

Abstract
Interleukin (IL)-6 is produced at the site of inflammation and plays a key role in the acute phase response as defined by a variety of clinical and biological features such as the production of acute phase proteins. IL-6 in combination with its soluble receptor sIL-6Rα, dictates the transition from acute to chonic inflammation by changing the nature of leucocyte infiltrate (from polymorphonuclear neutrophils to monocyte/macrophages). In addition, IL-6 exerts stimulatory effects on T- and B-cells, thus favoring chronic inflammatory responses. Strategies targeting IL-6 and IL-6 signaling led to effective prevention and treatment of models of rheumatoid arthritis and other chronic inflammatory diseases.
Introduction
Inflammation is a complex defence mechanism in which leucocytes migrate from the vasculature into damaged tissues to destroy the agents that potentially can cause tissue injury. Acute inflammation is a limited beneficial response, particularly during infectious challenge, whereas chronic inflammation is a persistent phenomenon that can lead to tissue damage. One hallmark of acute inflammation is that initially the leucocyte infiltrate is mostly neutrophilic, but after 24 to 48 hours monocytic cells predominate [1-3]. In contrast, chronic inflammation is histologically associated with the presence of mononuclear cells, such as macrophages and lymphocytes [1,2].
Although several explanations have been suggested, the mechanisms that control the transition from neutrophil to monocyte recruitment during the transformation from acute to chronic inflammation are poorly understood. It is possible that the IL-6/soluble IL-6 receptor α (sIL-6Rα) complex plays an important role in this transition.
IL-6 has a dual effect; at some levels it acts as a defence mechanism but in chronic inflammation it is rather proinflammatory.
Interleukin-6 in acute inflammation
The acute phase response includes changes in the concentrations of many plasma proteins, which are known as the acute phase proteins (Figure 1), as well as numerous behavioural, physiological, biochemical and nutritional changes [4]. The acute phase proteins have been defined as a set of plasma proteins with concentrations that increase (positive acute phase proteins) or decrease (negative acute phase proteins) by at least 25% in inflammatory disorders [4,5] (Figure 1). The changes in concentrations of acute phase proteins are largely due to changes in their production by hepatocytes.
Figure 1.
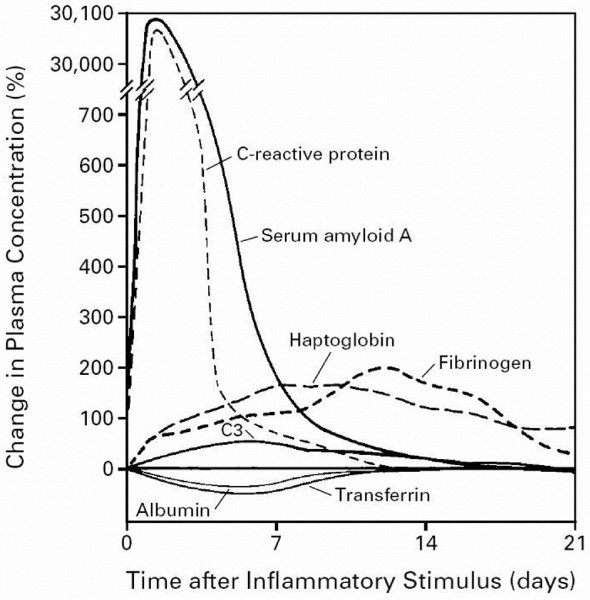
Kinetic of acute-phase protein production. Shown are the characteristic patterns of change that occur in plasma concentrations of some acute phase proteins after a moderate inflammatory stimulus. Reprinted, with permission, from [5]. Copyright © 1987 Elsevier.
The cytokines that are produced during inflammatory processes, and that participate in them, are stimulators of the production of acute phase proteins. These inflammation associated cytokines include IL-6, IL-1β, tumour necrosis factor-α, interferon-γ, transforming growth factor-β [6] and, possibly, IL-8 [7]. They are produced by a variety of cell types, but the most important sources are macrophages and monocytes at inflammatory sites.
