

Abstract
Amyloid beta (Aβ) is a major causative agent of Alzheimer disease (AD). This neurotoxic peptide is generated as a result of the cleavage of the Amyloid-Precursor-Protein (APP) by the action of β-secretase and γ-secretase. The neurotoxicity was previously thought to be the result of aggregation. However, recent studies suggest that the interaction of Aβ with numerous cell surface receptors such as N-methyl-D-aspartate (NMDA), receptor for advanced glycosylation end products (RAGE), P75 neurotrophin receptor (P75NTR) as well as cell surface proteins such as the cellular prion protein (PrPc) and heparan sulfate proteoglycans (HSPG) strongly enhances Aβ induced apoptosis and thereby contributes to neurotoxicity. This review focuses on the molecular mechanism resulting in Aβ-shedding as well as Aβ-induced apoptotic processes, genetic risk factors for familial AD and interactions of Aβ with cell surface receptors and proteins, with particular emphasis on the cellular prion protein. Furthermore, comparisons are drawn between AD and prion disorders and the role of laminin, an extracellular matrix protein, glycosaminoglycans and the 37 kDa/67 kDa laminin receptor (LRP/LR) have been highlighted with regards to both neurodegenerative diseases.
Key words: Alzheimer disease, amyloid β, apoptosis, 37 kDa/67 kDa laminin receptor, prion proteins
Alzheimer disease (AD), primarily defined by psychiatrist Alois Alzheimer in 1906, is a neurodegenerative disorder and currently exhibits a prevalence that “doubles approximately every five years from 0.5% at the common age of onset-65 years old.”1 This disease is the most common form of dementia afflicting the elderly and at present affects in excess of 37 million people globally2 and it is predicted that 100 million people will be living with the disease by 2050.3
AD has received mounting scientific interest and has stimulated tireless research endeavours not only due to the complex mechanism by which it is caused; the multitude of contributing factors and contradictions which have arisen between hypotheses and acquired results, but also due to the rise in life expectancies4 owing to the advent of modern medicine, which has socio-economic implications particularly in terms of strain placed upon national health systems.
Clinical Symptoms of Alzheimer Disease
AD related symptoms have been reported to occur in three stages. The initial symptoms include cognitive dysfunction, impaired short-term memory as well as language impairment and behavioural disturbances including paranoia, confusion, hallucinations and aggression.4,5 Ultimately, AD patients exhibit motoric disturbances and a loss in co-ordination and encounter difficulties in performing simple daily tasks. Disease progression, although variable in the chronology of symptom development and severity, is proposed to occur over a decade.3
Neuropathology
Neuritic plaques are comprised primarily of amyloid β (Aβ) peptides, predominantly the 42 amino acid isoform designated Aβ42 as it exhibits a greater propensity for aggregation6 owing to an increased number of non-polar hydrophobic amino acid residues—isoleucine and alanine. These slow forming extracellular protein aggregates are localized around the synaptic terminals and axons of neurons in the limbic and associated cortices of the brain,7 occurring first in the fronto cortex and progressing to the cortical regions.8
Neurofibrillary tangles are intracellular clusters of straight filaments of approximately 10 nm which appear helical when viewed under the electron microscope.3 The filaments are composed of hyperphosporylated τ protein (a microtubule associated protein), which is also associated with the onset of frontotemporal dementia.9 The abnormal phosphorylation of τ, which results in pathological consequences, may in turn be dependent on the presence of Aβ oligomers. It has been proposed that Aβ triggers p35 gene activation; the protein product is auto-catalytically cleaved to yield a p25 fragment which functions both as a transcription activator of the gene encoding for cyclin-dependent kinase 5 (Cdk5)—a τ phosphorylating kinase.10,11 In addition, p25 forms a cytoplasmic complex with the cdk5 protein.
τ neurofibrillary tangles have been reported to occur prior to Aβ plaque deposition and the tangle load has been shown to correlate more closely to the severity of the dementia than the plaque level.12–14 However, these tangles are not solely associated with AD and are instead characteristic pathological features of an array of neurodegenerative diseases and other disorders such as subacute sclerosing panencephalitis.4 Aβ neuritic plaques, despite being present in healthy and AD diseased brains as well as patients suffering from dementia with Lewy bodies (DLB), are not the major pathological traits of any other disease.4 Furthermore, mutations in the τ gene cause frontotemporal dementia as opposed to AD15 whereas mutations which result in an overproduction of Aβ42 (as shall be detailed below) cause familial AD. As a result hereof, it has been hypothesized that Aβ peptides are the central causative agents in AD.
The Molecular Basis of Alzheimer Disease
The amyloid precursor protein (APP) (Fig. 1) is a ubiquitous single transmembrane protein expressed in the cells of interest in neurodegenerative diseases namely neurons and astrocytes. The protein is encoded by an APP gene located on chromosome 2116 and owing to alternative m-RNA splicing is present in three isoforms: APP695, APP751 and APP770,17 the former being expressed in higher levels in neuronal tissues.18 APP is post-translationally modified through the addition of N and O-linked sugar moieties. APP, largely through the action of sAPPα (a cleavage product of the non-amyloidogenic pathway), is vital for normal brain development, long-term potentiation (LTP) and learning.19 APP is rapidly metabolized within 45–60 min of expression via two pathways namely: a non-pathogenic non-amyloidogenic pathway and a Aβ synthesizing amyloidogenic pathway (Fig. 2). It must be emphasized that both pathways are present in normal healthy individuals and AD is caused either through the disproportionate favoring of the amyloidogenic cleavage or the retardation of the Aβ turnover rate.17,20
Figure 1.

Schematic representation of the APP and the enzymatic cleavage sites located within the amyloid β sequence. (A) This transmembrane protein may be present in multiple isoforms APP695, APP751 and APP770, the latter is represented here. The regions of interest depicted in the diagram are: A signal peptide (yellow box) comprising 17 amino acid residues which ensures the protein is correctly transported to the cell surface; a 56 amino acid Kunitz-type serine protease inhibitor domain (KPI-green box) and the Aβ sequence.4 In addition, the sites of post-translational modifications such as N- and O-linked sugars (NCH2O and OCH2O), phosphate (PO4) and sulphate (SO4) groups are shown. (B) The 40–42 amino acid Aβ sequence is highlighted above-the first 28 amino acid residues are polar and located on the extracellular domain of APP whilst the remaining residues are located within the 23 aa APP transmembrane domain and are non-polar. The enzymatic cleavage sites of β secretase, α secretase and γ secretase are depicted (Adapted from reference 112).
Figure 2.

The proteolytic processing of the APP and its cleavage products. The amyloid precursor protein may be metabolized through two pathways. The first, depicted on the left, is termed the non-amyloidogenic pathways. This pathway involves the enzymatic cleavage by an α-secretase (presumably a member of the ADAM family) after residue 687, followed by γ-secretase mediated cleavage of the remaining carboxyl terminal fragment (CTF83), generating sAPPα, p3 and AICD (not shown), respectively. The amyloidogenic pathway (right) entails β-secretase mediated cleavage after residue 671, thereby releasing sAPPβ and the resultant CTF99 is cleaved by γ-secretase which results in Aβ shedding. The miscellaneous receptor, which may influence the process or serve as a receptor for either Aβ cleavage products, is hypothesized to be the 37 kDa/67 kDa Laminin Receptor Precursor/Laminin Receptor (LRP/LR) (adapted from reference 32).
