.2 | 3.2 ± 0.8 |
| 22 | 7 | 25 | Ca/K | 140 | 9.89 | 16.5 | 3.7 ± 1.3 | 22.6 ± 3.1 |
| 23 | 9 | 30 | Ca/Na | 190 | 6 | 11 | 2.5 ± 0.85 | 4.1 ± 1.4 |
| 24 | 8 | 25 | Ca/K | 90 | 12.02 | 16.5 | 1.7 ± 3.7 | 3.7 ± 0.9 |
| 25 | 8 | 35 | Mg/Na | 190 | 18 | 5.5 | 2.8 ± 1.6 | 13.4 ± 5.3 |
| 26 | 9 | 25 | Mg/K | 190 | 10.59 | 5.5 | 1.8 ± 0.7 | 4.7 ± 0.3 |
| 27 | 7 | 30 | Ca/Na | 140 | 12.54 | 16.5 | 4.1 ± 2.6 | 3.4 ± 1.6 |
| 28 | 7 | 25 | Mg/Na | 190 | 6 | 5.5 | 2.8 ± 1.4 | 8.7 ± 0.06 |
| 29 | 8 | 25 | Mg/K | 90 | 18 | 11 | 2.4 ± 0.8 | 10.7 ± 2.5 |
| 30 | 9 | 25 | Ca/K | 140 | 6 | 5.5 | 1.6 ± 1.5 | 2.6 ± 0.3 |
| 31 | 7 | 35 | Mg/Na | 190 | 6 | 16.5 | 4.5 ± 1.3 | 3.0 ± 1.1 |
| 32 | 9 | 35 | Mg/Na | 140 | 18 | 11 | 1.3 ± 0.6 | 2.8 ± 0.4 |
| 33 | 7 | 30 | Ca/Na | 140 | 18 | 16.5 | 5.6 ± 1.1 | 17.8 ± 10.0 |
| 34 | 8 | 30 | Mg/K | 190 | 6 | 16.5 | 1.8 ± 0.9 | 3.2 ± 0.7 |
| 35 | 7 | 35 | Ca/K | 90 | 12.05 | 5.5 | 13.3 ± 4.2 | 24.7 ± 1 |
| 36 | 9 | 30 | Ca/Na | 90 | 6 | 5.5 | 1.8 ± 1.4 | 3.7 ± 0.22 |
aPercentage editing activity is expressed as an average; n = 2–5, except where noted (1).
FIGURE 2.

Optimization of RNA editing by GST-APOBEC1. Primer extension analysis showing the editing of synthetic apoB mRNA by GST-APOBEC1, in the in vitro conversion assay. UAA denotes the edited codon and CAA shows the unedited codon. A representative sample of experiments is shown, and the assay conditions 13–36 are detailed in Table 1▶.
APOBEC1 is a thermostable enzyme
In the initial screening experiments, editing was most elevated at 35°C and low ionic strength, at the limits of the experimental conditions tested. To examine editing at higher temperatures and lower ionic strength, additional experiments were designed in a similar fashion (Table 2▶; expts 37–51). A Harden Sloane design (Table 2▶; expts 52–94) incorporating a new range of pH conditions was performed to refine the pH, temperature, ionic strength, and divalent cation concentration for optimal editing. The buffering system was replaced with PIPES or MES, with optimal buffering ranges of 6.1–7.5 and 5.5–6.7, respectively. The addition of PIPES or MES did not affect editing. Experiments were only performed with the addition of potassium and calcium, as these ions influenced editing more favorably in the initial screen than did sodium and magnesium. The substrate RNA was maintained at the lowest concentration from the screen (5.5 nM). Its variation had no apparent effect on editing in the screens, and we show below that it is saturating. The results were integrated with the previous data and analyzed (Table 2▶).
TABLE 2.
Optimization experiment performed to optimize the editing of mRNA by GST-APOBEC1 and establish response surfaces
| Sample | pH | Temperature °C | Ionic strength mM | Divalent ion mM | −ACF editinga% | +ACF editinga % |
|---|---|---|---|---|---|---|
| 37 | 6.9 | 50 | 92.92 | 18 | 19.0 ± 2.8 | 14.3 (1) |
| 38 | 7 | 50 | 65 | 6 | 20.7 ± 1.0 | 14.7 (1) |
| 39 | 7.5 | 50 | 105 | 18 | 19.8 ± 4.2 | 8.93 ± 0 |
| 40 | 7.5 | 50 | 81.99 | 18 | 23.9 ± 1.0 | ND |
| 41 | 7.1 | 50 | 105 | 6 | 22.6 ± 3.5 | ND |
| 42 | 6.5 | 50 | 105 | 18 | 20.8 ± 6.8 | ND |
| 43 | 6.5 | 50 | 85.17 | 11.03 | 26.2 ± 7.2 | ND |
| 44 | 6.9 | 60 | 92.92 | 18 | 8.0 ± 0.8 | ND |
| 45 | 7 | 60 | 65 | 6 | 14.9 ± 4.2 | ND |
| 46 | 7.5 | 60 | 105 | 18 | 8.1 ± 3.6 | ND |
| 47 | 7.5 | 60 | 81.99 | 12.05 | 10.5 ± 1.9 | ND |
| 48 | 7.1 | 60 | 105 | 6 | 11.7 ± 3.3 | ND |
| 49 | 6.5 | 60 | 105 | 18 | 11.0 ± 1.0 | 9.6 (1) |
| 50 | 6.5 | 60 | 85.17 | 11.3 | 14.0 ± 1.0 | ND |
| 51 | 5.9 | 60 | 25 | 18 | 3.5 ± 4.4 | 1.8 ± 0.8 |
| 52 | 6.9 | 42 | 92.92 | 9.89 | 20.6 ± 1.8 | 23.2 ± 34 |
| 53 | 6.5 | 35 | 65 | 6 | 11.7 ± 1.7 | 27.0 ± 3.6 |