IL-6 is the chief stimulator of the production of most acute phase proteins [8], whereas the other implicated cytokines influence subgroups of acute phase proteins. However, in mice rendered incapable of expressing IL-6 (knockout mice), the role played by IL-6 in stimulating the production of acute phase proteins depends on the nature or site of the inflammatory stimulus; the response is largely inhibited in IL-6 knockout mice injected with turpentine, but it is normal when bacterial lipopolysaccharide is the inflammatory stimulus [9]. This finding indicates that lipopolysaccharide causes the production of other cytokines that are capable of stimulating the production of acute phase proteins [9].
Several cytokines, particularly IL-6, stimulate the production of acute phase proteins in response to varied stimuli. The patterns of cytokine production and of the acute phase response differ in different inflammatory conditions. Acute phase changes reflect the presence and intensity of inflammation, and they have long been used as a clinical guide to diagnosis and management.
Using IL-6 gene knockout mice, Xing and coworkers [10] identified an anti-inflammatory component to the action of IL-6 in both local and systemic acute inflammatory responses elicited by local lung or systemic exposure to endotoxin. They demonstrated that IL-6 is critical in controlling the extent of local and systemic acute inflammatory responses, particularly the level of proinflammatory cytokines in the local and systemic compartments.
Overall, in a normal host, one function of inducible IL-6 during acute responses is to suppress the level of proinflammatory cytokines without compromising the level of anti-inflammatory cytokines [10,11]. In addition, IL-6 stimulates the production of IL-1 receptor antagonist, which is an anti-inflammatory mediator [12]. IL-6 therefore can have a protective effect.
The role of interleukin-6 in the development of chronic inflammation
Given the actions of IL-6 to return the host to a homeostatic state, it is clear that IL-6 operates to control the extent of tissue inflammatory responses. In chronic diseases, typically exemplified by immune stressors such as chronic intracellular infections and tumours, IL-6 not only serves as an inducer of acute phase reactions but also is an important player in eliciting cellular immune responses to affected cells and mucosal humoral responses directed against reinfection [13-18].
To be beneficial, the inflammatory reaction must be acute, destroying an injurious agent within a short period of time and in a localized area, while inducing an immune response. This is achieved through a complex series of events characterized by local leucocyte recruitment, death and migration [19]. The transition from neutrophil to monocyte recruitment assists in the efficient destruction of the inciting agent by the combined destructive and phagocytic actions of neutrophils and inflammatory macrophages. However, this transition also assists the resolution of inflammation through eliminating neutrophils and initiating an immune response.
IL-6 elicits not only acute phase reactions but also the development of specific cellular and humoral immune responses, including end-stage B cell differentiation, immunoglobulin secretion and T cell activation. The main switch from acute to chronic inflammation is the recruitment of monocytes to the area of inflammation. IL-6 is important to the transition between acute and chronic inflammation [11].
IL-6 plays a rather unexpected role in leucocyte recruitment in vivo. A complex of IL-6 and sIL-6Rα can activate endothelial cells to secrete IL-8 and monocyte chemoattractant protein (MCP)-1, and induce expression of adhesion molecules [20]. Notably, IL-6 activation of endothelial cells occurs in vivo, although these cells, like other stromal cells, express the gp130 transducing protein of the IL-6 receptor complex but not the 80 kDa ligand binding subunit, IL-6Rα; in vitro, such activation could only be achieved when IL-6 was combined with soluble recombinant IL-6Rα. Expression of IL-6Rα is limited in humans to leucocyte and hepatocyte membranes [20,21], but it can be shed from the neutrophil membrane as a soluble form, which is found at high concentrations in neutrophil enriched inflammatory fluids [22]. The sIL-6Rα combines with IL-6 to bind gp130 on the membranes of stromal cells, and activates these cells in a mechanism termed trans-signalling (Figure 2).
Figure 2.