Amyloid Precursor Protein Processing
The non-amyloidogenic pathway is initiated by the α-secretase mediated cleavage of APP between residues APP687 and APP688,21 which resides in the Aβ sequence thereby precluding Aβ formation and shedding. The released amino terminal ectodomain fragment is termed sAPPα (Fig. 2). The remaining 83aa carboxyl terminal fragment (CTF83) is subsequently cleaved by γ-secretase thereby generating a soluble 3 kDa p3 fragment and a 57–59 aa amyloid intracellular domain (AICD) (Fig. 2) which exhibits transcriptional regulatory abilities as shall be discussed below (Fig. 3).
Figure 3.
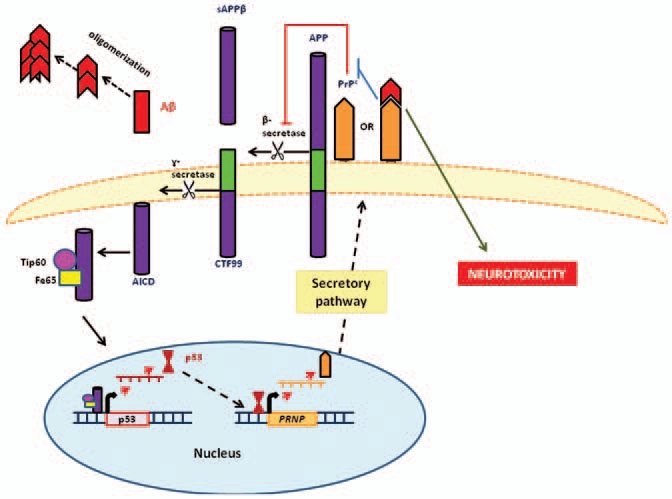
Feedback loop for the prion protein (PrPc) mediated regulation of APP. The amyloid intracellular domain (AICD) amyloidogenic APP cleavage product, indirectly upregulates prion protein (PrPc) expression through p53 gene activation. PrPc consequently hampers β-secretase (BACE1) activity thereby reducing APP amyloidogenic processing and Aβ synthesis. In the presence of Aβ-oligomers, which preferentially bind to PrPc, PrPc is unable to inhibit β-secretase (BACE1) activity. This reduces the degree of regulatory control exerted on the amyloidogenic process resulting in increased levels of potentially toxic Aβ oligomers (adapted from reference 81).
In contrast to the mechanism detailed above, the amyloidogenic pathway is initiated by β-secretase cleavage of APP (Fig. 2). This cleavage occurs 16 aa residues toward the amino terminal of the α-secretase cleavage site, between residues APP671 and APP672,18 generating sAPPβ. The resultant CTF99 is also subjected to γ-secretase cleavage which produces a 4 kDa soluble Aβ monomer and AICD (Fig. 2). It is noteworthy to add that γ-secretase cleavage at positions APP712 and APP713 generate Aβ40 while cleavage at APP714 results in Aβ42 (Fig. 1).22 Furthermore, as has been mentioned above, Aβ generation is a normal physiological process and Aβ levels in the cerebrospinal fluid and plasma of healthy individuals are in the 3–8 nM range23 and 500 pM respectively.24
Associated Enzymes
α-Secretase.
The proteolytic abilities of this plasma membrane protein are attributable to the activities of multiple disintegrin and metalloproteinases (ADAMs) and ADAM 10, 17 and 19 are suggested to be of particular importance.25–27
β-Secretase.
The predominant β-secretase in neural tissue is the β-site APP cleaving enzyme 1 (BACE1), although BACE2 isoforms are also present to a lesser extent.28 The expression of these enzymes is upregulated in response to cellular stress in the form of oxidative stress, ischemia and the depletion of utilisable energy29,30 and thus in AD patients as such stresses are augmented in diseased tissues. It must be noted that β-secretase is located in lipid raft domains of the plasma membrane and requires glycosaminoglycans to mediate effective cleavage,1 characteristics analogous to those of the 37 kDa/67 kDa Laminin Receptor Precursor/Laminin Receptor (LRP/LR).31,32
γ-Secretase.
γ-Secretase is a protein complex consisting of at least four proteins namely: presenilin 1 or 2 (aspartyl proteases which perform the catalytic function), nicastin (Nct), anterior pharynx defective-1 (Aph-1)/A and presenilin enhancer 2 (Pen 2).33 Nct and Aph-1 are required for substrate recognition34 which allow the enzyme to cleave in the order of 50 substrates.35 The enzymatic components of this complex, the presenilins are transmembrane proteins with 6–9 transmembrane domains and both the amino and carboxy termini are cytoplasmic. Mutations in the PSEN genes encoding these proteins result in familial AD which is characterized by the early onset of typical symptoms, as shall be detailed below.
Familial Alzheimer Disease
AD may be inherited as an autosomal dominant disease, with the disease following Mendelian inheritance patterns in merely 5% of AD cases.3 Familial AD is characterized by the onset of pathological symptoms before the age of 6536 and is the result of mutations in three genetic loci (genes) namely: APP on chromosome 21 and the presenilin encoding genes (PSEN1 and PSEN2) on chromosomes 14 and 1, respectively.37
APP gene.
Twenty three missense mutations and two recessive mutations (a duplication and a trinucleotide deletion) have been reported in references 36 and 38 at various locations. The common consequence of all APP mutations is enhanced Aβ generation (be it Aβ40, Aβ42 or both)—the mechanism by which this is achieved, be it through the upregulation of APP expression, α-secretase inhibition or β-secretase stimulation, varies with each mutation.39
PSEN 1 gene.
Within this gene 178 mutations, largely missense, have been identified and these are the most prevalent AD-related mutations in the general public.36 The mutations are mainly localized near the transmembrane domains and hydrophilic loops.40 β-secretase cleavage is affected by such mutations often resulting in an increased Aβ42/Aβ40 ratio either through enhanced synthesis of the more hydrophobic isoform24 or a reduction in Aβ40 generation.41
PSEN 2 gene.
Mutations within this gene are less common as only 14 mutations have been reported to date.36 However, the consequences of such mutations are indistinguishable from those described above with regards to PSEN1 mutations.
Thus, all mutations leading to familial AD share a common modus operandi—each mutation serves to significantly augment the concentration of Aβ42 isoform thereby providing additional support for the prevailing hypothesis that Aβ peptides are the causative agents of AD and consequently the central focus in the development of AD therapeutics.