| 54 | 7.5 | 35 | 77.96 | 12.02 | 9.3 ± 1.7 | 18.3 ± 2.2 |
| 55 | 7.1 | 35 | 105 | 18 | 11.3 ± 2.5 | 17.2 ± 0.9 |
| 56 | 7.5 | 28 | 99.84 | 10.59 | 5.4 ± 1.3 | 13.8 ± 0.3 |
| 57 | 7.1 | 28 | 73.77 | 12.54 | 6.8 ± 0.28 | 15.3 ± 1.1 |
| 58 | 7 | 35 | 86.81 | 6 | 11.8 ± 1.7 | 14.9 ± 0.3 |
| 59 | 7.5 | 28 | 105 | 18 | 4.6 ± 0.3 | 14.8 ± 1.3 |
| 60 | 6.5 | 28 | 87.35 | 6 | 6.7 ± 0.2 | 15.0 ± 0.5 |
| 61 | 7 | 42 | 65 | 6 | 18.7 ± 0.5 | 25.6 ± 4.9 |
| 62 | 6.5 | 33 | 102.52 | 18 | 10.7 ± 1.3 | 22.3 ± 3.6 |
| 63 | 7.5 | 42 | 105 | 18 | 19.4 ± 0.6 | 11.0 ± 2.8 |
| 64 | 6.9 | 35 | 93.1 | 6 | 12.7 ± 1.1 | 16.4 ± 1.1 |
| 65 | 6.9 | 28 | 81.28 | 12.05 | 7.2 ± 1.3 | 20.6 (1) |
| 66 | 7.5 | 42 | 81.99 | 6 | 18.5 ± 2.0 | 17.8 ± 0.9 |
| 67 | 7.1 | 42 | 105 | 18 | 16.0 ± 2.1 | 16.5 ± 1.6 |
| 68 | 6.5 | 42 | 105 | 17.26 | 19.4 ± 0.9 | 21.9 ± 1.4 |
| 69 | 7.5 | 33 | 97.61 | 11.03 | 12.2 ± 0.3 | 15.1 ± 0.5 |
| 70 | 6.5 | 42 | 85.17 | 15.54 | 18.8 ± 0.2 | 24.5 ± 0.1 |
| 71 | 6.5 | 28 | 105 | 18 | 6.9 ± 0.7 | 18.1 ± 1.3 |
| 72 | 5.5 | 49 | 25 | 9.89 | 4.7 ± 1.6 | 3.3 ± 0.3 |
| 73 | 6.5 | 49 | 57.4 | 6 | 19.0 ± 1.4 | 11.4 ± 3.0 |
| 74 | 6.1 | 47 | 125 | 12.02 | 14.2 ± 3.2 | 10.2 ± 1.6 |
| 75 | 6.5 | 36 | 104.53 | 18 | 10.7 ± 3.6 | 17.3 ± 0.9 |
| 76 | 6.1 | 36 | 25 | 10.59 | 9.8 ± 1.8 | 17.3 ± 1.3 |
| 77 | 6.0 | 48 | 79.53 | 12.54 | 12.7 ± 1.7 | 8.1 ± 0.3 |
| 78 | 6.5 | 36 | 125 | 6 | 7.4 ± 0.4 | 17.9 ± 1.8 |
| 79 | 5.5 | 36 | 25 | 18 | 4.1 ± 1.1 | 8.3 ± 1.4 |
| 80 | 6.0 | 60 | 25 | 6 | 3.3 ± 0.5 | 4.4 ± 0.3 |
| 81 | 5.5 | 44 | 118.81 | 6 | 5.9 ± 1.2 | 4.0 ± 1.3 |
| 82 | 5.0 | 60 | 125 | 18 | 2.6 ± 0.3 | 1.9 ± 0.8 |
| 83 | 5.9 | 47 | 75.17 | 18 | 13.6 ± 0.14 | 7.6 ± 1.3 |
| 84 | 5.9 | 36 | 63.21 | 6 | 10.5 ± 2.8 | 13.8 ± 2.0 |
| 85 | 6.5 | 60 | 66.94 | 12.05 | 7.4 ± 1.3 | 6.9 ± 0.9 |
| 86 | 6.1 | 60 | 125 | 6 | 5.3 ± 1.1 | 4.7 ± 2.0 |
| 87 | 5.5 | 60 | 125 | 18 | 2.9 ± 0.02 | 2.7 ± 0.6 |
| 88 | 6.5 | 44 | 25 | 17.26 | 17.4 ± 2.5 | 17.8 ± 0.5 |
| 89 | 5.5 | 60 | 75.42 | 11.03 | 5.2 ± 3.1 | 2.6 ± 1.5 |
| 90 | 5.5 | 36 | 125 | 15.54 | 6.3 ± 0.3 | 8.84 ± 0.3 |
| 91 | 5.0 | 49 | 104.53 | 18 | 3.0 ± 0.7 | 2.2 ± 1.9 |
| 92 | 5.0 | 36 | 125 | 9.89 | 2.9 ± 1.4 | 3.2 ± 1.6 |
| 93 | 5.0 | 60 | 66.94 | 6 | 3.1 ± 1.2 | 2.9 ± 1.3 |
| 94 | 5.0 | 44 | 25 | 12.02 | 2.7 ± 0.9 | 2.0 ± 1.1 |
aPercentage editing activity is expressed as an average; n = 2–5, except where noted (1).
Temperature had the major effect on editing efficiency, which increased with higher temperatures (Fig. 3A▶). Other factors contribute less significantly to the scatter seen in the data. To estimate the relative effects of temperature and other factors, we fitted a variety of response-surface models to the editing data derived by multivariate regression as described in Materials and Methods, and selected optimal models. To characterize the role of ACF in the editing reaction, a majority of in vitro editing assays used for the enzyme alone were repeated in the presence of purified ACF (Tables 1▶, 2▶). These assays used a threefold molar ratio excess of ACF based on the assumption that GST-APOBEC1 should exist as a dimer of 104 kDa and GST-ACF is determined to be 90 kDa. The assays were carried out and analyzed as previously described, and the results with APOBEC1 alone and with ACF were analyzed by response-surface modeling. Temperature and pH had negative quadratic coefficients, giving rise to a joint maximum, while the ionic strength and divalent ion concentration were linear. Temperature proved to have a higher order effect requiring a cubic term in the modeling, as described in the Materials and Methods section. Setting the latter terms from Tables 3A▶ and 3B▶ to typical values, the resulting response-surface equations were:
FIGURE 3.

