

Abstract
A central question in Alzheimer's disease (AD) research is what role β-amyloid peptide (Aβ) plays in synaptic dysfunction. Synaptic activity increases Aβ secretion, potentially inhibiting synapses, but also decreases intraneuronal Aβ, protecting synapses. We now show that levels of secreted Aβ fall with time in culture in neurons of AD-transgenic mice, but not wild-type mice. Moreover, the ability of synaptic activity to elevate secreted Aβ and reduce intraneuronal Aβ becomes impaired in AD-transgenic but not wild-type neurons with time in culture. We demonstrate that synaptic activity promotes an increase in the Aβ-degrading protease neprilysin at the cell surface and a concomitant increase in colocalization with Aβ42. Remarkably, AD-transgenic but not wild-type neurons show reduced levels of neprilysin with time in culture. This impaired ability to secrete Aβ and reduce intraneuronal Aβ has important implications for the pathogenesis and treatment of AD.
Introduction
A defining neuropathological feature of Alzheimer's disease (AD) is the aberrant accumulation of β-amyloid peptide (Aβ). Aβ accumulation can lead to alterations in synapses and memory (Selkoe, 2002; Almeida et al., 2005; Hsieh et al., 2006). The site(s) and mechanism(s) whereby Aβ initiates dysfunction of synapses in AD are of major interest. Secreted, extracellular Aβ has traditionally been viewed as the source of Aβ-induced toxicity to synapses in AD, since addition of Aβ1–42 impairs synaptic function (Cleary et al., 2005; Shankar et al., 2008). In contrast, picomolar levels of extracellular Aβ were recently shown to enhance synaptic plasticity (Puzzo et al., 2008). Remarkably, synaptic activity increases levels of secreted, extracellular Aβ (Kamenetz et al., 2003; Cirrito et al., 2005). Since the default network of the brain is particularly prone to the development of AD, it has been hypothesized that brain regions with the highest baseline metabolic activity are prone to AD because of high amounts of secreted Aβ (Cirrito et al., 2008; Palop and Mucke, 2010). However, it is unclear why such elevated levels of secreted Aβ from default network activity cause problems only with aging. Moreover, at-risk individuals for AD show reduced brain activity decades before clinical symptoms (Reiman et al., 2004), which might predict reduced Aβ secretion. There is increasing support for an alternative scenario focusing on aberrant intracellular accumulation of Aβ within vulnerable neurons (Gouras et al., 2010). In fact, we recently demonstrated that Aβ-related synapse damage and memory impairment in AD-transgenic mice correlated with this intracellular pool of Aβ but not with plaques (Tampellini et al., 2010).
We now provide evidence for reduced Aβ secretion with time in culture in AD-transgenic but not wild-type neurons. Furthermore, we show that synaptic activity is able to reduce levels of intracellular Aβ in AD-transgenic neurons at 12 but not at 19 days in vitro (DIV). We demonstrate that levels of neprilysin are reduced in AD-transgenic but not wild-type neurons with time in culture. Finally, we provide mechanistic evidence consistent with neprilysin leading to degradation of Aβ42 at the cell surface with synaptic activity.
Materials and Methods
Neuronal cultures.
Primary neuronal cultures were prepared from cortices and hippocampi of embryonic day 15 Tg2576 (AD-transgenic) (Hsiao et al., 1996) and wild-type littermate mouse embryos, as described previously (Tampellini et al., 2009). Tg2576 male and wild-type female mice (Jackson Laboratories) were bred to generate the embryos. Mice were used in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Antibodies.
For immunofluorescence, the following antibodies were used.: neprilysin (H-321; Santa Cruz Biotechnology), early endosomal antigen-1 (BD Transduction Laboratories), tumor susceptibility gene 101 (GeneTex), amyloid precursor protein (APP) intracellular domain (AICD; Covance), postsynaptic density-95 (PSD-95; Millipore), Aβ42 (C terminus; Covance). For Western blot, the following antibodies were used: human-specific Aβ/APP 6E10 (Covance), murine and human Aβ/APP 4G8 (Covance), rabbit polyclonal APP (C terminus) 369, phospho-CaMKII (Millipore), total CaMKII (Millipore). Secondary antibodies were conjugated to Alexa Fluor-488 or -546 (Invitrogen) or horseradish peroxidase (GE Healthcare).
Treatments.
For glycine-induced long term potentiation (g-LTP), neurons were treated as described previously (Tampellini et al., 2009). In experiments on neprilysin and Aβ42 surface colocalization during g-LTP stimulation, 50 μm thiorphan was added to neurons during the 15 min incubation with or without glycine; 50 μm thiorphan was then added during the following 1 h chase to prevent Aβ degradation.
ELISA analysis.
To measure Aβ secretion from primary neurons (10 cm dish) at steady state, media were replaced with 1 ml of fresh neurobasal medium and collected after 5 h. To measure Aβ secretion with or without g-LTP, LTP buffer was collected after the 1 h chase. Concentrations of Aβ1–40 and Aβ1–42 were measured using the respective ELISA kits (Biosource) for mouse (wild-type neurons) or human (AD-transgenic neurons) Aβ.