Other Genetic Risk Factors
A single risk factor, the ε4 allele of the apolipoprotein E (APOE) gene on chromosome 19q is the only susceptibility factor which has consistently been reported to predispose people to late-onset AD.42 However, recent genetic studies have revealed an array of potential risk genes at specific chromosomal regions, particularly those on chromosomes 6, 9, 10, 12 and 21.43 Such novel risk genes include CLU which encodes for clusterin and CR1 encoding for the complement C3b protein and PICALM encoding for the phosphatidylinositol binding clathrin assembly protein.36 These genes are suggested to function in Aβ transport (escort) and endocytosis (CLU), Aβ clearance (CR1)44 and APP recycling (PICALM). Moreover, the PRNP gene encoding for the cellular prion protein (PrPc) and the possible Met129Val homozygosity thereby associated have also been described as potential risk factors.45,46
Focusing on the Causative Agent—the Aβ Peptide
Aβ peptides are amphiphilic 38–43 amino acid peptides34 as the first 28 aa residues are polar and the remaining residues are non-polar in nature8 (Fig. 1). As a result hereof, the peptides exhibit great differences in polarity at neutral pH and thus show a high propensity for aggregation.47,48 The unfolded Aβ monomers may associate to form dimers, trimers and higher order oligomers which may ultimately form insoluble fibrils and plaques—a process termed nucleation dependent polymerization. This process in vitro is highly sensitive to experimental conditions and in vivo, such oligomers are predominantly dimeric as opposed to larger insoluble oligomers which are produced synthetically.49
Revisiting the Aβ Model
Neuritic plaques were initially regarded as the basis of AD. However, this is no longer the prevailing thought due to poor correlation between the degree of dementia and the anatomical plaque load.50 The presence of said plaques in healthy individuals;51 the onset of cognitive impairment in mice prior to plaque deposition52 and the presence of intracellular Aβ oligomers suggesting that the extracellular deposits may not be the sole contributors to disease establishment. Current hypotheses suggest that enhanced levels of Aβ42 or elevated Aβ42/Aβ40 peptide ratios increase the probability of developing Alzheimer disease (in line with the effects of the mutations stated above).8 However, a remarkably larger body of research provides evidence for the latest hypothesis that neurotoxicity is mainly associated with Aβ oligomers, particularly dimers.53
Effects of Toxic Aβ Oligomers
It is suspected that Aβ oligomers are primarily deposited at small blood vessels and interactions between the peptides and receptors of the endothelial cells of the blood vessel wall activates inflammatory responses and cytokine release. These may ultimately lead to the destruction of the blood vessels, consequently reducing oxygen supply to neurons (rendering them ischemic) and finally resulting in neuronal death.54 In the presence of Aβ oligomers neural associated-cells such as astrocytes and microglia would in addition activate the afore stated responses and the complement cascade.54 Furthermore, Aβ-cell surface receptor interactions have been reported to cause oxidative damage by enhancing the synthesis of reactive oxygen species (ROS) such as superoxide and hydrogen peroxide54 which consequently contributes to protein degradation and lipid (such as myelin) oxidation, thereby slowing the rate of signal transmission55 as well as the consolidation of new information, retrieval of memories and motor functions. Furthermore, these Aβ-receptor interactions induce the activation of apoptotic pathways and are thus an additional mechanism through which Aβ mediates its neurotoxicity. Moreover, the interaction of Aβ oligomers with cell-surface receptors such as metabotrophic glutamate receptors (mGluR5)1,56 and N-methyl-D-aspartate receptors (NMDA),1,57 disrupts ion homeostasis-particularly with respect to calcium ions (Ca2+). These interactions allow increased Ca2+ to enter neurons, resulting in excitotoxicity, synaptic dysfunction and neuronal death.58
Aβ Interactions
Aβ oligomers may be directly incorporated into the plasma membrane,59–62 neuronal membranes and those of lysosomes, Golgi apparatus and endoplasmic reticulum.63 In the former, Aβ is located in the lipid raft domains (the same cellular location as of 37 kDa/67 kDa LRP/LR) and disrupts membrane structure and function by forming Ca2+ channels which deregulates homeostasis.
Aβ interacts with a variety of proteins on the surfaces of neuronal and glial cells (Table 1). These include APP, NMDA receptors, integrins, α7 nicotininc acetylcholine receptors (α7nAchR), p75 neurotrophin receptors (p75NTR), collagen-like Alzheimer amyloid plaque component precursor/collagen type XXV (CLAC-P/ColXXV), the receptor for advanced glycosylation end products (RAGE), serpin-enzyme complex receptor (SEC-R), insulin receptors, scavenger receptors on microglial cells and heparin sulphate proteoglycans (HSPGs).63 The outcome of some of these Aβ-receptor/protein binding interactions are neuroprotective while the majority are toxic.63 It is, however, noteworthy to add that the majority of these receptors—AAP, p75NTR, CLAC-P/ColXXV, RAGE, SEC-R and integrins—are transmembrane receptors63 as is the 37 kDa/67 kDa LRP/LR. Furthermore, τ has been proposed to be central in mediating Aβ toxicity.64,65
Table 1.
Membrane associated proteins to which Aβ may bind and result in neurotoxic consequences63
| Amyloid Precursor Protein (APP) |
| N-methyl-D-aspartate receptor (NMDA-R) |
| α7 Nicotininc Acetylcholine receptor (α7nAChR) |
| P75 Neurotrophin receptor (P75NTR) |
| Integrins (particularly α5β1) |
| Receptor for advanced glycosylation end products (RAGE) |
| Insulin receptor |
| Formyl peptide receptor-like-1 (FPRL1) |
| Scavenger receptors-classes A, B, BI |
| Heparan Sulfate Proteoglycans |
Laminin, a 850 kDa glycoprotein component of the basal membrane, functions in cell adhesion and basement membrane assembly. This cross-shaped glycoprotein is composed of three disulfide-bonded chains: α (of which there are five isoforms ranging from 200–400 kDa), β (a single 220 kDa isoform) and γ (a single isoform of 210 kDa).66 The α-chain contains the Aβ binding site and the protein has also been reported to bind, via its IKAV neurite inducing site, to APP.67 The Aβ-laminin interaction has been demonstrated to promote neurite outgrowth68 and inhibit fibrillogenesis.69
The Prion Protein: A Central Factor in Alzheimer Disease
Prion proteins, encoded by the PRNP gene located on chromosome 20,70 are cellular proteinaceous particles which have been implicated in pathogenesis, particularly with regards to the fatal neurodegenerative diseases termed transmissible spongiform encephalopathies (TSEs).31,32,113 Two forms of the protein exist: the normal cellular prion protein (PrPc) and the infectious form (PrPSc), the latter is considered the causative agent of TSEs.71 PrPc is composed of 250 amino acids containing an N-terminal domain and N-linked oligosaccharides, four proline and glycine-rich octarepeat regions and a C-terminal glycosyl-phosphatidylinositol (GPI) anchor through which the protein is bound to the lipid raft domains of the plasma membrane. PrPc constitutively cycles between the cell membrane and intra-cellular compartments via the endocytic pathway—a process mediated by the 37 kDa/67 kDa Laminin Receptor Precursor,72 a high affinity receptor for PrPc73 and PrPSc.72,74–76 The prion variants share a common amino acid sequence and differ primarily with regards to secondary structure (PrPSc exhibits greater β sheet proportion than PrPc-consistent with its higher aggregation propensity) and proteinase K digestion (PrPSc resists degradation while PrPc does not).71,77
PrPc has a multitude of physiological functions70 and the majority of these functions are dependent on the interaction of the prion protein with an array (>70) of proteins. These include ApoE, APP, HSPGs, RAGE, p75 NTR, amyloid β (proteins of importance with regards to Alzheimer disease as discussed above) and the 37 kDa/67 kDa laminin receptor precursor (LRP/LR).73 The specific PrPc-protein interactions and the functions thereof are beyond the scope of this paper but have been recently reviewed in reference 70. Owing to the potential role of PrPc in Alzheimer disease and the central role of the PrPc and PrPSc-LRP/LR interaction in TSEs31 it may be proposed that a link between the two neurodegenerative diseases and more specifically the amyloid β and LRP/LR components may exist and it is plausible that PrPc may facilitate possible AB-LRP/LR interactions.
Prion Protein's Influence on Alzheimer Disease
The cellular prion protein (PrPc) has been identified as a high affinity receptor for Aβ oligomers78 and residues PrP23–27 and PrP95–110 serve as Aβ binding sites.79 The receptor specifically binds to Aβ oligomers as opposed to Aβ monomers or fibrils.79 As the oligomers are considered to be the neurotoxic species, a PrPc-Aβ interaction may influence synaptoxicity. However, due to contradictory results as to whether Aβ oligomers induce their toxic effects in a PrPc-dependent or independent manner, the functional role of these binding interactions has not yet been firmly established.1 Conversely PrPc has been implicated in APP processing and has been reported to hamper β-secretase cleavage of APP thereby reducing Aβ generation (Fig. 3).80 Furthermore, PrPc has been shown to protect neurons against AD-promoting oxidative stress. Thus, PrPc may serve a neuroprotective role in neuronal tissue.