Response surface modeling of APOBEC1 editing with and without ACF. (A) The ensemble of all assays plotted against temperature with (green) and without (blue) the auxiliary factor ACF. (B) pH versus temperature level surfaces for the multivariate analytical models (equations 1) obtained by fitting the editing data in (A) as functions of pH, temperature, ionic strength, and Ca++ concentration. Profiles of the temperature dependence at the optimum pH value above the plots show the effects of the cubic terms in temperature. The coloring is as in (A). Normal probability plots to either side are described in Materials and Methods. They compare fitted residuals with (green dots) and without (red dots) the cubic term in temperature. Deviations from straight lines indicate serious systematic errors when the cubic term is omitted.
TABLE 3A.
Statistics for the regression of the optimization models for APOBEC1: No ACF regression model
| Variable | Coefficient | STD error | T | p (2 tail) |
|---|---|---|---|---|
| Constant | 6.9 | 15.7 | 0.44 | 0.6600 |
| PH | 23 | 2.25 | 10.43 | 0.10E-14 |
| D | 0.17 | 0.13 | 1.34 | 0.182 |
| T | −6.8 | 1.09 | −6.22 | 0.36E-08 |
| I | 55 | 34.9 | 1.58 | 0.115 |
| SCAL | −6.2 | 0.78 | −7.92 | 0.26E-12 |
| RNA_2 | −1.5 | 0.50 | −3.04 | 0.0027 |
| T*RNA_2 | 0.049 | 0.017 | 2.97 | 0.0034 |
| PH*PH | −1.5 | 0.16 | −9.66 | 0.10E-14 |
| D*D | −0.0096 | 0.0058 | −1.66 | 0.0992 |
| T*T | 0.18 | 0.029 | 6.23 | 0.34E-08 |
| I*I | −310 | 148.4 | −2.09 | 0.039 |
| T*T*T | −0.0016 | 0.0002 | −7.32 | 0.87E-11 |
| Analysis of variance | |||||
|---|---|---|---|---|---|
| Source | Sum-of- squares | df | Mean-square | F-ratio | p |
| Dependent variable: Q−ACF editing activity in the absence of ACF. | |||||
| Number of experiment (including duplicates), 188. | |||||
| Multiple R, 0.926. Squared multiple R, 0.86. | |||||
| Adjusted squared multiple R, 0.85. Standard error of estimate: 2.70. | |||||
| adf, degrees of freedom. | |||||
| bp, probability of F-ratio test. | |||||
| Regression | 7677.4 | 12 | 639.78 | 87.61 | 0.999E-15 |
| Residual | 1277.9 | 175 | 7.30 | ||
TABLE 3B.
Statistics for the regression of the optimization models for APOBEC1: +ACF-regression model
| Variable | Coefficient | STD error | T | p (2 tail) |
|---|---|---|---|---|
| Constant | −161 | 33 | −4.87 | 0.27E-05 |
| PH | 29 | 4.1 | 7.17 | 0.27E-10 |
| D | −0.65 | 0.24 | −2.70 | 0.0078 |
| T | 5.7 | 2.0 | 2.78 | 0.0061 |
| I | 322 | 114 | 2.82 | 0.0054 |
| SCAL | −4.4 | 1.1 | −4.01 | 0.93E-04 |
| RNA_2 | −1.6 | 1.3 | −1.25 | 0.21 |
| D*T | 0.010 | 0.0064 | 1.60 | 0.11 |
| T*RNA_2 | 0.093 | 0.034 | 2.74 | 0.0068 |
| I*RNA_2 | −9.3 | 4.1 | −2.27 | 0.0246 |
| PH*PH | −2.1 | 0.28 | −7.20 | 0.24E-10 |
| T*T | −0.16 | 0.054 | −2.94 | 0.0038 |
| I*I | −1040 | 310.0 | −3.40 | 0.0009 |
| T*T*T | 0.0011 | 0.0004 | 2.59 | 0.011 |
| Analysis of Variance | |||||
|---|---|---|---|---|---|
| Source | Sum-of-squares | df | Mean-square | F-ratio | p |
| Dependent variable: Q+ACF editing activity in the presence of ACF. | |||||
| Number of experiment (including duplicates), 171. | |||||
| Multiple R, 0.81. Squared multiple R, 0.65. | |||||
| Adjusted squared multiple R, 0.620. Standard error of estimate: 4.54. | |||||
| adf, degrees of freedom. | |||||
| bp, probability of F-ratio test. | |||||
| Regression | 5999.1 | 13 | 461.47 | 22.38 | 0.999E-15 |
| Residual | 3237.4 | 157 | 20.62 | ||
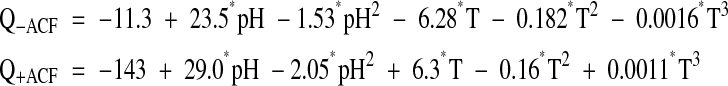 |
(1) |
Temperature-pH level surfaces of these models are plotted in Figure 3B▶. Optimal activity for APOBEC1 alone is observed at 45°C, pH 7.46, and 80 mM ionic strength.
GST-ACF complements APOBEC1 much less at higher temperatures
GST-ACF complements GST-APOBEC1 editing activity substantially at physiological temperatures (Fig. 3B▶). The complementation of APOBEC1 RNA editing activity by the addition of ACF is maximal at 30°C, pH 7.0, and 90 mM ionic strength. These conditions are consistent with those previously described for editing by partially purified APOBEC1 and auxiliary factors (Greeve et al. 1991). The optimal pH has been reduced from pH 8.0 to a lower pH 7.0, but the conclusion that apoB mRNA editing requires low ionic strength remains unchanged.