Possible role played by IL-6 in the shift from acute to chronic inflammation. Stage 1: following acute inflammatory response, IL-6 can bind with sIL-6R. Stage 2: trans-signalling through gp130 leads to monocyte recruitment. Stage 3: prolonged IL-6 leads to neutrophilic apoptosis, phagocytosis and mononuclear accumulation at the site of injury. IL, interleukin; JAK, Janus activated kinase; MCP, monocyte chemoattractant protein; PMN, polymorphonuclear neutrophil; sIL-6R, soluble IL-6 receptor.
The IL-6/IL-6Rα complex favours the transition from neutrophil to monocyte in inflammation [22,23]. The transition from neutrophil to monocyte accumulation may be secondary to a shift in the type of chemokine produced by stromal cells, inflammatory macrophages or neutrophils [11]. Neutrophils stimulated with inflammatory cytokines for several hours will selectively produce MCP-1 and not IL-8 [24]. MCP-1 accumulation does not desensitize cells and leads to late monocyte recruitment. Endothelial (or stromal cell) activation by proinflammatory molecules leads to secretion of platelet activating factor, IL-8 and IL-6, as well as leuco-endothelial adhesion molecule expression. Chemoattractants, originating from the endothelium or other cell sources, recruit neutrophils and induce IL-6Rα shedding from their membranes. The combination of IL-6Rα with IL-6 enables ligation to gp130 on the endothelial cell membrane and increases both IL-6 and MCP-1 endothelial (or stromal) cell secretion but not IL-8, favouring a transition from neutrophil to monocyte recruitment [22,23] (Figure 2). IL-6 signalling through sIL-6Rα controls leucocyte infiltration. In vitro studies confirm that sIL-6Rα mediated events regulate both chemokine and adhesion molecule expression. As a result, IL-6 trans-signalling controls the intermediary factors that are involved in resolving inflammation. A disruption in control of leucocyte trafficking, for example by continuous IL-6 production, may therefore be significant at the onset of chronic disease. IL-6 appears to influence dramatically the nature of the immune response by dictating the recruitment, activation and apoptotic clearance of individual leucocyte subpopulations (e.g. neutrophils, monocytes and lymphocytes) [25].
A transition from neutrophil to monocyte accumulation at the site of inflammation suggests that there is a progression of events that leads not only to monocyte recruitment but also to disappearance of neutrophils. Neutrophils are central cells in the defence of an organism against injury, notably infection, through their capacity to synthesize oxygen metabolites and to liberate various enzymes. However, these agents can also be toxic to normal surrounding tissues and potentially induce inflammatory diseases. As a result, there is rapid negative regulation. Apoptotic neutrophils expressing new membrane antigens are recognized by various receptors on macrophages, leading to phagocytosis (Figure 2). This phagocytosis of apoptotic polymorphonuclear neutrophils by macrophages increases transforming growth factor-β and MCP-1 secretion and decreases IL-8 production, leading to a chemokine shift favouring monocyte recruitment. As neutrophils are depleted from the inflammatory site, blood monocytes, in contrast, accumulate and differentiate into inflammatory macrophages, which complete phagocytosis and destruction of the injurious agents [1-3]. The monocytes and macrophages then emigrate after several days in the local lymph nodes [26]. During this migratory process, monocytes differentiate into dendritic cells, upregulating HLA class II antigen membrane expression, and acquiring costimulatory molecules such as CD80 and CD86 [27]. These cells may then present antigenic peptides to lymphocytes, contributing to the generation of an immune response.
IL-6 exhibits two contrasting features. In models of chronic inflammatory diseases, such as collagen-induced arthritis, murine colitis, or experimental autoimmune encephalomyelitis, IL-6 is proinflammatory [28,29], whereas in models of acute inflammation IL-6 exhibits an anti-inflammatory profile [10].