Kellett and Hooper81 have proposed a potential negative feedback loop which controls PrPc expression and Aβ synthesis. PrPc impedes the activity of β-secretase, thereby restraining amyloidogenic APP processing and reducing the concentrations of Aβ and AICD.81 AICD is a transcription factor which regulates the expression of p53-which in turn regulates PRNP expression82-therefore, PrPc induced reduction of AICD ultimately results in downregulation of PRNP gene expression. It may be suggested that in AD patients, who exhibit increased levels of Aβ42 oligomers, the binding of these oligomers to PrPc may hamper the PrPc regulation of β-secretase through steric inhibition or through enhanced PrPc endocytosis and thereby prevent PrPc-β-secretase interactions.81
Commonalities between Alzheimer Disease and Prion Disorders
The structural commonalities between prion proteins and Aβ include conserved histidine metal binding domains which result in the synthesis of ROS when the proteins bind to copper (Cu2+) and zinc (Zn2+) ions;83,84 conserved tyrosines and methionine residues within the proteins' transmembrane regions and three GxxxG sequence repeats for transmembrane association.85
Comparable Processing and Post-Translational Modifications between APP and PrPc
PrPc may be released from the cell surface via two mechanisms-cleavage of the GPI anchor or cleavage by a member of the ADAM family (possibly ADAM10) between residues His111 and His112 which lie within the neurotoxic PrP106–PrP126 sequence (the region of PrPc central in initiating the conformational change from PrPc to PrPSc as well as the aggregation process).86–90 This is analogous to the non-amyloidogenic APP processing pathway. However, cleavage may also occur upstream—at residue 88 (in the event of mutations in the PRNP gene) and thereby yield a neurotoxic product—PrPSc. This is not unlike the upstream positioning of the β-secretase cleavage site which yields potentially pathogenic Aβ peptides. In both TSEs and AD, non-fibril forming peptides, PrP106–126,91 and Aβ oligomers are the neurotoxic agents initiating the disease. The resultant aggregates and plaques, although not the predominant agents, play a role in the pathogenic process.
In lieu of the above mentioned similarities in both the structure and neurotoxicity-inducing mechanisms of Aβ and PrPc, it may be proposed that receptors fundamental in one process may be involved in the other. One such receptor, which is central to TSE disease establishment is the 37 kDa/67 kDa LRP/LR.
The 37 kDa/67 kDa Laminin Receptor Precursor/Laminin Receptor
The 37 kDa/67 kDa LRP/LR is a multifunctional receptor (Fig. 4) located at the cell surface and is soluble in the cytoplasm and in the nucleus. At the cell surface the protein serves as a receptor for a variety of substrates including: ECM components such as elastin and laminin; viruses including Sindbis92 Dengue,93 Venezuelan equine encephalitis and Adeno-associated virus subtypes 2, 3, 8 and 9,31,32 as well as cellular73 and infectious prion proteins72,74,75 (Fig. 5). Additionally, the receptor is present in the cytoplasm where it functions in the maturation of the 40S ribosomal subunit and protein synthesis31,32 and in the nucleus in which it is associated with histones H2A, H2B and H4.94
Figure 4.

Schematic representation of the the functional domains of the 37 kDa/67 kDa Laminin Receptor Precursor/Laminin Receptor. This receptor, which is 295 amino acids in length, may be located at the cell surface, in the cytoplasm and the nucleus and displays different functional roles in each. A cell-surface associated form of the multi-functional protein is depicted here. The transmembrane domain of the receptor is located between amino acid residues LRP86–101. In this location the protein functions primarily as a receptor and encompasses four defined ligand-binding domains, including a prion protein binding domain (LRP161–180) and two laminin binding domains (LRP160–180 and LRP205–229), the latter functions as a heparin binding domain as well and an IgG-antibody binding domain (LRP272–280) (adapted from reference 32).
Figure 5.

A schematic representation of the two classical apoptotic pathways in mammalian cells. The extrinsic is triggered at the cell surface by ligand (CD95L, FASL, TNFα) binding to death receptors (CD95, FASR, TNFR) and the ultimate formation of a death inducing signaling complex (DISC). Conversely the intrinsic pathway involves alterations in mitochondrion permeability as a result of intracellular signals such as DNA damage and oxidative stress. The activation of the aforementioned pathways leads to the cleavage and activation of initiator caspases 8 and caspases 9 respectively. These in turn activate the effector caspases 3 which facilitates DNA fragmentation and cytoskeletal protein degradation-leading to the morphological and physical features characteristic of apoptotic cells (adapted from reference 96).
LRP/LR is a transmembrane (TM) receptor (TMR) which is located at the cholesterol-rich lipid rafts and may employ HSPGs to mediate its binding interactions.31,32 Aβ oligomers, as previously stated, have been shown to bind to an array of TM receptors and HSPGs or are alternatively inserted to lipid raft domains of the plasma membrane. Furthermore, the 37 kDa/67 kDa LRP/LR binds laminin (to which Aβ oligomers similarly bind), as well as PrPc (a protein to which Aβ oligomers bind with high affinity). Thus, owing to the fact that Aβ and 37 kDa/67 kDa LRP/LR share a common cellular location and binding partners, as well as the likeness between the pathogenic agents and mechanisms in TSEs and AD, a relationship between Aβ and 37 kDa/67 kDa LRP/LR cannot be excluded. Moreover, a binding interaction between these proteins, be it direct or indirect, seems plausible and thereby warrants investigation.
The neurotoxicity of Aβ oligomers, especially with regards to the induction of oxidative stress and the deregulation of Ca2+ homeostasis may further be mediated by apoptosis.
Apoptosis
Apoptosis, primarily described by Kerr et al. in 1972, is a normal physiological process of particular importance during development, immune responses, tissue remodeling and the maintenance of normal cellular homeostasis.95 Apoptotic events are regulated by multi-complex pathways which, although able to induce apoptosis independently of one another, are integrated and “cross talk” in the event that the apoptotic signal requires amplification to ensure the cell is committed to apoptosis.96 These pathways are the extrinsic (death receptor) and intrinsic (mitochondrial) pathways (Fig. 5). An additional pathway, the granzyme/perforin pathway has also been proposed but shall not be discussed here.
The critical components of the said pathways/cascades are the caspases. These are highly conserved, hetero-tetrameric cysteine proteases which cleave aspartic residues and are synthesized as inactive zymogens (pro-caspases). Fourteen such caspases have been identified, seven of which are involved in apoptosis and the remaining (including caspases 1, 4 and 5) function in cytokine activation and inflammatory responses. Caspases may be subdivided into two groups-initiator caspases (caspases 2, 8, 9 and 10) which function at the beginning of the pathways and serve to cleave and activate the effector caspases (caspases 3, 6 and 7) which in turn cleave and activate cellular target protein and thereby induce apoptosis.