Steady-state kinetics of apoB RNA editing by APOBEC1 and ACF
Once it became clear that near optimal editing conditions had been determined for editing by APOBEC1 alone and in the presence of ACF, we proceeded to test the effect of the purified cofactor ACF using steady-state kinetic measurements. Four Michaelis-Menten experiments were performed—two at 30°C and two at 41°C—the temperature at which editing with and without ACF appeared to be comparable (Fig. 3B▶) in the presence and absence of ACF, at the optimal conditions determined previously. The results, plotted in Figure 4▶, indicate that ACF abolished the strong temperature-dependence observed for APOBEC1 alone. At the lower temperature, the addition of ACF had a pronounced effect on the maximal velocity. To clarify the effects of ACF, Michaelis-Menten parameters were estimated for all experiments by a variety of algorithms, including the Eadie-Hofstee linearization fitting of the velocity data and substrate concentrations to a rectangular hyperbola (Fig. 4▶; Table 4▶).
FIGURE 4.

Determination of Km and Vmax. (A) Michaelis-Menton plots of kinetic data for RNA editing by GST-APOBEC1 in the absence and presence of ACF. Relative Vmax is measured as percentage editing rather than specific activity. The gray vertical bar denotes the minimum concentration used in the Arrhenius analysis of Figure 5▶. (B) Eadie-Hofstee plot of the kinetic data. Percentage editing (V) plotted against V/substrate concentration. Triangles show editing with GST-APOBEC1 only, and circles show editing with GST-APOBEC1 plus GST-ACF. Open and closed symbols denote editing performed at 30°C and 41°C, respectively. Regression statistics are shown for the determination of kcat and Km from both analyzes.
TABLE 4.
The effect of ACF on APOBEC1 steady-state kinetic parameters
| 30°C | 41°C | ||
|---|---|---|---|
| Km (nM) | −ACF | 0.21 | 0.22 |
| +ACF | 0.017 | 0.027 | |
| Relative Vmax % | −ACF | 17 | 29 |
| +ACF | 44 | 41 | |
| Vmax,rel/Km | −ACF | 81 | 130 |
| +ACF | 2600 | 1500 |
These estimates are rough by comparison to the standard set for conventional enzyme assays, owing to the rather ambitious nature of this effort. However, the qualitative behavior is unmistakeably that of steady-state catalysis, and there was essentially no difference in the quality of the fit or estimated error for either the resulting kinetic parameters.
Stimulation of editing by the auxiliary factor at 30°C involves roughly a 12-fold reduction in Km, combined with a 2.5-fold increase in Vmax for a 30-fold increase in Vmax/ Km. Similar, but reduced effects are seen at 41°C. The 32-fold stimulation of the apparent second-order rate constant at 30°C is reduced roughly threefold at higher temperatures.
Arrhenius analysis of the temperature dependence of editing ±ACF
As suggested by the multivariate optimization, the principle effect of ACF is to significantly broaden the temperature dependence of the activation energy required for editing. As the editing assays used for optimization were performed at near saturating substrate concentrations ([RNA] = 5 nM >> 0.2 nM), and hence, represent Vmax values, an Arrhenius analysis of this effect is possible, using measurements made at different temperatures in the Harden-Sloane optimization (Fig. 5▶). In the absence of ACF, the enthalpy of activation obtained from the slope in Figure 5▶, Ea–kT = 13.1 kcal/mole, is similar to that for deamination of cytidine by ECCDA (14.9 kcal/mole; Snider et al. 2000). This suggests that the binding site for the RNA substrate is essentially intact in the APOBEC1, and that once bound to the pyrimidine moiety, deamination proceeds mechanistically much as for the nucleoside deaminase. Stimulation by ACF lowers this activation enthalpy for editing to roughly 2.8 kcal/mole, primarily by improving the apparent binding affinity.
FIGURE 5.

Temperature dependence of deamination ±ACF. Arrhenius-like plots derived from editing measurements at different temperatures. All points were determined at saturating RNA concentrations, as indicated by plots in Figure 4▶. Scatter in the data arise from variations in pH, divalent ion concentration, and ionic strength, and, in the case of the +ACF data, variation in the assembly of the editosome complex itself.
DISCUSSION
Highly purified GST-APOBEC1 protein is homogenous, and has significant RNA editing activity, even in the absence of added auxiliary factors. We have characterized and optimized APOBEC1 editing activity with respect to pH dependence, mono- and divalent cation requirement, substrate concentration, and temperature dependence. Maximum RNA editing by APOBEC1 depends on three factors: pH, ionic strength, and temperature.
We observed that GST-APOBEC1 remains active well above normal physiological temperatures with optimal editing occurring at 45°C and editing activity still evident at 60°C. This is an unusual feature of a eukaryotic enzyme, and is similar to that observed for the proteosome isolated from bovine lens (Wagner and Margolis 1993). Recently, the homotetrameric Bacillus calolyticus cytidine deaminase was purified, and characterization of this enzyme revealed this enzyme thermostable with optimal deamination occurring at 70°C–75°C (Woo et al. 2002). The amino acid sequence conservation across the entire family of cytidine deaminases suggests that in addition to the catalytic mechanism, enzyme characteristics such as thermostability may also be conserved throughout this protein family.
Moreover, we describe here the first serious effort to understand the enzymology of APOBEC1, using an assay previously thought capable only of qualitative use. As such, our results are capable only of approximate definitions of parameters—Km, Vmax, and ΔH‡, which for other enzymes are more readily and more precisely determined. We believe, however, that these estimates are nevertheless quite informative. We provide evidence here for the first time that APOBEC1 can edit apoB mRNA without the addition of any auxiliary factors, together with evidence concerning the role of the auxiliary factor, ACF.
Interpreting such a rich set of new data, however, presents both opportunities and challenges. We have already noted that the variance of editing measurements with ACF reflect as yet undetermined factors, likely related to the assembly of the editing complex of APOBEC1, ACF, and RNA. The system is complicated by other factors, including possible conformational changes in all components, especially in the RNA substrate, and their respective and possibly interacting sensitivities to temperature.