Western blot.
Neuron lysates were prepared as described previously (Tampellini et al., 2009). Membranes were immunoblotted with antibodies 6E10 or 369, and intensities were quantified using Scion Image software (NIH).
Immunofluorescence.
Immunofluorescence and its quantification were performed as described previously (Tampellini et al., 2009). Localization of surface neprilysin with Aβ42 and subcellular markers was determined using a colocalization algorithm (Leica Application Suite 1.8.2 software). To count AICD-positive nuclei, all nuclei were marked with Hoechst stain. Counts of AICD-positive and total nuclei were performed with MetaMorph on 6–10 fields per coverslip at 20× magnification.
Statistical analysis.
Statistical comparisons were made using two-tailed unpaired t tests with significance placed at p < 0.05.
Results
We previously reported progressive intraneuronal Aβ42 accumulation, alterations in endosomal trafficking, and AD-like synapse alterations in AD-transgenic neurons with time in culture, analogous to changes seen in vivo in AD-transgenic mouse brains with aging (Takahashi et al., 2004; Almeida et al., 2005, 2006). In this study, we examined whether time in culture affects Aβ secretion in AD-transgenic neurons. In wild-type neurons, levels of secreted Aβ1–40 and Aβ1–42 remained unchanged between 12 and 19 DIV (Fig. 1A, left). In contrast, in AD-transgenic neurons, levels of secreted Aβ1–40 and Aβ1–42 fell by 33 ± 4% and 39 ± 9%, respectively, between 12 and 19 DIV (Fig. 1A, right). We next examined levels of intraneuronal Aβ with time in culture. There was a 46 ± 17% increase in levels of intraneuronal Aβ42 in AD-transgenic neurites at 19 compared with 12 DIV, as quantified by confocal immunofluorescence microscopy (Fig. 1B). In contrast, levels of intraneuronal Aβ42 were unchanged in wild-type neurons at 19 compared with 12 DIV (Fig. 1C,D). Interestingly, the levels of APPα C-terminal fragments (αCTFs) fell by 52 ± 2% between 12 and 19 DIV in AD-transgenic neurons, whereas levels of APPβ C-terminal fragments (βCTFs) and full-length APP remained unchanged (Fig. 1E, left). Thus, amyloidogenic processing of APP, as reflected by the βCTF/αCTF ratio, increased approximately twofold from 12 to 19 DIV in AD-transgenic neurons. Levels of αCTFs and βCTFs were both comparably decreased in wild-type neurons at 19 compared with 12 DIV (Fig. 1E, right), and therefore the βCTF/αCTF ratio did not change.
Figure 1.
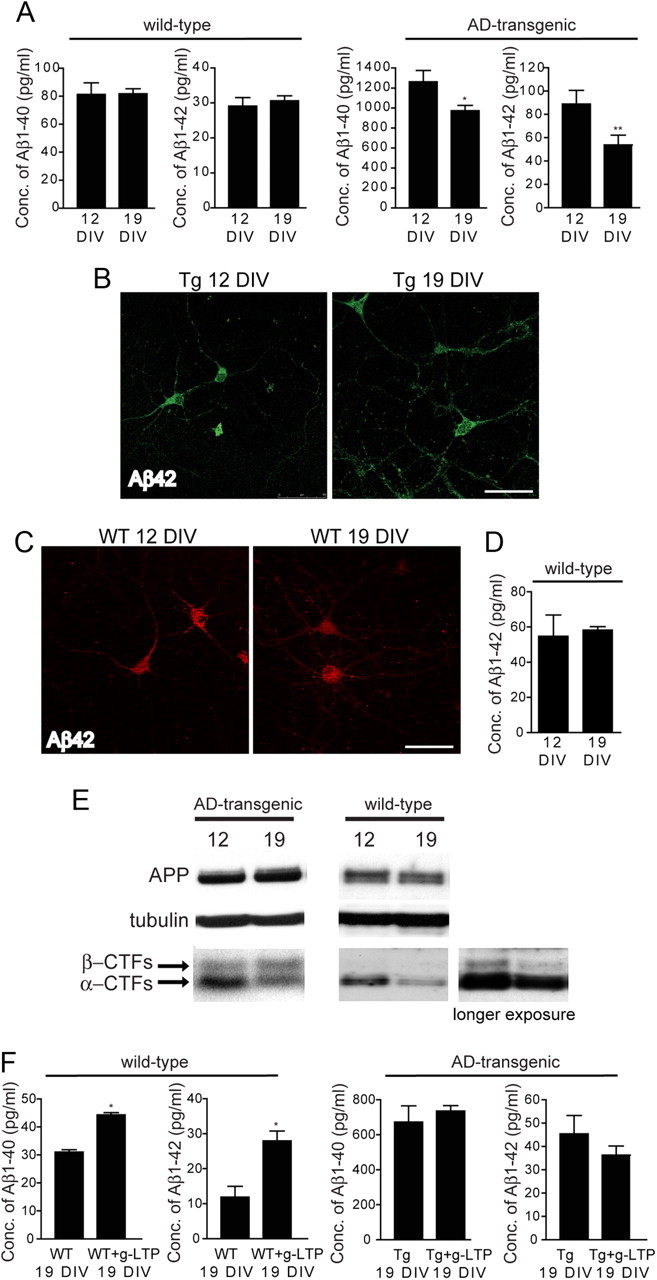
Decreased Aβ secretion in AD-transgenic (Tg) but not wild-type (WT) neurons with time in culture. A, Levels of Aβ1–40 and Aβ1–42 were assayed by ELISA in media of AD-transgenic or wild-type neurons. While wild-type neurons secrete comparable amounts of Aβ peptides at 12 and 19 DIV (n = 6; left), AD-transgenic neurons showed decreased levels of Aβ1–40 and Aβ1–42 in media at 19 compared with 12 DIV (n = 10; *p < 0.05, **p < 0.01; right). B, Levels of intraneuronal