The extrinsic pathway (Fig. 5) is induced through signal protein-receptor interactions at the cell membrane, including FasL/FasR, tumor necrosis factor (TNF)α/TNFR1 (receptor), Apo3L/DR3, Apo2L/DR4 and Apo2L/DR5.96,97 The transmembrane receptors exhibit cytosolic death domains which facilitates the recruitment of adaptor proteins with the corresponding death domains as well as death effector domains. The latter domain in turn allows for the recruitment of multiple initiator procaspases (procaspase 8 and/or 10) and the resultant complex is termed the death inducing signaling complex (DISC). Within DISC, the high concentration of procaspases allows for the procaspases to auto-catalytically cleave and thereby activate each other and the activated enzymes subsequently activate effector caspases (usually caspases 3).95,96
Intracellular death signals and extracellular survival signals induce the intrinsic pathway in a receptor-independent manner. The intrinsic pathway centres around alterations in the permeability of the mitochondrial membrane and consequently the release of Ca2+ and cytochrome c (a component of the mitochondrial electron transport chain). This pathway is regulated by approximately 25 members of the Bcl-2 protein family. These proteins may either be anti-apoptotic (by sequestering Apaf-1 and inhibiting procaspase 9 activation) such as Bcl-2, Bcl-x, Bcl-XL, Bcl-XS, Bcl-w and BAG or pro-apoptotic by promoting mitochondrial permeability through the formation of membrane channels such as Bax, Bak, Bid, Bad, Bim, Bik, Blk, Puma and Noxa.95 Released cytochrome c binds to the cytosolic apoptotic protease activating factor-1 (Apaf-1) forming a heptamer termed the apoptosome, which subsequently recruits procaspase 9 which, upon cleavage-induced activation, activates caspases 3.
It is noteworthy to add that the MAP kinase, JNK and p53, activate Bim and Bax, Puma and Noxa respectively and are thus pro-apoptotic98,99 p53 has also been reported to induce the expression of death receptors and thus promote apoptosis.100,101
The extrinsic pathway may induce the intrinsic pathway to amplify the apoptotic signal and does so through Bid. Activated caspase 8 cleaves Bid and the truncated form of this protein inhibits Bcl-2 (an anti-apoptotic protein) and thereby facilitates Apaf-1 induced procaspase 9 activation.95
Cellular Death
Activation of caspases 3, 6 and 7 ultimately leads to the morphological and biochemical alterations associated with apoptosis.95 Caspase 3 activation results in the cleavage of the inhibitor of the caspases activated deoxyribonuclease (ICAD), thereby releasing the suppression of this enzyme and thus resulting in DNA fragmentation.95 Furthermore, the effector caspases cleave cytoskeletal and associated proteins such as actin and gelsolin. The resultant disruption of the cytoskeleton consequently causes cellular shrinkage, cell membrane blebbing, disintegration of the cell into apoptotic bodies as well as defects in transport, division and signal transduction.95
Furthermore apoptosis may, in addition to caspase-mediated mechanisms, occur via processes independent of these cysteine proteases. One such route of programmed cell death (PCD) involves the nuclear translocation of the Apoptosis-inducing factor (AIF), a 57 kDa mitochondrial flavoprotein, upon stimulation by death signals. In the nuclear compartment AIF induces chromatin condensation and DNA fragmentation, the hallmarks of apoptosis, in a caspase independent manner.102 An homologue of AIF, termed the AIF-homologous mitochondrion-associated inducer of death (AMID), similarly induces caspase-independent apoptosis.103 In addition the mitochondrial release of endonuclease G has been reported to mediate caspase-independent DNA fragmentation.104
Evidence of Apoptosis in Alzheimer Disease
Alzheimer disease is characterized by enhanced oxidative stress and intracellular Ca2+ concentration-both of which may serve as stimuli to induce the intrinsic pathway.95,105,106 Mutations in presenilins (particularly presenilin 2) may not only lead to familial Alzheimer disease but also render neurons more vulnerable to apoptosis. Further evidence for apoptosis in Alzheimer disease includes the detection of elevated levels of caspases 3, p53 and Bax in neurons of AD patients.107 Aβ induces apoptosis through a number of mechanisms largely through the interaction of these peptides with cell surface receptors. Aβ activates microglial cells, through interaction with the scavenger receptors present on their surfaces (Table 1),108 and this interaction results in the enhanced expression of TNFα (a ligand of the TNF death receptor) and thereby triggers the extrinsic pathway.109 Aβ interactions with neuronal cell surface receptors (p75NTR, FAS, TNFR1, RAGE, APP) (Table 1) may not only induce the production of reactive oxygen species but may also, through signal transduction cascades, induce the expression of caspases and pro-apoptotic genes such as p53, p35 and tumor necrosis factors110 as well as enhance mitochondrial permeability. It must be emphasised that these pro-apoptotic interactions need not necessarily involve death receptors as receptors lacking these domains (such as APP, RAGE, 37 kDa/67 kDa LRP/LR) may indirectly lead to apoptosis through such signaling pathways.
Apoptotic Potential of the Aβ-LRP/LR Interaction
The 37 kDa/67 kDa LRP/LR is a critical cell surface receptor with regards to the maintenance of cell viability and siRNA mediated downregulation of this receptor has been reported to induce apoptosis in transformed liver cells (Hep3B).111 The significance of the receptor may be proposed to be a result of receptor interactions with cognate ligands and/or receptor mediated signaling. Thus the possibility that Aβ-LRP/LR interactions may inhibit these conventional interactions and thereby reduce the receptor's ability to promote cell viability may not be excluded. Furthermore, strong evidence suggests that Aβ-cell surface receptor interactions may be directly or indirectly apoptotic.110 Therefore, owing to structural similarity and common cellular localization between the 37 kDa/67 kDa LRP/LR and receptors such as RAGE, P75NTR and others, if an Aβ-LRP/LR interaction is identified, further investigation of the possible apoptotic inducing potential of this Aβ-LRP/LR interaction is warranted as it may contribute to neurodegeneration and may be an alternative target for Alzheimer disease therapeutics.
Conclusion
Aβ induced neuronal apoptosis is mediated through the interactions of Aβ with the plasma membrane as well as cell surface proteins and receptors. The prion protein plays a vital role in Aβ aggregation and serves as a link between prion disorders and Alzheimer disease. The similarities between these neurodegenerative disorders offer alternatives for the development of therapeutic tools to target both diseases.
Abbreviations
- Aβ
amyloid beta/beta amyloid peptide
- AD
Alzheimer disease
- AICD
amyloid intracellular domain
- APP
amyloid precursor protein
- BACE
β-site APP cleaving enzyme
- HSPGs
heparan sulphate proteoglycans
- LRP/LR
non-integrin laminin receptor precursor
- LTP
long-term potentiation
- mGluR5
metabotrophic glutamate receptor
- NMDA
N-methyl-D-aspartate receptor
- p75NTR
p75 neurotrophin receptor
- PrPc
cellular prion protein isoform
- PrPSc
infectious prion protein isoform
- RAGE
receptor for advanced glycosylation end products
- TSE
transmissible spongiform encephalopathies
Disclosure of Potential Conflicts of Interest
Any opinion, findings and conclusions or recommendations express in this material are those of the author(s) and therefore the NRF do not accept any liability in regard thereto.
Financial Support
This work is based upon research supported by the National Research Foundation (NRF), the Republic of South Africa (RSA) and the Medical Research Council (MRC), the Republic of South Africa (RSA).