We can only sketch here some of the more reasonable possibilities, while pointing out sources of ambiguity and avenues that it may be fruitful to pursue in more detail. Not surprisingly, many of the issues arise from the unusual temperature dependences of the editing with and without ACF, and the manner in which our results differ from the behavior of enzymes in general (Wolfenden et al. 1999). We have noted above the similarity of the APOBEC1 activation enthalpy to that of the analogous ECCDA catalyzed reaction. In fact, activation enthalpies for nearly all enzyme-catalyzed reactions all fall within a remarkably narrow range (Wolfenden et al. 1999). Thus, although this value is reassuring evidence that our data are valid, it is not, by itself, particularly surprising.
More surprising is that the Km for the APOBEC1 alone is independent of temperature. Generally, enzyme binding interactions weaken as temperature increases, and to the same extent as the catalytic rate constant increases, so that the apparent second-order rate constants for many enzymes are more or less temperature-independent.
It is also surprising that the activation enthalpy of the ACF-assisted reaction is so modest. One possibility is that the threefold stoichiometric excess used in our assays is not sufficient to saturate the editing complex, reducing the ACF effect, especially at higher temperatures. This could arise because assembly of the active complex is diminished, either because ACF is unstable at higher temperatures, or because its affinity for RNA substrate and/or APOBEC1 weakens at higher temperatures. One or more of these effects could mean that the editing rates measured at high temperature reflect Vmax/Km, and not Vmax, as is assumed in Figure 5▶. The ACF fusion protein itself denatures at Tm ~57°C (Fig. 6▶), so it is unlikely that denaturation plays a role in stimulation of APOBEC1 editing by the ACF complex in the higher temperature range of 45°C–55°C. It seems to be important, therefore, to examine the [ACF] dependence of its activation of editing.
FIGURE 6.

Thermal denaturation of ACF. Thermal denaturation of the GST-ACF fusion protein was monitored by circular dichroism and fitted as described in Materials and Methods.
Taken together, the editing dependencies of the APOBEC1 protein itself present an interesting paradox. Not only is the protein itself obviously stable at elevated temperatures, the apparent second-order rate constant, Vmax/Km, actually increases by 60% at 41°C. To see how unusual a result this is, one can refer to the temperature dependence of the related nucleoside deaminase, E. coli cytidine deaminase, ECCDA. The catalytic rate constant, kcat, for ECCDA increases with temperature as does that of APOBEC1, and by a comparable amount, 2.5× versus 1.7×, between 30°C and 40°C (Snider et al. 2000). However, in ECCDA, that increase is accompanied by a parallel increase in Km, so the apparent second-order rate constant is nearly independent of temperature.
The role of ACF is apparently only to increase Vmax modestly, while decreasing Km more dramatically (10–12-fold). A reasonable, although certainly not unique interpretation developed by analogy with work on ECCDA is suggested by the fact that the loop that binds to the ribose in ECCDA and provides an “anchor” contributing substantially to catalysis (Snider et al. 2000), is entirely missing from APOBEC1 (Navaratnam et al. 1998). Modeling of the APOBEC1 structure, based on the ECCDA structure reveals a substantial opening into which the RNA substrate fits. Mutation of E91 within that loop disrupts the most important residue in ECCDA’s interaction with the ribose, and results in a reduction in kcat by 10−5. The modest effect of ACF on kcat suggests that APOBEC1 itself is nearly fully competent in catalysis once the substrate is bound. This suggests that the RNA sugar phosphate backbone, which likely replaces the loop that binds to the ribose, can anchor the ribose in a catalytically productive manner in the absence of ACF.
Data presented here do not point to an unambiguous interpretation of the effects of temperature or ACF on APO-BEC1 editing activity, either of which might result from altered RNA substrate, enzyme, or both. Nevertheless, the thermostability of APOBEC1 itself and the dominant effect of ACF in reducing Km at both low and high temperatures (Table 4▶; Fig. 5▶) suggest that one likely role for ACF is to bind to an RNA conformation that binds productively to APOBEC1, thereby increasing the effective substrate RNA concentration. Details of this proposal are shown in Figure 7▶. The active conformation is shown here as a flipping out of C6666; thus, the active RNA conformation is in equilibrium with an inactive conformation. Higher temperatures shift the equilibrium constant for the conformational change, and the RNA substrate more readily assumes the active conformation promoted by ACF binding. At lower temperatures, the effect of ACF therefore would be more pronounced. Moreover, a higher effective substrate concentration at higher temperatures would be consistent with the expected, but cryptic, weakening of the APOBEC1 RNA affinity, thereby leaving the Km essentially unchanged. The conformational changes that are proposed to occur during editing will only be confirmed by the acquisition of the structure of the APOBEC1/ACF/apoB RNA editing complex by NMR or X-ray crystallography.
FIGURE 7.

Proposed (schematic) configurations of the apoB mRNA during editing. The amino group (NH2) of cytidine 6666 probably needs to be “flipped out” and exposed to assume the correct orientation for editing. At 30°C, APOBEC1 can edit the apoB RNA weakly in the absence of ACF. The relative Km values ±ACF suggest that the majority of the RNA is in conformation A, with a small proportion, perhaps 10%, in the correct orientation (B), allowing APOBEC1 access to C6666 (1). The maximal catalytic activity of APOBEC1 is at 45°C, suggesting that a change in the RNA secondary structure occurs at this temperature (C). A larger proportion of the cytidine will be in the “flipped out” conformation, allowing greater access by APOBEC1 (2). This editing is more comparable to that catalyzed by APOBEC1 plus ACF at 30°C (3), and the presence of ACF probably causes a similar change in RNA structure to that seen at 45°C. Cytidine 6666 is indicated, and the “mooring” sequence is highlighted in blue. ACF and APOBEC1 are shown in green and red, respectively, with the RRMs and active sites shaded.
Finally, as homogenous GST-APOBEC1 protein can edit apoB mRNA without the addition of ACF, we suggest that this mimics, in vitro, the phenomenon of “hyperediting” or nonspecific editing observed in transgenic mice overexpressing APOBEC1. These animals develop hepatocellular carcinomas, and it has been suggested that this is a direct result of the increased expression of APOBEC1. To date, only one other RNA, NAT1, has been reported to be edited in these mice (Yamanaka et al. 1997). However, if overexpression of APOBEC1 outstrips the regulatory capacities of factors like ACF and GRY-RBP, then its ability to edit RNA substrates would likely lead to more widespread activity.