Aβ42 were increased by 46 ± 17% in AD-transgenic neurites at 19 compared with 12 DIV (n = 4; p < 0.05). C, D, Levels of intraneuronal Aβ42 were unchanged in wild-type neurons at 19 compared with 12 DIV, as quantified by confocal immunofluorescence (C) and ELISA (D) (n = 4). E, Left, Levels of αCTFs were decreased by 52 ± 2%, while levels of βCTFs and full-length APP were unchanged in AD-transgenic neurons at 19 compared with 12 DIV (n = 4; p < 0.01). Right, Levels of αCTFs were decreased by 32 ± 9% (n = 5; p < 0.05), while levels of βCTFs (longer exposure) showed a trend for a 49 ± 16% decrease (p = 0.074) in wild-type neurons at 19 compared with 12 DIV. F, At 19 DIV, AD-transgenic neurons failed to enhance secretion of both Aβ1–40 and Aβ1–42 during g-LTP (n = 6, right). In contrast, wild-type neurons were still able to increase Aβ1–40 and Aβ1–42 secretion during g-LTP (n = 4; *p < 0.05, left). Conc., Concentration. Scale bars, 50 μm.
It has been shown that Aβ secretion is enhanced by synaptic activation (Kamenetz et al., 2003; Cirrito et al., 2005; Tampellini et al., 2009). Given the fall in steady-state secretion of Aβ in AD-transgenic neurons with time in culture, we investigated whether Aβ secretion was impaired during synaptic activity in AD-transgenic neurons with time in culture. g-LTP was used to stimulate neurons (Ehlers, 2003) at 12 and 19 DIV followed by Aβ ELISA of the conditioned media. We previously demonstrated that g-LTP increases Aβ secretion in AD-transgenic neurons at 12 DIV (Tampellini et al., 2009). We now show that, although g-LTP increases Aβ secretion in wild-type neurons at 19 DIV, it failed to increase Aβ secretion in AD-transgenic neurons at 19 DIV (Fig. 1F).
We reported that synaptic activation reduced intracellular Aβ42 and restored levels of PSD-95 back to wild-type levels in AD-transgenic neurons at 12 DIV (Tampellini et al., 2009). To investigate whether these effects are modulated by aging in vitro, we induced g-LTP in 19 DIV AD-transgenic neurons and quantified levels of intraneuronal Aβ and PSD-95. At 19 DIV, g-LTP reduced levels of intraneuronal Aβ42 by 23 ± 4% in wild-type neurons, but there was no decrease in AD-transgenic neurons as determined by confocal immunofluorescence (Fig. 2A). These data were confirmed by ELISA (Fig. 2B) and Western blot (Fig. 2C) of neuronal lysates. We confirmed that 12 DIV lysates showed decreased levels of intraneuronal Aβ in g-LTP-treated compared with -untreated AD-transgenic neurons (Fig. 2C; lanes 1 and 2, longer exposure). Activity-induced reduction in levels of intraneuronal Aβ promoted recovery of PSD-95 puncta to wild-type levels in AD-transgenic neurons at 12 DIV (Tampellini et al., 2009). In contrast, levels of PSD-95 did not recover back to wild-type levels in g-LTP-treated compared with -untreated 19 DIV AD-transgenic neurons (Fig. 2D), although there was still an increase compared with untreated AD-transgenic neurons.
Figure 2.