References
- 1.Forloni G, Balducci C. beta-amyloid oligomers and prion protein: Fatal attraction? Prion. 2011;5:10–15. doi: 10.4161/pri.5.1.14367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mount C, Downton C. Alzheimer disease: progress or profit? Nat Med. 2006;12:780–784. doi: 10.1038/nm0706-780. [DOI] [PubMed] [Google Scholar]
- 3.Palmer AM. Neuroprotective therapeutics for Alzheimer's disease: progress and prospects. Trends Pharmacol Sci. 2011;32:141–147. doi: 10.1016/j.tips.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 4.Selkoe DJ. Alzheimer's disease: genes, proteins and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 5.Burns A, Iliffe S. Alzheimer's disease. BMJ. 2009;338:158. doi: 10.1136/bmj.b158. [DOI] [PubMed] [Google Scholar]
- 6.Jarrett JT, Berger EP, Lansbury P., Jr The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 7.Dickson DW. The pathogenesis of senile plaques. J Neuropathol Exp Neurol. 1997;56:321–339. doi: 10.1097/00005072-199704000-00001. [DOI] [PubMed] [Google Scholar]
- 8.Pimplikar SW. Reassessing the amyloid cascade hypothesis of Alzheimer's disease. Int J Biochem Cell Biol. 2009;41:1261–1268. doi: 10.1016/j.biocel.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iqbal K, Liu F, Gong CX, Grundke-Iqbal I. Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res. 2010;7:656–664. doi: 10.2174/156720510793611592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- 11.Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 12.Braak H, Braak E, Bohl J, Bratzke H. Evolution of Alzheimer's disease related cortical lesions. J Neural Transm Suppl. 1998;54:97–106. doi: 10.1007/978-3-7091-7508-8_9. [DOI] [PubMed] [Google Scholar]
- 13.Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- 14.Josephs KA, Whitwell JL, Ahmed Z, Shiung MM, Weigand SD, Knopman DS, et al. Beta-amyloid burden is not associated with rates of brain atrophy. Ann Neurol. 2008;63:204–212. doi: 10.1002/ana.21223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stanford PM, Halliday GM, Brooks WS, Kwok JB, Storey CE, Creasey H, et al. Progressive supranuclear palsy pathology caused by a novel silent mutation in exon 10 of the tau gene: expansion of the disease phenotype caused by tau gene mutations. Brain. 2000;123:880–893. doi: 10.1093/brain/123.5.880. [DOI] [PubMed] [Google Scholar]
- 16.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 17.Selkoe DJ. Cell biology of the amyloid beta-protein precursor and the mechanism of Alzheimer's disease. Annu Rev Cell Biol. 1994;10:373–403. doi: 10.1146/annurev.cb.10.110194.002105. [DOI] [PubMed] [Google Scholar]
- 18.Haass C, Hung AY, Selkoe DJ. Processing of beta-amyloid precursor protein in microglia and astrocytes favors an internal localization over constitutive secretion. J Neurosci. 1991;11:3783–3793. doi: 10.1523/JNEUROSCI.11-12-03783.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ring S, Weyer SW, Kilian SB, Waldron E, Pietrzik CU, Filippov MA, et al. The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral and electrophysiological abnormalities of APP-deficient mice. J Neurosci. 2007;27:7817–7826. doi: 10.1523/JNEUROSCI.1026-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, et al. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson JP, Esch FS, Keim PS, Sambamurti K, Lieberburg I, Robakis NK. Exact cleavage site of Alzheimer amyloid precursor in neuronal PC-12 cells. Neurosci Lett. 1991;128:126–128. doi: 10.1016/0304-3940(91)90775-o. [DOI] [PubMed] [Google Scholar]
- 22.Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, et al. Isolation and quantification of soluble Alzheimer's beta-peptide from biological fluids. Nature. 1992;359:325–327. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- 23.Motter R, Vigo-Pelfrey C, Kholodenko D, Barbour R, Johnson-Wood K, Galasko D, et al. Reduction of beta-amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer's disease. Ann Neurol. 1995;38:643–648. doi: 10.1002/ana.410380413. [DOI] [PubMed] [Google Scholar]
- 24.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 25.Fahrenholz F, Gilbert S, Kojro E, Lammich S, Postina R. Alpha-secretase activity of the disintegrin metalloprotease ADAM 10. Influences of domain structure. Ann NY Acad Sci. 2000;920:215–222. doi: 10.1111/j.1749-6632.2000.tb06925.x. [DOI] [PubMed] [Google Scholar]
- 26.Asai M, Hattori C, Szabo B, Sasagawa N, Maruyama K, Tanuma S, et al. Putative function of ADAM9, ADAM10 and ADAM17 as APP alpha-secretase. Biochem Biophys Res Commun. 2003;301:231–235. doi: 10.1016/s0006-291x(02)02999-6. [DOI] [PubMed] [Google Scholar]
- 27.Tanabe C, Hotoda N, Sasagawa N, Sehara-Fujisawa A, Maruyama K, Ishiura S. ADAM19 is tightly associated with constitutive Alzheimer's disease APP alpha-secretase in A172 cells. Biochem Biophys Res Commun. 2007;352:111–117. doi: 10.1016/j.bbrc.2006.10.181. [DOI] [PubMed] [Google Scholar]
- 28.Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, et al. BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional and synaptic functions. J Neurosci. 2005;25:11693–11709. doi: 10.1523/JNEUROSCI.2766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, et al. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60:988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guglielmotto M, Aragno M, Autelli R, Giliberto L, Novo E, Colombatto S, et al. The upregulation of BACE1 mediated by hypoxia and ischemic injury: role of oxidative stress and HIF1alpha. J Neurochem. 2009;108:1045–1056. doi: 10.1111/j.1471-4159.2008.05858.x. [DOI] [PubMed] [Google Scholar]
- 31.Omar A, Jovanovic K, Da Costa Dias B, Gonsalves D, Moodley K, Caveney R, et al. Patented biological approaches for the therapeutic modulation of the 37 kDa/67 kDa laminin receptor. Expert Opin Ther Pat. 2011;21:35–53. doi: 10.1517/13543776.2011.539203. [DOI] [PubMed] [Google Scholar]
- 32.Mbazima V, Da Costa Dias B, Omar A, Jovanovic K, Weiss SF. Interactions between PrP(c) and other ligands with the 37-kDa/67-kDa laminin receptor. Front Biosci. 2010;15:1150–1163. doi: 10.2741/3667. [DOI] [PubMed] [Google Scholar]
- 33.Wolfe MS. Inhibition and modulation of gamma-secretase for Alzheimer's disease. Neurotherapeutics. 2008;5:391–398. doi: 10.1016/j.nurt.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chow VW, Mattson MP, Wong PC, Gleichmann M. An overview of APP processing enzymes and products. Neuromolecular Med. 2010;12:1–12. doi: 10.1007/s12017-009-8104-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wakabayashi T, De Strooper B. Presenilins: members of the gamma-secretase quartets, but part-time soloists too. Physiology. 2008;23:194–204. doi: 10.1152/physiol.00009.2008. [DOI] [PubMed] [Google Scholar]
- 36.Bettens K, Sleegers K, Van Broeckhoven C. Current status on Alzheimer disease molecular genetics: from past, to present, to future. Hum Mol Genet. 2010;19:4–11. doi: 10.1093/hmg/ddq142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 38.Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- 39.Hardy J. Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci. 1997;20:154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- 40.Cruts M, Van Broeckhoven C. Presenilin mutations in Alzheimer's disease. Hum Mutat. 1998;11:183–190. doi: 10.1002/(SICI)1098-1004(1998)11:3<183::AID-HUMU1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 41.Kumar-Singh S, Theuns J, Van Broeck B, Pirici D, Vennekens K, Corsmit E, et al. Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum Mutat. 2006;27:686–695. doi: 10.1002/humu.20336. [DOI] [PubMed] [Google Scholar]