MATERIALS AND METHODS
Plasmid constructs
GST-APOBEC1 cDNA was amplified from pGEX4T3 containing APOBEC1 (Amersham Biosciences) and cloned into the EcoRI/ SalI sites of pFastbac-1 (Invitrogen). Plasmid DNA was transposed into E. coli DH10Bac (Invitrogen) to generated bacmid DNA. ACF cDNA was cloned into pGEX6P1 (Amersham Biosciences).
Expression of recombinant proteins
APOBEC1 was expressed in Sf9 cells as an N-terminal GST fusion protein. Recombinant virus with multiplicity of infection of 2 was used to infect Sf9 cells and cells harvested 72 h postinfection. ACF cloned into pGEX6P1 was transformed into E. coli BL21 cells. Fusion proteins were affinity purified using glutathione sepharose 4B (Amersham Biosciences) following the manufacturers instructions and further purified by size-exclusion chromatography.
Soluble protein extracts were prepared from Sf9 cells by homogenization followed by 1-min sonication in HEPES buffer (25 mM HEPES pH 8, 1 mM EDTA, 5 mM β-mercaptoethanol, 0.01% Thesit and 10% [v/v] glycerol) supplemented with 1% Triton X100 (v/v), 1/1000th volume Trasylol™ (Bayer), and 1 mM phenylmethylsulfonyl fluoride (PMSF). The soluble fraction was collected by centrifugation at 12,000 × g for 20 min and rocked overnight at 4°C with glutathione sepharose 4B (Amersham Biosciences) following the manufacturer’s instructions. The resin was washed with 10 column volumes of ice-cold HEPES buffer without glycerol, 10 column volumes of HEPES buffer containing 0.5 M NaCl, and finally, with 5 column volumes of buffer containing 1 M NaCl. The column was then eluted four times with 5 column volumes of elution buffer (20 mM reduced glutathione, 5 mM β-mercaptoethanol, 1 mM Tris-HCl pH 8, 0.1% Triton X-100, and 2 M urea). The fractions were collected and individually dialyzed in three steps against 300 volumes of HEPES buffer containing 1 M urea, 0.5 M urea, and then buffer alone. The aliquots were then concentrated over polyethylene glycol (PEG) M.W. 20,000 and applied to a Superdex-75 column (Amersham Biosciences).
GST-ACF was expressed in E. coli. Cells were grown to an O.D. of 0.8 and induced with 0.1 mM IPTG for 3 h at 37°C. GST-ACF was affinity purified using glutathione sepharose 4B (Amersham Biosciences) and further purified via size-exclusion chromatography using a Sephadex 200.
Protein concentrations were determined using bovine serum albumin for a standard curve, using the Bio-Rad protein assay.
Conversion and primer extension analysis
In vitro conversion and primer extension analysis of apoB RNA editing was carried out as previously described (Navaratnam et al. 1995) using a 208-base (nucleotides 6510–6717) rat apoB mRNA. ApoB mRNA substrate was edited in the presence of GST-APOBEC1 (250 ng) and GST-ACF (0–750 ng) in a volume of 20 μL, under the conditions indicated. Quantification of editing was performed by laser densitometric scanning (Storm Phosphoimager, Molecular Dynamics) and data analyzed using ImageQuant software.
Thermal denaturation analysis
Thermal denaturation of the GST-ACF fusion protein was carried out using a Pistar-180 CDF circular dichorism spectrometer (Applied Physics, Ltd.) equipped with temperature ramping capability. Samples (three independent, one of which is shown in Fig. 6▶) were initially incubated at 40°C and ramped to 80°C in 1 degree intervals, ±0.2°C with a settling time of 30 sec. The denaturation curve was fitted to the six-parameter empirical equation (Edgell et al. 2003),
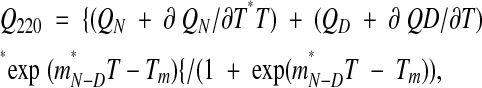 |
where Tm is the melting temperature midpoint, QN and QD are the values of the circular dichroism of the native and denatured states, σ QN/∂T and ∂ QD/∂T are the respective slopes of the curves before and after the transition, mN-D is the slope of the curve during the transition, and Tm is the midpoint of the thermal transition. The Tm for the three samples averaged 57.1 ± 0.53°C.
Experimental designs for screening and optimization of factors affecting APOBEC1 editing
We approached the optimization problem in a manner similar to that used for increasing the yield of in vitro transcription with T7 RNA polymerase (Yin and Carter 1996). Incomplete factorial sampling was utilized to optimize the RNA editing by GST-APOBEC1 alone, and then in the presence of purified GST-ACF, to compare the optima with and without auxiliary factor. The experimental design, which assumes that a desired result is, a priori, equally likely at any point within a range of values for a number of different parameters varied simultaneously, was originally introduced for determining protein crystallization conditions (Carter and Carter 1979). In brief, we first did a broad screen using an incomplete factorial design to identify factors having significant impact on editing activity and subsequently examined factors identified in the initial screen using response surface methods based on a minimum prediction variance design (Hardin and Sloane 1993).
Data analysis and modeling
Variations in APOBEC1 activity were analyzed as linear functions of the independent experimental variables using multiple regression and the analysis of variance as implemented in the multivariate statistical packages SYSTAT V.5.2.1. (SPSS Inc.) and JMP v.4.0 (SAS Institute). Different experimental conditions were evaluated by stepwise multiple regression, and the significant predictors of APOBEC1 editing activity were subjected to standard least-squares regression. These were then retained and varied in subsequent designs.
Editing experiments were carried out over an extended time period, using five different preparations of enzyme with differing activities. This situation was less optimal than using comparable amounts of active enzyme in each experiment. As the experimental conditions always provided a significant number of overlapping experiments done under otherwise identical conditions, we used overlapping experiments to calculate a linear scale constant for experiments done with different batches. The assumption of such linear scaling was checked several times by verifying that the regression models gave similar coefficients from different batches, and by comparing the fitting statistics obtained for the combined and partial data sets.