Chronic inflammation, interleukin-6 and disease
At the beginning of acute inflammation, IL-6 mediates the acute phase responses. When its activity as a proinflammatory cytokine persists, acute inflammation turns into chronic inflammation that includes immune responses. In chronic inflammation, IL-6 has a detrimental role that favours mononuclear cell accumulation at the site of injury, through continuous MCP-1 secretion, angioproliferation and anti-apoptotic functions on T cells [30]. This may increase serum levels of IL-6 and provide the basis for the amplification step of chronic inflammatory proliferation. Plasmacytosis and hyperplasia of synovial cells in the joints of patients with rheumatoid arthritis (RA) are a typical example of chronic inflammatory proliferation. In autoimmune diseases, IL-6 not only maintains inflammation but also modifies the immune responses.
Levels of circulating IL-6 are elevated in several inflammatory diseases including RA (as mentioned above), systemic juvenile idiopathic arthritis, systemic lupus erythematosus, ankylosing spondylitis, psoriasis and Crohn's disease. In RA there is a correlation between IL-6 levels in synovial fluid and markers of inflammation. In systemic juvenile idiopathic arthritis there is a correlation between disease activity and IL-6 levels. In lupus IL-6 levels are elevated, but in this setting there is no relation to levels of C-reactive protein. IL-6 levels in ankylosing spondylitis, psoriasis and Crohn's disease are also elevated and correlate with markers of disease activity. Table 1 summarizes some of the research that has been conducted in various disease models, using IL-6 deficient mice and blockade of IL-6 signalling.
Table 1.
IL-6 signalling blockade in different disease models
| Finding | Ref. | Comment |
|---|---|---|
| Experimental arthritis | ||
| IL-6 is required for the development of collagen induced arthritis | [31] | IL-6-/- and IL-6+/+ mice |
| IL-6 plays a key role in the development of antigen induced arthritis | [32] | IL-6-/- mice |
| Blockade of IL-6 receptor ameliorates joint disease in murine collagen induced arthritis | [33] | |
| Soluble IL-6 receptor governs IL-6 activity in antigen-induced arthritis: blockade of arthritis severity by soluble gp130 | [34] | IL-6-/- mice |
| Colitis | ||
| Blockade of IL-6 trans-signalling suppresses T cell resistance against apoptosis in chronic intestinal inflammation | [30] | Neutralization of sIL-6R by a gp130-Fc fusion protein |
| IL-6 is required for the development of Th1 cell mediated murine colitis | [35] | C.B-17-scid mice transferred with CD45RBhigh CD4+ T anti-IL-6R mAb (MR16-1), used in a murine model of colitis |
| Modulating signaling | ||
| CIS3/SOCS3/SSI3 plays a negative role in STAT3 activation and intestinal inflammation | [36] | Development of colitis as well as STAT3 activation was significantly reduced in IL-6 deficient mice |
| Induction of SOCS3/CIS3 as a therapeutic strategy for treating inflammatory arthritis | [37] | Recombinant adenovirus carrying the CIS3 cDNA injected periarticularly into the ankle joints of mice with antigen induced arthritis or collagen induced arthritis |
| Models of infection | ||
| IL-6 deficient mice are susceptible to: | ||
| Listeria monocytogenes | [38] | |
| Toxoplasma gondii | [39] | |
| Candida albicans | [40] |
CIS, cytokine-induced SH2 protein; IL, interleukin; mAb, monoclonal antibody; sIL-6R, soluble IL-6 receptor; SOCS, suppressors of cytokine signaling; SSI, STAT-induced STAT inhibitors; STAT, signal transducer and activator of transcription; Th, T-helper.
Conclusion
IL-6 has been shown to be a key player in chronic inflammation, and IL-6 levels are elevated in inflammatory diseases in humans. Expression of IL-6 is enhanced at the site of inflammation, and blockade of IL-6 and IL-6 signalling is effective at prevention and treatment in models of inflammatory diseases (including arthritis and colitis).
Overall, IL-6 is important – indeed, critical under certain conditions – to the development of persistent inflammation. Therefore, it would be appropriate to continue to investigate IL-6 blockade as an approach to treatment of chronic inflammatory diseases.