- 42.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bertram L, Tanzi RE. Alzheimer's disease: one disorder, too many genes? Hum Mol Genet. 2004;13:135–141. doi: 10.1093/hmg/ddh077. [DOI] [PubMed] [Google Scholar]
- 44.Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63:168–174. doi: 10.1001/archpsyc.63.2.168. [DOI] [PubMed] [Google Scholar]
- 45.Dermaut B, Croes EA, Rademakers R, Van den Broeck M, Cruts M, Hofman A, et al. PRNP Val129 homozygosity increases risk for early-onset Alzheimer's disease. Ann Neurol. 2003;53:409–412. doi: 10.1002/ana.10507. [DOI] [PubMed] [Google Scholar]
- 46.Riemenschneider M, Klopp N, Xiang W, Wagenpfeil S, Vollmert C, Muller U, et al. Prion protein codon 129 polymorphism and risk of Alzheimer disease. Neurology. 2004;63:364–366. doi: 10.1212/01.wnl.0000130198.72589.69. [DOI] [PubMed] [Google Scholar]
- 47.Bitan G, Vollers SS, Teplow DB. Elucidation of primary structure elements controlling early amyloid beta-protein oligomerization. J Biol Chem. 2003;278:34882–34889. doi: 10.1074/jbc.M300825200. [DOI] [PubMed] [Google Scholar]
- 48.Teplow DB, Lazo ND, Bitan G, Bernstein S, Wyttenbach T, Bowers MT, et al. Elucidating amyloid beta-protein folding and assembly: A multidisciplinary approach. Acc Chem Res. 2006;39:635–645. doi: 10.1021/ar050063s. [DOI] [PubMed] [Google Scholar]
- 49.Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 51.Nordberg A. Amyloid plaque imaging in vivo: current achievement and future prospects. Eur J Nucl Med Mol Imaging. 2008;35:46–50. doi: 10.1007/s00259-007-0700-2. [DOI] [PubMed] [Google Scholar]
- 52.Lesne S, Kotilinek L, Ashe KH. Plaque-bearing mice with reduced levels of oligomeric amyloid-beta assemblies have intact memory function. Neuroscience. 2008;151:745–749. doi: 10.1016/j.neuroscience.2007.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marchesi VT. Alzheimer's dementia begins as a disease of small blood vessels, damaged by oxidative-induced inflammation and dysregulated amyloid metabolism: implications for early detection and therapy. FASEB J. 2011;25:5–13. doi: 10.1096/fj.11-0102ufm. [DOI] [PubMed] [Google Scholar]
- 55.Bartzokis G. Alzheimer's disease as homeostatic responses to age-related myelin breakdown. Neurobiol Aging. 2011;32:1341–1371. doi: 10.1016/j.neurobiolaging.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, et al. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron. 2010;66:739–754. doi: 10.1016/j.neuron.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bi X, Gall CM, Zhou J, Lynch G. Uptake and pathogenic effects of amyloid beta peptide 1–42 are enhanced by integrin antagonists and blocked by NMDA receptor antagonists. Neuroscience. 2002;112:827–840. doi: 10.1016/s0306-4522(02)00132-x. [DOI] [PubMed] [Google Scholar]
- 58.Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stefani M. Biochemical and biophysical features of both oligomer/fibril and cell membrane in amyloid cytotoxicity. FEBS J. 2010;277:4602–4613. doi: 10.1111/j.1742-4658.2010.07889.x. [DOI] [PubMed] [Google Scholar]
- 60.Sepulveda FJ, Parodi J, Peoples RW, Opazo C, Aguayo LG. Synaptotoxicity of Alzheimer beta amyloid can be explained by its membrane perforating property. PLoS One. 2010;5:11820. doi: 10.1371/journal.pone.0011820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qiu L, Lewis A, Como J, Vaughn MW, Huang J, Somerharju P, et al. Cholesterol modulates the interaction of beta-amyloid peptide with lipid bilayers. Biophys J. 2009;96:4299–4307. doi: 10.1016/j.bpj.2009.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sakurai T, Kaneko K, Okuno M, Wada K, Kashiyama T, Shimizu H, et al. Membrane microdomain switching: a regulatory mechanism of amyloid precursor protein processing. J Cell Biol. 2008;183:339–352. doi: 10.1083/jcb.200804075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Verdier Y, Penke B. Binding sites of amyloid beta-peptide in cell plasma membrane and implications for Alzheimer's disease. Curr Protein Pept Sci. 2004;5:19–31. doi: 10.2174/1389203043486937. [DOI] [PubMed] [Google Scholar]
- 64.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 65.Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 66.Beck K, Hunter I, Engel J. Structure and function of laminin: anatomy of a multidomain glycoprotein. FASEB J. 1990;4:148–160. doi: 10.1096/fasebj.4.2.2404817. [DOI] [PubMed] [Google Scholar]
- 67.Kibbey MC, Jucker M, Weeks BS, Neve RL, Van Nostrand WE, Kleinman HK. beta-Amyloid precursor protein binds to the neurite-promoting IKVAV site of laminin. Proc Natl Acad Sci USA. 1993;90:10150–10153. doi: 10.1073/pnas.90.21.10150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Koo EH, Park L, Selkoe DJ. Amyloid beta-protein as a substrate interacts with extracellular matrix to promote neurite outgrowth. Proc Natl Acad Sci USA. 1993;90:4748–4752. doi: 10.1073/pnas.90.10.4748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Castillo GM, Lukito W, Peskind E, Raskind M, Kirschner DA, Yee AG, et al. Laminin inhibition of beta-amyloid protein (Abeta) fibrillogenesis and identification of an Abeta binding site localized to the globular domain repeats on the laminin a chain. J Neurosci Res. 2000;62:451–462. doi: 10.1002/1097-4547(20001101)62:3<451::AID-JNR15>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 70.Mehrpour M, Codogno P. Prion protein: From physiology to cancer biology. Cancer Lett. 2010;290:1–23. doi: 10.1016/j.canlet.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 71.Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morel E, Andrieu T, Casagrande F, Gauczynski S, Weiss S, Grassi J, et al. Bovine prion is endocytosed by human enterocytes via the 37 kDa/67 kDa laminin receptor. Am J Pathol. 2005;167:1033–1042. doi: 10.1016/S0002-9440(10)61192-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gauczynski S, Peyrin JM, Haik S, Leucht C, Hundt C, Rieger R, et al. The 37-kDa/67-kDa laminin receptor acts as the cell-surface receptor for the cellular prion protein. EMBO J. 2001;20:5863–5875. doi: 10.1093/emboj/20.21.5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gauczynski S, Nikles D, El-Gogo S, Papy-Garcia D, Rey C, Alban S, et al. The 37-kDa/67-kDa laminin receptor acts as a receptor for infectious prions and is inhibited by polysulfated glycanes. J Infect Dis. 2006;194:702–709. doi: 10.1086/505914. [DOI] [PubMed] [Google Scholar]
- 75.Kolodziejczak D, Da Costa Dias B, Zuber C, Jovanovic K, Omar A, Beck J, et al. Prion interaction with the 37-kDa/67-kDa laminin receptor on enterocytes as a cellular model for intestinal uptake of prions. J Mol Biol. 2010;402:293–300. doi: 10.1016/j.jmb.2010.06.055. [DOI] [PubMed] [Google Scholar]
- 76.Da Costa Dias B, Jovanovic K, Weiss SF. Alimentary prion infections: Touchdown in the intestine. Prion. 2011;5:6–9. doi: 10.4161/pri.5.1.14283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci. 2001;24:519–550. doi: 10.1146/annurev.neuro.24.1.519. [DOI] [PubMed] [Google Scholar]