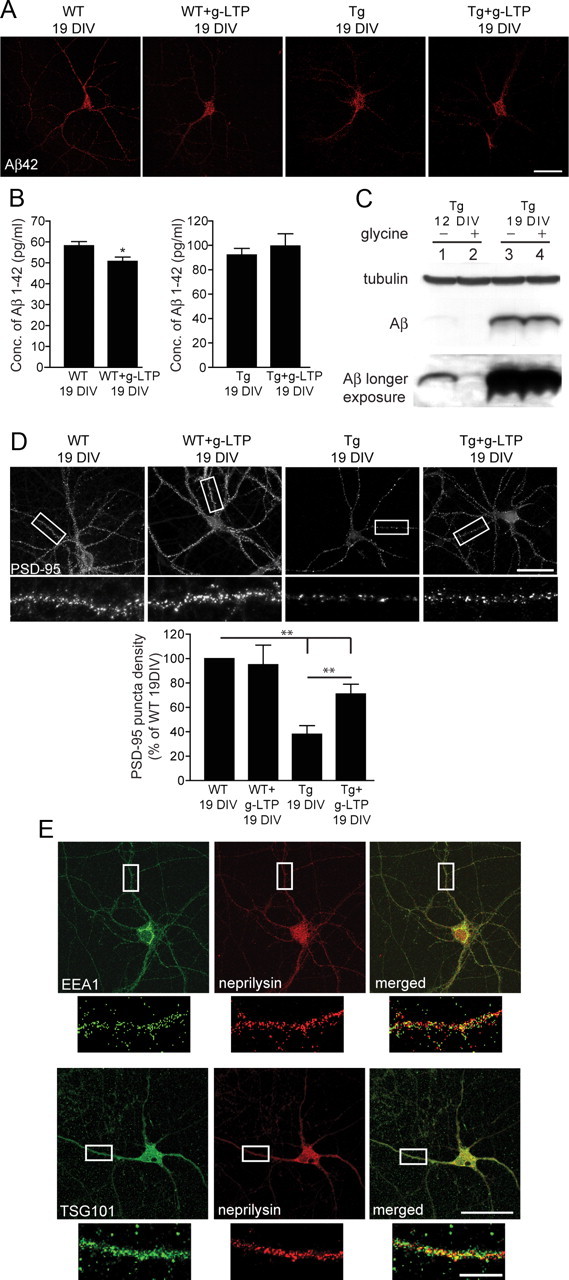
Impaired ability for synaptic activity to reduce intraneuronal Aβ42 and protect synapses in AD-transgenic neurons with time in culture. A, g-LTP reduced levels of intraneuronal Aβ42 by 23 ± 4% in wild-type (WT; left) but not AD-transgenic (Tg; right) neurons at 19 DIV, as determined by confocal immunofluorescence (n = 4; p < 0.01). B, Aβ1–42 ELISA of lysates from g-LTP compared with untreated neurons at 19 DIV revealed reduced levels of Aβ1–42 in wild-type but not in AD-transgenic neurons (n = 4; *p < 0.05). C, Western blot of cell lysates demonstrated reduced levels of intraneuronal Aβ in g-LTP-treated compared with -untreated AD-transgenic neurons at 12 (lanes 1 and 2, longer exposure) but not 19 (lanes 3 and 4) DIV (n = 3). D, PSD-95 puncta increased in g-LTP-activated AD-transgenic neurons at 19 DIV, although they failed to reach wild-type levels (n = 5; **p < 0.01). E, Wild-type neurons (12 DIV) showed a 74 ± 21% greater relative colocalization of neprilysin with the MVB/late endosomal marker tumor susceptibility gene 101 (TSG101) than the early endosomal marker early endosomal antigen-1 (EEA1) (n = 3; p < 0.01). Conc., Concentration. Scale bars: A, D, E, 50 μm; E, inset, 10 μm.
We showed that activity-dependent reduction in intraneuronal Aβ is dependent on the Aβ-degrading protease neprilysin (Tampellini et al., 2009). Data on the subcellular localization of neprilysin showed marked relative colocalization of neprilysin predominantly with late endosomes (Fig. 2E). To investigate the mechanism of neprilysin-dependent Aβ42 clearance with synaptic activity, levels of neprilysin were quantified as a function of synaptic activation. g-LTP did not change total levels of neprilysin (data not shown); however, it did increase levels of surface neprilysin by 37 ± 7% in stimulated compared with unstimulated wild-type neurons (Fig. 3A). Surface neprilysin also increased in stimulated compared with nonstimulated AD-transgenic neurons (Fig. 3B). We next explored whether the increased localization of neprilysin to the cell surface could be responsible for the augmented neprilysin-dependent Aβ42 degradation. To test this hypothesis, g-LTP was induced in AD-transgenic neurons in the presence of the neprilysin inhibitor thiorphan to prevent loss of Aβ42 labeling from degradation. g-LTP increased the relative colocalization of neprilysin with Aβ42 at the cell surface by 54 ± 21% (Fig. 3C).
Figure 3.