Results from Hardin and Sloane response surface designs (Hardin and Sloane 1993) were analyzed using the same software, but the models fitted were generally quadratic polynomials, the simplest functions that model local optima. Fitting both without and with ACF was unusual in that we had to include a cubic term in the temperature dependence, T3, to remove systematic fitting errors. In the absence of the cubic term, normal probability plots of expected versus observed residuals for both regressions are noticeably curved (Fig. 3B▶, red dots) implying that they are not normally distributed, hence reflecting systematic error in the models. Including the cubic term increased the Pearson R2 by about 5 percentage points in both cases, and produced essentially straight probability plots (Fig. 3B▶, green dots).
Stationary points for the fitted functions were then obtained by partial differentiation and solving the simultaneous equations that resulted from setting the gradients equal to zero. All such calculations were carried out in JMP v.4.0 (SAS Institute). Graphs were produced using SYSTAT v.5.2.1 (SPSS Inc.), JMP v.4.0 (SAS Institute Inc.), and KaleidaGraph v.3.0.5 (Synergy Software).
The final R2 values for the models plotted in Figure 3B▶ were 0.86 and 0.65. Although R2 provides a direct estimate for the percentage of variation in the data that can be explained in terms of the model, the corresponding quantity, (1 – R2), does not represent the relative amounts of predictive information missing from the two models. The R2 decreases with the square of the intrinsic experimental error (Edgell et al. 2003). Editing results with the ACF cofactor proved to be noisier than those in its absence. The reproducibility from triplicate experiments was used to evaluate the coefficients of variation (the ratio of the standard deviation for replicate measurements to their average) and to provide estimates of ~0.11 and and 0.21 for the intrinsic experimental errors in the absence and presence of ACF.
The increase in the inherent experimental “noise” when using ACF is consistent with the fact that these experiments involved something akin to assembly of a partial “editosome,” which doubtlessly introduces additional complications not entirely recognized or properly controlled. Moreover, it also means that even appropriate models will not fit as well with these data as corresponding models fitted to data for editing by APOBEC1 alone. The error estimates are compatible with R2 values of ~0.9 and 0.64 if all systematic variation were explained by the models. This consistency and the linear normal probability plots of both residuals provide strong support for the fitted models in Figure 3▶.
Acknowledgments
We thank Ashutosh Tripathy at the University of North Carolina (UNC) Macromolecular Interaction facility for assistance with the temperature-ramping CD spectrometer. Work at UNC was supported by NIGMS 48519.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.7490704.
REFERENCES
- Anant, S. and Davidson, N.O. 2000. An AU-rich sequence element (UUUN[A/U]U) downstream of the editied C in apolipoprotein B mRNA is a high-affinity binding site for Apobec-1: Binding of Apobec-1 to this motif in the 3′ untranslated region of c-myc increases mRNA stability. Mol. Cell. Biol. 20 1982–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anant, S., Macginnitie, A.J., and Davidson, N.O. 1995. Apobec-1, the catalytic subunit of the mammalian apolipoprotein B mRNA editing enzyme, is a novel RNA-binding protein. J. Biol. Chem. 270 14762–14767. [PubMed] [Google Scholar]
- Backus, J.W. and Smith, H.C. 1991. Apolipoprotein B mRNA sequences 3′ of the editing site are necessary and sufficient for editing and editosome assembly. Nucleic Acids Res. 19 6781–6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts, L., Xiang, S., Short, S.A., Wolfenden, R., and Carter Jr., C.W. 1994. Cytidine deaminase. The 2.3 Å crystal structure of an enzyme: Transition-state analog complex. J. Mol. Biol. 235 635–656. [DOI] [PubMed] [Google Scholar]
- Bhattacharya, S., Navaratnam, N., Morrison, J.R., Scott, J., and Taylor, W.R. 1994. Cytosine nucleoside/nucleotide deaminases and apolipoprotein B mRNA editing. Trends Biochem. Sci. 19 105–106. [DOI] [PubMed] [Google Scholar]
- Blanc, V., Henderson, J.O., Kennedy, S., and Davidson, N.O. 2001. Mutagenesis of apobec-1 complementation factor reveals distinct domains that modulate RNA binding, protein–protein interaction with apobec-1, and complementation of C to U RNA-editing activity. J. Biol. Chem. 276 46386–46393. [DOI] [PubMed] [Google Scholar]
- Bostrom, K., Garcia, Z., Poksay, K.S., Johnson, D.F., Lusis, A.J., and Innerarity, T.L. 1990. Apolipoprotein B mRNA editing—Direct determination of the edited base and occurrence in non-apolipoprotein B-producing cell-lines. J. Biol. Chem. 265 22446–22452. [PubMed] [Google Scholar]
- Carter Jr., C.W. and Carter, C.W. 1979. Protein crystallization using incomplete factorial experiments. J. Biol. Chem. 254 12219–12223. [PubMed] [Google Scholar]
- Chen, S.-H., Habib, G., Yang, C.Y., Gu, Z.W., Lee, B.R., Weng, S.A., Silberman, S.R., Cai, S.J., Deslypere, J.P., Rosseneu, M., et al. 1987. Apolipoprotein B-48 is the product of a messenger RNA with an organ-specific in-frame stop codon. Science 238 363–366. [DOI] [PubMed] [Google Scholar]
- Chen, S.-H., Li, X.X., Liao, W.S., Wu, J.H., and Chan, L. 1990. RNA editing of apolipoprotein B mRNA. Sequence specificity determined by in vitro coupled transcription editing. J. Biol. Chem. 265 6811–6816. [PubMed] [Google Scholar]