Abbreviations
IL = interleukin; mAb = monoclonal antibody; MCP = monocyte chemoattractant protein; RA = rheumatoid arthritis; sIL-6Rα = soluble IL-6 receptor α.
Competing interests
CG has received consulting fees from Roche.
Acknowledgements
Cem Gabay is supported by a Swiss National Science Foundation grant (320000-107592).
References
- Ryan GB, Majno G. Acute inflammation. A review. Am J Pathol. 1977;86:183–276. [PMC free article] [PubMed] [Google Scholar]
- Melnicoff MJ, Horan PK, Morahan PS. Kinetics of changes in peritoneal-cell populations following acute inflammation. Cell Immunol. 1989;118:178–191. doi: 10.1016/0008-8749(89)90367-5. [DOI] [PubMed] [Google Scholar]
- Doherty DE, Downey GP, Worthen GS, Haslett C, Henson PM. Monocyte retention and migration in pulmonary inflammation. Requirement for neutrophils. Lab Invest. 1988;59:200–213. [PubMed] [Google Scholar]
- Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448–454. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- Gitlin JD, Colten HR. In: Lymphokines. Pick F, editor. Vol. 14. Academic Press; 1987. Molecular biology of acute phase plasma proteins; pp. 123–153. [Google Scholar]
- Kushner I. Regulation of the acute phase response by cytokines. Perspect Biol Med. 1993;36:611–622. doi: 10.1353/pbm.1993.0004. [DOI] [PubMed] [Google Scholar]
- Wigmore SJ, Fearon KCH, Maingay JP, Lai PBS, Ross JA. Interleukin-8 can mediate acute-phase protein production by isolated human hepatocytes. Am J Physiol. 1997;273:E720–E726. doi: 10.1152/ajpendo.1997.273.4.E720. [DOI] [PubMed] [Google Scholar]
- Gauldie J, Richards C, Harnish D, Lansdorp P, Baumann H. Interferon β2/B-cell stimulatory factor type 2 shares identity with monocyte-derived hepatocyte-stimulating factor and regulates the major acute phase protein response in liver cells. Proc Natl Acad Sci USA. 1987;84:7251–7255. doi: 10.1073/pnas.84.20.7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fattori E, Cappelletti M, Costa P, Sellitto C, Cantoni L, Carelli M, Faggioni R, Fantuzzi G, Ghezzi P, Poli V. Defective inflammatory response in interleukin 6-deficient mice. J Exp Med. 1994;180:1243–1250. doi: 10.1084/jem.180.4.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Z, Gauldie J, Cox G, Baumann H, Jordana M, Lei XF, Achong MK. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest. 1998;101:311–320. doi: 10.1172/JCI1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplanski G, Marin V, Montero-Julian F, Mantovani A, Farnarier C. IL-6: a regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends Immunol. 2003;24:25–29. doi: 10.1016/S1471-4906(02)00013-3. [DOI] [PubMed] [Google Scholar]
- Gabay C, Smith MF, Eidlen D, Arend WP. Interleukin 1 receptor antagonist (IL-1Ra) is an acute-phase protein. J Clin Invest. 1997;99:2930–2940. doi: 10.1172/JCI119488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, Zinkernagel R, Bleuthmann H, Kohler G. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature. 1994;368:339–342. doi: 10.1038/368339a0. [DOI] [PubMed] [Google Scholar]
- Xing Z, Braciak T, Jordana M, Croitoru K, Graham FL, Gauldie J. Adenovirus-mediated cytokine gene transfer at tissue sites: overexpression of IL-6 induces lymphocytic hyperplasia in the lung. J Immunol. 1994;153:4059–4069. [PubMed] [Google Scholar]