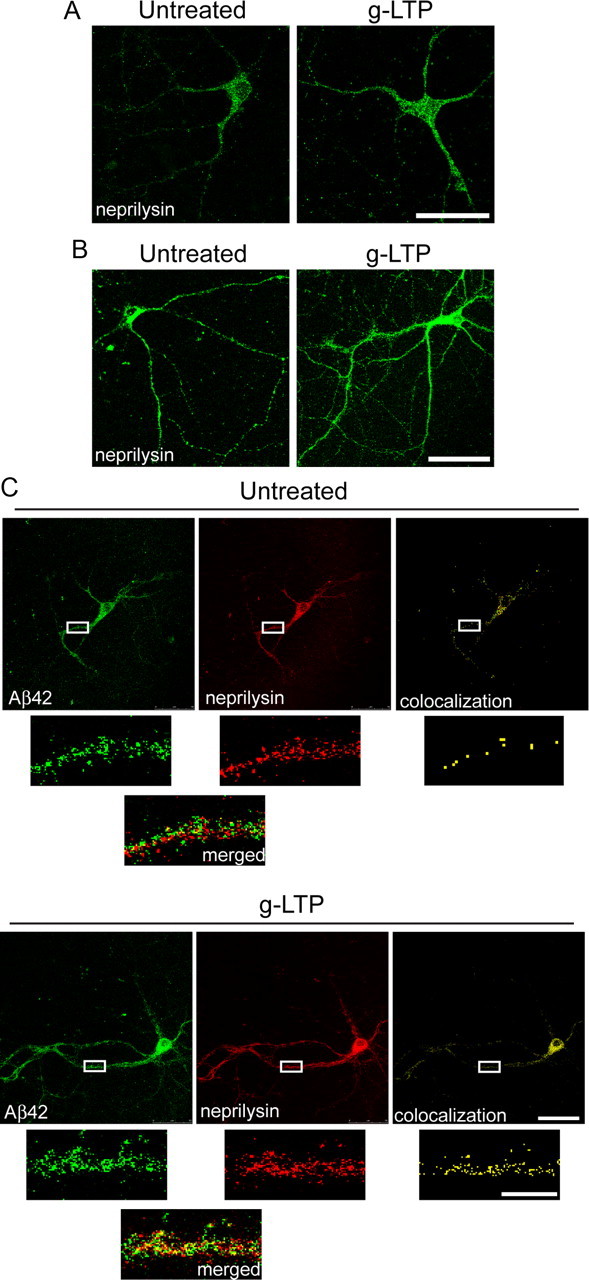
Synaptic activation increases neprilysin surface levels and colocalization with Aβ42. A, g-LTP increased levels of surface neprilysin by 37 ± 7% in g-LTP-treated compared with -untreated wild-type neurons at 12 DIV (n = 6; p < 0.01). B, g-LTP also increased levels of surface neprilysin in g-LTP-treated compared with untreated AD-transgenic neurons at 12 DIV (n = 3; p < 0.01). C, g-LTP increased the relative colocalization of neprilysin with Aβ42 by 54 ± 21% at the cell surface in AD-transgenic neurons at 12 DIV (n = 3; p < 0.01). Scale bars: A–C, 50 μm; C, insets, 10 μm.
Since synaptic activation failed to reduce levels of intraneuronal Aβ in AD-transgenic neurons at 19 DIV, we next investigated whether levels of neprilysin were altered in AD-transgenic neurons with time in culture. Remarkably, levels of neprilysin were decreased by 18 ± 8% in 19 compared with 12 DIV AD-transgenic but not wild-type neurons, as quantified by confocal immunofluorescence (Fig. 4A). Previous work reported that neprilysin is regulated by the AICD (Pardossi-Piquard et al., 2005). To investigate whether reduction of neprilysin might be related to reduced levels of AICD in the nucleus with time in culture, we double-labeled nuclei of 12 and 19 DIV AD-transgenic neurons with Hoechst stain and an AICD-specific antibody. Remarkably, the number of AICD-positive nuclei was decreased by 27 ± 6% in 19 compared with 12 DIV AD-transgenic neurons (Fig. 4B), suggesting reduced synthesis of neprilysin with time in culture.
Figure 4.