- Chester, A., Scott, J., Anant, S., and Navaratnam, N. 2000. RNA editing: Cytidine to uridine conversion in apolipoprotein B mRNA. Biochim. Biophys. Acta 1494 1–13. [DOI] [PubMed] [Google Scholar]
- Davies, M.S., Wallis, S.C., Driscoll, D.M., Wynne, J.K., Williams, G.W., Powell, L.M., and Scott, J. 1989. Sequence requirements for apolipoprotein B RNA editing in transfected rat hepatoma cells. J. Biol. Chem. 264 13395–13398. [PubMed] [Google Scholar]
- Driscoll, D.M., Wynne, J.K., Wallis, S.C., and Scott, J. 1989. An in vitro system for the editing of apolipoprotein B mRNA. Cell 58 519–525. [DOI] [PubMed] [Google Scholar]
- Driscoll, D.M., Lakhe-Reddy, S., Oleksa, L.M., and Martinez, D. 1993. Induction of RNA editing at heterologous sites by sequences in apolipoprotein B mRNA. Mol. Cell. Biol. 13 7288–7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgell, M.H., Sims, D.A., Pielak, G.J., and Yi, F. 2003. High precision, high throughput stability determinations facilitated by robotics and a semiautomated tiotrating fluorimeter. Biochemistry 42 7587–7593. [DOI] [PubMed] [Google Scholar]
- Greeve, J., Navaratnam, N., and Scott, J. 1991. Characterization of the apolipoprotein B mRNA editing enzyme: No similarity to the proposed mechanism of RNA editing in kinetoplastid protozoa. Nucleic Acids Res. 13 3569–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardin, R.H. and Sloane, N.J.A. 1993. A new approach to the construction of optimal designs. J. Stat. Plann. Inference 37 339–369. [Google Scholar]
- Hersberger, M. and Innerarity, T.L. 1998. Two efficiency elements flanking the editing site of cytidine 6666 in the apolipoprotein B mRNA support mooring-dependent editing. J. Biol. Chem. 273 9435–9442. [DOI] [PubMed] [Google Scholar]
- Hersberger, M., Patarroyo-White, S., Arnold, K.S., and Innerarity, T.L. 1999. Phylogenetic analysis of the apolipoprotein B mRNA-editing region. J. Biol. Chem. 274 34590–34597. [DOI] [PubMed] [Google Scholar]
- Knott, T.J., Pease, R.J., Powell, L.M., Wallis, S.C., Rall Jr., S.C., Innerarity, T.L., Blackhart, B., Taylor, W.H., Marcel, Y.L., Milne, R., et al. 1986. Complete protein sequence and identification of structural domains of human apolipoprotein B. Nature 323 734–738. [DOI] [PubMed] [Google Scholar]
- Lellek, H., Kirsten, R., Diehl, I., Apostel, F., Buck, F., and Greeve, J. 2000. Purification and molecular cloning of a novel essential component of the apolipoprotein B mRNA editing enzyme-complex. J. Biol. Chem. 275 19848–19856. [DOI] [PubMed] [Google Scholar]
- Mehta, A. and Driscoll, D.M. 2002. Identification of domains in apobec-1 complementation factor required for RNA binding and apolipoprotein-B mRNA editing. RNA 8 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta, A., Kinter, M.T., Sherman, N.E., and Driscoll, D.M. 2000. Molecular cloning of Apobec-1 complementation factor, a novel RNA-binding protein involved in the editing of Apolipoprotein B mRNA. Mol. Cell. Biol. 20 1846–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell, L.M., Wallace, S.C., Pease, R.J., Edwards, Y.H., Knott, T.J., and Scott, J. 1987. A novel form of tissue-specific RNA processing produces apolipoprotein-B48 in intestine. Cell 50 831–840. [DOI] [PubMed] [Google Scholar]
- Richardson, N., Navaratnam, N., and Scott, J. 1998. Secondary structure for the apolipoprotein B mRNA editing site—AU-binding proteins interact with a stem loop. J. Biol. Chem. 273 31707–31717. [DOI] [PubMed] [Google Scholar]
- Scott, J. 1995. A place in the world for RNA editing. Cell 81 833–836. [DOI] [PubMed] [Google Scholar]
- Shah, R.R., Knott, T.J., LeGros, J.E., Navaratnam, N., Greeve, J.C., and Scott, J. 1991. Sequence requirements for the editing of apolipoprotein B mRNA. J. Biol. Chem. 266 16301–16304. [PubMed] [Google Scholar]
- Snider, M.J., Gaunitz, S., Ridgway, C., Short, S.A., and Wolfenden, R. 2000. Temperature effects on the catalytic efficiency, rate enhancement, and transition state affinity of cytidine deaminase, and the thermodynamic consequences for catalysis of removing a substrate “anchor.” Biochemistry 39 9746–9753. [DOI] [PubMed] [Google Scholar]
- Teng, B.-B., Burant, C.F., and Davidson, N.O. 1993. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science 260 1816–1819. [DOI] [PubMed] [Google Scholar]
- Wagner, B.J. and Margolis, J.W. 1993. Thermal stability and activation of bovine lens multicatalytic proteinase complex (proteasome). Arch. Biochem. Biophys. 307 146–152. [DOI] [PubMed] [Google Scholar]
- Wolfenden, R., Snider, M.J., Ridgway, C., and Miller, B. 1999. The temperature dependence of enzyme rate enhancements. J. Am. Chem. Soc. 121 7419–7420. [Google Scholar]
- Woo, J.H., Heo, N.J., Ghim, S.G., and Song, B.H. 2002. Purification and characterization of thermostable cytidine deaminase encoded by the Bacillus caldolyticus cdd gene. Enzyme Microb. Technol. 30 153–160. [Google Scholar]
- Yamanaka, S., Poksay, K.S., Arnold, K.S., and Innerarity, T.L. 1997. A novel translational repressor mRNA is edited extensively in livers containing tumors caused by the transgene expression of the apoB mRNA-editing enzyme. Genes & Dev. 11 321–333. [DOI] [PubMed] [Google Scholar]
- Yang, C.H., Chen, S.H., Gianturco, S.H., Bradley, W.A., Sparrow, J.T., Tanimua, M., Li, W.H., Sparrow, D.A., DeLoof, H., and Rosseneu, M. 1986. Sequence, structure, receptor binding domains and internal repeats of human apolipoprotein B100. Nature 323 738–742. [DOI] [PubMed] [Google Scholar]
- Yin, Y. and Carter Jr., C.W. 1996. Incomplete factorial and response surface methods in experimental design: Yield optimization of tRNATrp from in vitro T7 RNA polymerase transcription. Nucleic Acids Res. 24 1279–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]