- Romani L, Mencacci A, Cenci E, Spaccapelo R, Toniatti C, Puccetti P, Bistoni F, Poli V. Impaired neutrophil response and CD41 T helper cell 1 development in interleukin-6-deficient mice infected with Candida albicans. J Exp Med. 1996;183:1345–1355. doi: 10.1084/jem.183.4.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzek MC, Miller AH, Opal SM, Pearce BD, Biron CA. Characterization of early cytokine responses and an interleukin-6-dependent pathway of endogenous glucocorticoid induction during murine cytomegalovirus infection. J Exp Med. 1997;185:1185–1192. doi: 10.1084/jem.185.7.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay AJ, Husband AJ, Ramshaw IA, Bao S, Matthaei KI, Koehler G, Kopt M. The role of IL-6 in mucosal IgA antibody responses in vivo. Science. 1994;264:561–563. doi: 10.1126/science.8160012. [DOI] [PubMed] [Google Scholar]
- Mule JJ, Custer MC, Travis WD, Rosenberg SA. Cellular mechanisms of the antitumor activity of recombinant IL-6 in mice. J Immunol. 1992;148:2622–2629. [PubMed] [Google Scholar]
- Buckley CD, Pilling D, Lord JM, Akbar AN, Scheel-Toellner D, Salmon M. Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends Immunol. 2001;22:199–204. doi: 10.1016/S1471-4906(01)01863-4. [DOI] [PubMed] [Google Scholar]
- Romano M, Sironi M, Toniatti C, Polentarutti N, Fruscella P, Ghezzi P, Faggioni R, Luini W, van Hinsbergh V, Sozzani S. et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity. 1997;6:315–325. doi: 10.1016/S1074-7613(00)80334-9. [DOI] [PubMed] [Google Scholar]
- Desgeorges A, Gabay C, Silacci P, Novick D, Roux-Lombard P, Grau G, Dayer JM, Vischer T, Guerne PA. Concentrations and origins of soluble interleukin 6 receptor-alpha in serum and synovial fluid. J Rheumatol. 1997;24:1510–1516. [PubMed] [Google Scholar]
- Hurst SM, Wilkinson TS, McLoughlin RM, Jones S, Horiuchi S, Yamamoto N, Rose-John S, Fuller GM, Topley N, Jones SA. IL-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity. 2001;14:705–714. doi: 10.1016/S1074-7613(01)00151-0. [DOI] [PubMed] [Google Scholar]
- Marin V, Montero-Julian FA, Gres S, Boulay V, Bongrand P, Farnarier C, Kaplanski G. The IL-6/soluble IL-6Ra autocrine loop of endothelial activation as an intermediate between acute and chronic inflammation: an experimental model involving thrombin. J Immunol. 2001;167:3435–3442. doi: 10.4049/jimmunol.167.6.3435. [DOI] [PubMed] [Google Scholar]
- Yamashiro S, Kamohara H, Yoshimura T. MCP-1 is selectively expressed in the late phase by cytokine-stimulated human neutrophils: TNF-a plays a role in maximal MCP-1 mRNA expression. J Leukoc Biol. 1999;65:671–679. doi: 10.1002/jlb.65.5.671. [DOI] [PubMed] [Google Scholar]
- Jones SA, Richards PJ, Scheller J, Rose-John S. IL-6 transsignaling: the in vivo consequences. J Interferon Cytokine Res. 2005;25:241–253. doi: 10.1089/jir.2005.25.241. [DOI] [PubMed] [Google Scholar]
- Bellingan GJ, Caldwell H, Howie SE, Dransfield I, Haslett C. In vivo fate of the inflammatory macrophage during the resolution of inflammation. Inflammatory macrophages do not die locally but emigrate to the draining lymph nodes. J Immunol. 1996;157:2577–2585. [PubMed] [Google Scholar]
- Randolph GJ, Beaulieu S, Lebecque S, Steinman RM, Muller WA. Differentiation of monocytes into dendritic cells in a model of transendothelial trafficking. Science. 1998;282:480–483. doi: 10.1126/science.282.5388.480. [DOI] [PubMed] [Google Scholar]
- Alonzi T, Fattori E, Lazzaro D, Costa P, Probert L, Kollias G, De Benedetti F, Poli V, Ciliberto G. Interleukin-6 is required for the development of collagen-induced arthritis. J Exp Med. 1998;187:461–468. doi: 10.1084/jem.187.4.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Yoshizaki K, Kishimoto T, Ito H. IL-6 is required for the development of Th-1 cell-mediated murine colitis. J Immunol. 2000;164:4878–4882. doi: 10.4049/jimmunol.164.9.4878. [DOI] [PubMed] [Google Scholar]