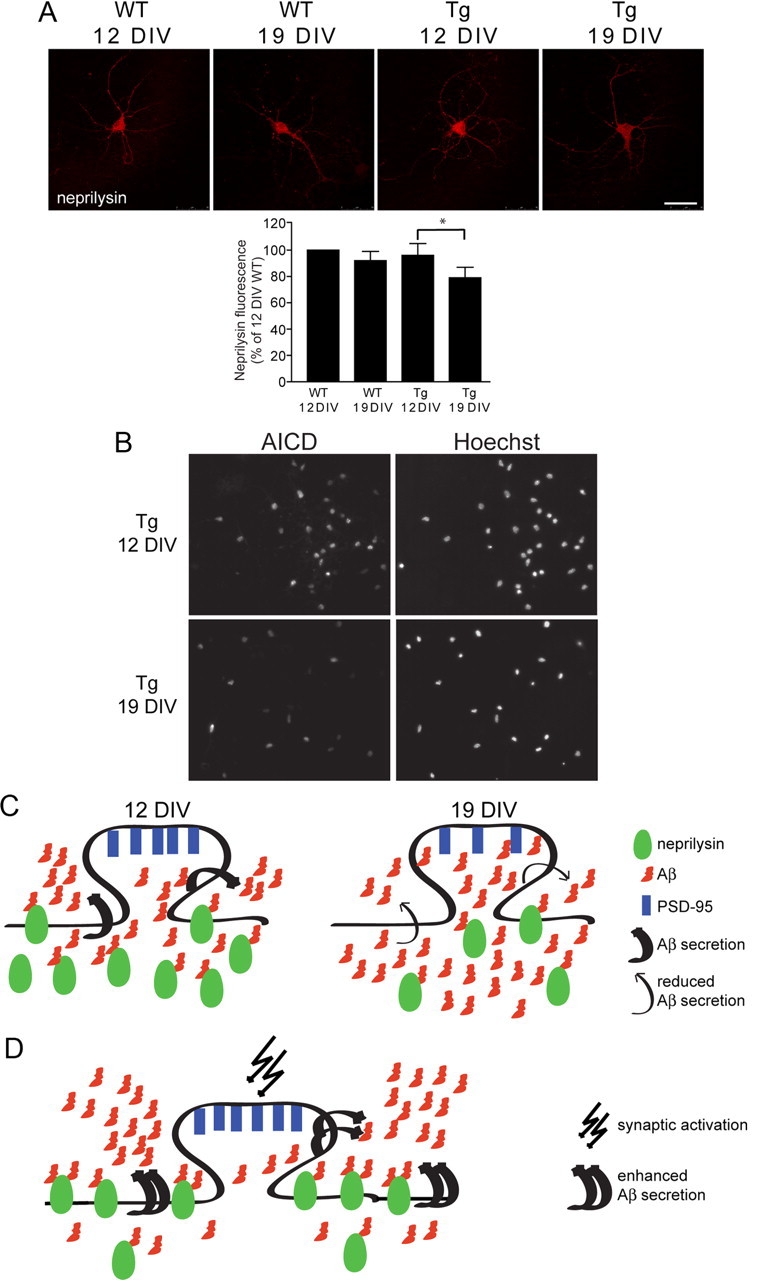
Decreased levels of neprilysin in AD-transgenic neurons with time in culture. A, Levels of neprilysin were decreased by 18 ± 8% in 19 compared with 12 DIV AD-transgenic neurons (n = 4; *p < 0.05; scale bar, 50 μm). In contrast, 19 compared with 12 DIV wild-type neurons did not show a decrease of neprilysin. B, The number of AICD-positive nuclei was decreased by 27 ± 6% in 19 compared with 12 DIV AD-transgenic neurons (n = 3; p < 0.05). C, The ability to secrete Aβ falls with time in culture in AD-transgenic neurons. Neprilysin levels fall with time in culture, suggesting a mechanism for progressive intracellular Aβ accumulation and loss of synaptic proteins (PSD-95) in aged AD-transgenic neurons. D, Synaptic activity enhances Aβ secretion, leads to neprilysin recruitment to the cell surface, and increases colocalization of neprilysin with Aβ42.
Discussion
The data presented here provide evidence for an impaired ability of familial AD-transgenic neurons to efficiently modulate levels of secreted and intraneuronal Aβ with time in culture. We demonstrate that the ability to secrete Aβ falls with time in culture in AD-transgenic but not wild-type neurons. Activity-induced Aβ secretion also falls with time in culture in AD-transgenic but not wild-type neurons. Moreover, with time in culture, AD-transgenic neurons become impaired in their ability to reduce intracellular Aβ and protect synapses in response to synaptic activity. We provide evidence that the mechanism by which synaptic activity reduces intracellular Aβ42 is dependent on the Aβ-degrading protease neprilysin. Synaptic activity leads to recruitment of neprilysin to the cell surface and increased colocalization with Aβ42. Finally, we show that neprilysin levels fall with time in culture in AD-transgenic but not wild-type neurons, providing a mechanism for the failure of synaptic activation to reduce intracellular Aβ in aged AD-transgenic neurons (Fig. 4C,D).
These data have important implications for AD. Decreased neuronal secretion of Aβ with aging in AD could explain previous data that baseline interstitial Aβ42 levels appear to be reduced in older compared with younger AD-transgenic mice (Cirrito et al., 2005). Reduced Aβ42 levels are also seen in CSF in human AD, even in subjects who are cognitively normal but later develop AD (Fagan et al., 2009). The traditional explanation for reduced CSF Aβ42 with AD is that secreted Aβ is decreased by sequestration to extracellular plaques. Decreased secretion would also contribute to reduced Aβ42 levels in CSF or interstitial fluid. Of note, other pathological conditions linked with AD, such as brain injury, also show reduced levels of extracellular Aβ (Brody et al., 2008) and increased intraneuronal Aβ (Gouras et al., 2010).