- 78.Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen S, Yadav SP, Surewicz WK. Interaction between human prion protein and amyloid-beta (Abeta) oligomers: role OF N-terminal residues. J Biol Chem. 2010;285:26377–26383. doi: 10.1074/jbc.M110.145516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Parkin ET, Watt NT, Hussain I, Eckman EA, Eckman CB, Manson JC, et al. Cellular prion protein regulates beta-secretase cleavage of the Alzheimer's amyloid precursor protein. Proc Natl Acad Sci USA. 2007;104:11062–11067. doi: 10.1073/pnas.0609621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kellett KA, Hooper NM. Prion protein and Alzheimer disease. Prion. 2009;3:190–194. doi: 10.4161/pri.3.4.9980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vincent B, Sunyach C, Orzechowski HD, George-Hyslop P, Checler F. p53-Dependent transcriptional control of cellular prion by presenilins. J Neurosci. 2009;29:6752–6760. doi: 10.1523/JNEUROSCI.0789-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bush AI. The metallobiology of Alzheimer's disease. Trends Neurosci. 2003;26:207–214. doi: 10.1016/S0166-2236(03)00067-5. [DOI] [PubMed] [Google Scholar]
- 84.Brown DR. Copper and prion disease. Brain Res Bull. 2001;55:165–173. doi: 10.1016/s0361-9230(01)00453-1. [DOI] [PubMed] [Google Scholar]
- 85.Barnham KJ, Cappai R, Beyreuther K, Masters CL, Hill AF. Delineating common molecular mechanisms in Alzheimer's and prion diseases. Trends Biochem Sci. 2006;31:465–472. doi: 10.1016/j.tibs.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 86.Vincent B, Paitel E, Saftig P, Frobert Y, Hartmann D, De Strooper B, et al. The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol ester-regulated normal cleavage of the cellular prion protein. J Biol Chem. 2001;276:37743–37746. doi: 10.1074/jbc.M105677200. [DOI] [PubMed] [Google Scholar]
- 87.Cisse MA, Sunyach C, Lefranc-Jullien S, Postina R, Vincent B, Checler F. The disintegrin ADAM9 indirectly contributes to the physiological processing of cellular prion by modulating ADAM10 activity. J Biol Chem. 2005;280:40624–40631. doi: 10.1074/jbc.M506069200. [DOI] [PubMed] [Google Scholar]
- 88.Jobling MF, Stewart LR, White AR, McLean C, Friedhuber A, Maher F, et al. The hydrophobic core sequence modulates the neurotoxic and secondary structure properties of the prion peptide 106–126. J Neurochem. 1999;73:1557–1565. doi: 10.1046/j.1471-4159.1999.0731557.x. [DOI] [PubMed] [Google Scholar]
- 89.Tagliavini F, Prelli F, Verga L, Giaccone G, Sarma R, Gorevic P, et al. Synthetic peptides homologous to prion protein residues 106–147 form amyloid-like fibrils in vitro. Proc Natl Acad Sci USA. 1993;90:9678–9682. doi: 10.1073/pnas.90.20.9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brown DR, Schmidt B, Kretzschmar HA. Role of microglia and host prion protein in neurotoxicity of a prion protein fragment. Nature. 1996;380:345–347. doi: 10.1038/380345a0. [DOI] [PubMed] [Google Scholar]
- 91.Salmona M, Malesani P, De Gioia L, Gorla S, Bruschi M, Molinari A, et al. Molecular determinants of the physicochemical properties of a critical prion protein region comprising residues 106–126. Biochem J. 1999;342:207–214. [PMC free article] [PubMed] [Google Scholar]
- 92.Wang KS, Kuhn RJ, Strauss EG, Ou S, Strauss JH. High-affinity laminin receptor is a receptor for Sindbis virus in mammalian cells. J Virol. 1992;66:4992–5001. doi: 10.1128/jvi.66.8.4992-5001.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thepparit C, Smith DR. Serotype-specific entry of dengue virus into liver cells: identification of the 37-kilodalton/67-kilodalton high-affinity laminin receptor as a dengue virus serotype 1 receptor. J Virol. 2004;78:12647–12656. doi: 10.1128/JVI.78.22.12647-12656.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kinoshita K, Kaneda Y, Sato M, Saeki Y, Wataya-Kaneda M, Hoffmann A. LBP-p40 binds DNA tightly through associations with histones H2A, H2B and H4. Biochem Biophys Res Commun. 1998;253:277–282. doi: 10.1006/bbrc.1998.9699. [DOI] [PubMed] [Google Scholar]
- 95.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 97.Peter ME, Krammer PH. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003;10:26–35. doi: 10.1038/sj.cdd.4401186. [DOI] [PubMed] [Google Scholar]
- 98.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 99.Henry H, Thomas A, Shen Y, White E. Regulation of the mitochondrial checkpoint in p53-mediated apoptosis confers resistance to cell death. Oncogene. 2002;21:748–760. doi: 10.1038/sj.onc.1205125. [DOI] [PubMed] [Google Scholar]
- 100.Hussein MR, Haemel AK, Wood GS. Apoptosis and melanoma: molecular mechanisms. J Pathol. 2003;199:275–288. doi: 10.1002/path.1300. [DOI] [PubMed] [Google Scholar]
- 101.Hu W, Kavanagh JJ. Anticancer therapy targeting the apoptotic pathway. Lancet Oncol. 2003;4:721–729. doi: 10.1016/s1470-2045(03)01277-4. [DOI] [PubMed] [Google Scholar]
- 102.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 103.Wu M, Xu LG, Li X, Zhai Z, Shu HB. AMID, an apoptosis-inducing factor-homologous mitochondrion-associated protein, induces caspase-independent apoptosis. J Biol Chem. 2002;277:25617–25623. doi: 10.1074/jbc.M202285200. [DOI] [PubMed] [Google Scholar]
- 104.van Loo G, Schotte P, van Gurp M, Demol H, Hoorelbeke B, Gevaert K, et al. Endonuclease G: a mitochondrial protein released in apoptosis and involved in caspase-independent DNA degradation. Cell Death Differ. 2001;8:1136–1142. doi: 10.1038/sj.cdd.4400944. [DOI] [PubMed] [Google Scholar]
- 105.Mattson MP, Tomaselli KJ, Rydel RE. Calcium-destabilizing and neurodegenerative effects of aggregated beta-amyloid peptide are attenuated by basic FGF. Brain Res. 1993;621:35–49. doi: 10.1016/0006-8993(93)90295-x. [DOI] [PubMed] [Google Scholar]
- 106.Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 107.Eckert A, Keil U, Marques CA, Bonert A, Frey C, Schussel K, et al. Mitochondrial dysfunction, apoptotic cell death and Alzheimer's disease. Biochem Pharmacol. 2003;66:1627–1634. doi: 10.1016/s0006-2952(03)00534-3. [DOI] [PubMed] [Google Scholar]
- 108.El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD. Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature. 1996;382:716–719. doi: 10.1038/382716a0. [DOI] [PubMed] [Google Scholar]
- 109.Tan J, Town T, Paris D, Mori T, Suo Z, Crawford F, et al. Microglial activation resulting from CD40-CD40L interaction after beta-amyloid stimulation. Science. 1999;286:2352–2355. doi: 10.1126/science.286.5448.2352. [DOI] [PubMed] [Google Scholar]
- 110.Small DH, Mok SS, Bornstein JC. Alzheimer's disease and Abeta toxicity: from top to bottom. Nat Rev Neurosci. 2001;2:595–598. doi: 10.1038/35086072. [DOI] [PubMed] [Google Scholar]
- 111.Susantad T, Smith DR. siRNA-mediated silencing of the 37/67-kDa high affinity laminin receptor in Hep3B cells induces apoptosis. Cell Mol Biol Lett. 2008;13:452–464. doi: 10.2478/s11658-008-0017-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chen Q, Schubert D. Presenilin-interacting proteins. Expert Rev Mol Med. 2002;4:1–18. doi: 10.1017/S1462399402005008. [DOI] [PubMed] [Google Scholar]
- 113.Da Costa Dias B, Weiss SF. A kiss of a prion: new implications for oral transmissibility. J Infect Dis. 2010;201:1615–1616. doi: 10.1086/652458. [DOI] [PubMed] [Google Scholar]