- Atreya R, Mudter J, Finotto S, Mullberg J, Jostock T, Wirtz S, Schutz M, Bartsch B, Holtmann M, Becker C. et al. Blockade of interleukin-6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in Crohn's disease and experimental colitis in vivo. Nat Med. 2000;6:583–588. doi: 10.1038/75068. [DOI] [PubMed] [Google Scholar]
- Alonzi T, Fattori E, Lazzaro D, Costa P, Probert L, Kollias G, De Benedetti F, Poli V, Ciliberto G. Interleukin 6 is required for the development of collagen-induced arthritis. J Exp Med. 1998;187:461–468. doi: 10.1084/jem.187.4.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima S, Saeki Y, Mima T, Sasai M, Nishioka K, Nomura S, Kopf M, Katada Y, Tanaka T, Suemura M. et al. Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proc Natl Acad Sci USA. 1998;95:8222–8226. doi: 10.1073/pnas.95.14.8222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi N, Mihara M, Moriya Y, Nishimoto N, Yoshizaki K, Kishimoto T, Takeda Y, Ohsugi Y. Blockage of interleukin-6 receptor ameliorates joint disease in murine collagen-induced arthritis. Arthritis Rheum. 1998;41:2117–2121. doi: 10.1002/1529-0131(199812)41:12<2117::AID-ART6>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Nowell MA, Richards PJ, Horiuchi S, Yamamoto N, Rose-John S, Topley N, Williams AS, Jones SA. Soluble IL-6 receptor governs IL-6 activity in experimental arthritis: blockade of arthritis severity by soluble glycoprotein 130. J Immunol. 2003;171:3202–3209. doi: 10.4049/jimmunol.171.6.3202. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Yoshizaki K, Kishimoto T, Ito H. IL-6 is required for the development of Th1 cell-mediated murine colitis. J Immunol. 2000;164:4878–4882. doi: 10.4049/jimmunol.164.9.4878. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Hanada T, Mitsuyama K, Yoshida T, Kamizono S, Hoshino T, Kubo M, Yamashita A, Okabe M, Takeda K. et al. CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3 activation and intestinal inflammation. J Exp Med. 2001;193:471–481. doi: 10.1084/jem.193.4.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shouda T, Yoshida T, Hanada T, Wakioka T, Oishi M, Miyoshi K, Komiya S, Kosai K, Hanakawa Y, Hashimoto K. et al. Induction of the cytokine signal regulator SOCS3/CIS3 as a therapeutic strategy for treating inflammatory arthritis. J Clin Invest. 2001;108:1781–1788. doi: 10.1172/JCI200113568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalrymple SA, Lucian LA, Slattery R, McNeil T, Aud DM, Fuchino S, Lee F, Murray R. Interleukin-6-deficient mice are highly susceptible to Listeria monocytogenes infection: correlation with inefficient neutrophilia. Infect Immun. 1995;63:2262–2268. doi: 10.1128/iai.63.6.2262-2268.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Rani S, Liesenfeld O, Kojima T, Lim S, Nguyen TA, Dalrymple SA, Murray R, Remington JS. Impaired resistance to the development of toxoplasmic encephalitis in interleukin-6-deficient mice. Infect Immun. 1997;65:2339–2345. doi: 10.1128/iai.65.6.2339-2345.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani L, Mencacci A, Cenci E, Spaccapelo R, Toniatti C, Puccetti P, Bistoni F, Poli V. Impaired neutrophil response and CD4+ T helper cell 1 development in interleukin 6-deficient mice infected with Candida albicans. J Exp Med. 1996;183:1345–1355. doi: 10.1084/jem.183.4.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]