Reduced Aβ secretion fits well with increasing evidence for early intraneuronal Aβ accumulation with AD pathogenesis, as reported in human AD, Down syndrome, and in AD-transgenic rodents (Gouras et al., 2000; D'Andrea et al., 2001; Wirths et al., 2001; Takahashi et al., 2002; Oddo et al., 2003; Cataldo et al., 2004; Echeverria et al., 2004; Lord et al., 2006; Oakley et al., 2006; Knobloch et al., 2007; Gandy et al., 2010). Intraneuronal Aβ accumulates and oligomerizes preferentially in distal processes even before plaques (Takahashi et al., 2004), and is associated with memory impairment, decreased synaptic plasticity, and subcellular pathology (Mori et al., 2002; Takahashi et al., 2002; Oddo et al., 2003; Almeida et al., 2005; Billings et al., 2005; Tomiyama et al., 2010). Intraneuronal Aβ42 localizes and accumulates preferentially in late endosomes, including multivesicular bodies (MVBs) (Takahashi et al., 2002; Langui et al., 2004; Almeida et al., 2006). Interestingly, neprilysin also localizes to late endosomes/MVBs (Fig. 2E). We show that neprilysin significantly relocates to the cell surface during synaptic activation. Neprilysin is a transmembrane neutral endopeptidase with the catalytic site at the C terminal side, which can be either extracellular or luminal. Since neprilysin works best at neutral pH, the cell surface would be the optimal location for it to degrade Aβ42. In fact, we observed that colocalization of neprilysin and Aβ42 was increased at the cell surface with synaptic activation. This pool of Aβ42 could derive from amyloidogenic APP processing occurring within synaptic endosomes with synaptic activity (Cirrito et al., 2008; Tampellini et al., 2009), followed by trafficking of Aβ to the cell surface (Rajendran et al., 2006). The finding that neprilysin normally declines with aging in synaptic layers of wild-type mouse brain (Iwata et al., 2002) supports the theory that it plays a critical role in synaptic accumulation of Aβ with aging and AD. We show that neprilysin is reduced in AD-transgenic but not wild-type neurons with time in culture, although it is possible that a decline would eventually occur later in wild-type neurons. Reduced neprilysin in AD-transgenic neurons with time in culture can explain the impaired ability to reduce intraneuronal Aβ, in particular Aβ42, with synaptic activity, since we previously reported the inability of synaptic activity to reduce Aβ42 in neprilysin knock-out neurons or when neprilysin activity was blocked by treatment with thiorphan (Tampellini et al., 2009). In addition, it has been shown that extracellular Aβ can upregulate intracellular Aβ (Yang et al., 1999; Tampellini et al., 2009), which could lead to a vicious cycle of Aβ-induced elevation and synapse damage.
Overall, this study provides novel evidence for reduced Aβ secretion and impaired ability to reduce intraneuronal Aβ with time in culture in AD-transgenic neurons, and further underscores the important role of neprilysin in regulating neuronal Aβ.
Footnotes
This work was supported by an Alzheimer's Association New Investigator Award (D.T.) and Zenith Award (G.K.G.), National Institute of Health Grants AG027140, AG028174, AG09464 (G.K.G.), and AG020729 (M.T.L.), and the Strong Research Environment Multipark (Multidisciplinary research in Parkinson's disease at Lund University). We thank Fangmin Yu and Eleanor Reilly for technical support.
The authors declare no competing financial interests.
References
- Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, Gouras GK. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis. 2005;20:187–198. doi: 10.1016/j.nbd.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Almeida CG, Takahashi RH, Gouras GK. Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J Neurosci. 2006;26:4277–4288. doi: 10.1523/JNEUROSCI.5078-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Brody DL, Magnoni S, Schwetye KE, Spinner ML, Esparza TJ, Stocchetti N, Zipfel GJ, Holtzman DM. Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science. 2008;321:1221–1224. doi: 10.1126/science.1161591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Petanceska S, Terio NB, Peterhoff CM, Durham R, Mercken M, Mehta PD, Buxbaum J, Haroutunian V, Nixon RA. Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol Aging. 2004;25:1263–1272. doi: 10.1016/j.neurobiolaging.2004.02.027. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, Holtzman DM. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008;58:42–51. doi: 10.1016/j.neuron.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- D'Andrea MR, Nagele RG, Wang HY, Peterson PA, Lee DH. Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer's disease. Histopathology. 2001;38:120–134. doi: 10.1046/j.1365-2559.2001.01082.x. [DOI] [PubMed] [Google Scholar]
- Echeverria V, Ducatenzeiler A, Dowd E, Jänne J, Grant SM, Szyf M, Wandosell F, Avila J, Grimm H, Dunnett SB, Hartmann T, Alhonen L, Cuello AC. Altered mitogen-activated protein kinase signaling, tau hyperphosphorylation and mild spatial learning dysfunction in transgenic rats expressing the beta-amyloid peptide intracellularly in hippocampal and cortical neurons. Neuroscience. 2004;129:583–592. doi: 10.1016/j.neuroscience.2004.07.036. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Head D, Shah AR, Marcus D, Mintun M, Morris JC, Holtzman DM. Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Ann Neurol. 2009;65:176–183. doi: 10.1002/ana.21559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandy S, Simon AJ, Steele JW, Lublin AL, Lah JJ, Walker LC, Levey AI, Krafft GA, Levy E, Checler F, Glabe C, Bilker WB, Abel T, Schmeidler J, Ehrlich ME. Days to criterion as an indicator of toxicity associated with human Alzheimer amyloid-beta oligomers. Ann Neurol. 2010;68:220–230. doi: 10.1002/ana.22052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouras GK, Tampellini D, Takahashi RH, Capetillo-Zarate E. Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer's disease. Acta Neuropathol. 2010;119:523–541. doi: 10.1007/s00401-010-0679-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata N, Takaki Y, Fukami S, Tsubuki S, Saido TC. Region-specific reduction of A beta-degrading endopeptidase, neprilysin, in mouse hippocampus upon aging. J Neurosci Res. 2002;70:493–500. doi: 10.1002/jnr.10390. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Knobloch M, Konietzko U, Krebs DC, Nitsch RM. Intracellular Abeta and cognitive deficits precede beta-amyloid deposition in transgenic arcAbeta mice. Neurobiol Aging. 2007;28:1297–1306. doi: 10.1016/j.neurobiolaging.2006.06.019. [DOI] [PubMed] [Google Scholar]
- Langui D, Girardot N, El Hachimi KH, Allinquant B, Blanchard V, Pradier L, Duyckaerts C. Subcellular topography of neuronal Abeta peptide in APPxPS1 transgenic mice. Am J Pathol. 2004;165:1465–1477. doi: 10.1016/s0002-9440(10)63405-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord A, Kalimo H, Eckman C, Zhang XQ, Lannfelt L, Nilsson LN. The arctic Alzheimer mutation facilitates early intraneuronal Abeta aggregation and senile plaque formation in transgenic mice. Neurobiol Aging. 2006;27:67–77. doi: 10.1016/j.neurobiolaging.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Mori C, Spooner ET, Wisniewsk KE, Wisniewski TM, Yamaguch H, Saido TC, Tolan DR, Selkoe DJ, Lemere CA. Intraneuronal Abeta42 accumulation in Down syndrome brain. Amyloid. 2002;9:88–102. [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardossi-Piquard R, Petit A, Kawarai T, Sunyach C, Alves da Costa C, Vincent B, Ring S, D'Adamio L, Shen J, Müller U, St George Hyslop P, Checler F. Presenilin-dependent transcriptional control of the Abeta-degrading enzyme neprilysin by intracellular domains of betaAPP and APLP. Neuron. 2005;46:541–554. doi: 10.1016/j.neuron.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Puzzo D, Privitera L, Leznik E, Fà M, Staniszewski A, Palmeri A, Arancio O. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008;28:14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, Simons K. Alzheimer's disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A. 2006;103:11172–11177. doi: 10.1073/pnas.0603838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia. Proc Natl Acad Sci U S A. 2004;101:284–289. doi: 10.1073/pnas.2635903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA, Gouras GK. Oligomerization of Alzheimer's beta-amyloid within processes and synapses of cultured neurons and brain. J Neurosci. 2004;24:3592–3599. doi: 10.1523/JNEUROSCI.5167-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tampellini D, Rahman N, Gallo EF, Huang Z, Dumont M, Capetillo-Zarate E, Ma T, Zheng R, Lu B, Nanus DM, Lin MT, Gouras GK. Synaptic activity reduces intraneuronal Abeta, promotes APP transport to synapses, and protects against Abeta-related synaptic alterations. J Neurosci. 2009;29:9704–9713. doi: 10.1523/JNEUROSCI.2292-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tampellini D, Capetillo-Zarate E, Dumont M, Huang Z, Yu F, Lin MT, Gouras GK. Effects of synaptic modulation on beta-amyloid, synaptophysin, and memory performance in Alzheimer's disease transgenic mice. J Neurosci. 2010;30:14299–14304. doi: 10.1523/JNEUROSCI.3383-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomiyama T, Matsuyama S, Iso H, Umeda T, Takuma H, Ohnishi K, Ishibashi K, Teraoka R, Sakama N, Yamashita T, Nishitsuji K, Ito K, Shimada H, Lambert MP, Klein WL, Mori H. A mouse model of amyloid {beta} oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J Neurosci. 2010;30:4845–4856. doi: 10.1523/JNEUROSCI.5825-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirths O, Multhaup G, Czech C, Blanchard V, Moussaoui S, Tremp G, Pradier L, Beyreuther K, Bayer TA. Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett. 2001;306:116–120. doi: 10.1016/s0304-3940(01)01876-6. [DOI] [PubMed] [Google Scholar]
- Yang AJ, Chandswangbhuvana D, Shu T, Henschen A, Glabe CG. Intracellular accumulation of insoluble, newly synthesized abetan-42 in amyloid precursor protein-transfected cells that have been treated with Abeta1–42. J Biol Chem. 1999;274:20650–20656. doi: 10.1074/jbc.274.29.20650. [DOI] [PubMed] [Google Scholar]