

Abstract
Although dietary, genetic, or disease-related excesses in urate production may contribute to hyperuricemia, impaired renal excretion of uric acid is the dominant cause of hyperuricemia in the majority of patients with gout. The aims of this review are to highlight exciting and clinically pertinent advances in our understanding of how uric acid is reabsorbed by the kidney under the regulation of urate transporter (URAT)1 and other recently identified urate transporters; to discuss urate-lowering agents in clinical development; and to summarize the limitations of currently available antihyperuricemic drugs. The use of uricosuric drugs to treat hyperuricemia in patients with gout is limited by prior urolothiasis or renal dysfunction. For this reason, our discussion focuses on the development of the novel xanthine oxidase inhibitor febuxostat and modified recombinant uricase preparations.
Introduction
Gout is a crystal deposition disease with clinical manifestations that include acute gouty arthritis, chronic gouty arthropathy, tophi, and renal functional impairment due to monosodium urate (MSU) crystal deposition; and urolithiasis and obstructive uropathy due to uric acid crystal deposition [1]. Gout ultimately results from inflammatory and/or degenerative responses to one or more derangements in the metabolism or physiology of urate, the obligatory end-product of human purine degradation [2]. In all untreated patients with gout, the body pool of urate exceeds normal, the level of serum urate is elevated, and the accompanying state of urate supersaturation predisposes to clinical events [3]. Persistent hyperuricemia (defined as a serum urate level >6.8 mg/dl) reflects extracellular fluid supersaturation for urate; it is simple to measure and is the primary risk factor for symptomatic gout. Although dietary, genetic, or disease-related excesses in urate production underlie hyperuricemia in some affected individuals [3], impaired renal excretion of uric acid is the dominant cause of hyperuricemia in the majority of patients with gout [1-3].
Urate physiology
A weak organic acid with a pKa1 of 5.75, uric acid is the final product of human purine metabolism. At the physiologic pH of 7.4 in extracellular fluid, the concentration of urate ion is approximately 50-fold that of the less soluble un-ionized uric acid. Because of the high concentration of sodium in extracellular fluid, urate is largely present as MSU; a consequence of this is that the appreciable solubility of urate ion (120 mg/dl at 37°C) is replaced by the much lower solubility of MSU (approximately 6.8 mg/dl). As urate concentrations increasingly exceed 6.8 mg/dl, the risk for urate crystal formation and precipitation increases. At pH 5.0 (often found in urine), undissociated uric acid predominates, with a solubility of approximately 10–15 mg/dl [3].
The human diet contains little urate. Urate is synthesized endogenously in the liver and, to a lesser extent, in the small intestine and circulates relatively free of protein binding (<4%), so that all, or nearly all, urate is filtered at the glomerulus before undergoing extensive net renal tubular reabsorption (see below). Purine ingestion, endogenous synthesis of purines from nonpurine precursors, and reutilization of preformed purine compounds are the sources of urate production, an overall process that under steady state conditions is balanced by uric acid disposal [4]. Daily renal uric acid excretion is equivalent to about two-thirds of daily production, and urate secretion into the small intestine, with breakdown of urate by gut bacteria (intestinal uricolysis), accounts for nearly all of the remainder of urate disposal [5].
Humans and certain other primate species lack expression of uricase [6], the enzyme that catalyzes conversion of urate to allantoin, which is a substantially more soluble product than urate and that is easily eliminated by renal excretion. Consequently, serum urate levels are several fold higher in normal humans than in rodents, for example. The body pool of urate in humans is normally composed entirely of soluble urate. In normal men and women the urate pools range from about 800 to 1500 mg and from about 500 to 1000 mg, respectively, with a daily turnover (the balanced production and disposal of urate) of about 0.6–0.7 pools/day [3,4]. Imbalance between the production and disposal of urate may result in expansion and supersaturation of the urate pool [3,4], sometimes resulting in urate crystal deposition and, ultimately, the formation of tophi, which may or may not be measurable in estimates of the miscible urate pool [3].
In about 90% of individuals with sustained hyperuricemia, impaired renal uric acid excretion is the dominant mechanism underlying expansion of the urate pool [1-3]. Important advances in our understanding of renal uric acid excretion are discussed below. Xanthine oxidase, the enzyme that catalyzes the terminal steps in urate production, namely oxidation of the purine bases hypoxanthine to xanthine and xanthine to uric acid, is a critical target of drug action in the treatment of hyperuricemia; this is also discussed below. Hyperuricemia may also be caused by excessive urate production alone or in combination with impaired renal uric acid excretion [1-3,7]. The pathways of purine metabolism [3], their normal regulation [8,9], and regulatory aberrations that lead to urate overproduction [2,3,8,9] are reviewed in detail elsewhere.
Impaired renal uric acid excretion in gout
Most individuals with hyperuricemia and impaired renal uric acid excretion have normal amounts of uric acid in their daily urine. However, excretion of normal amounts of uric acid in these individuals is accomplished only when serum urate levels are, on average, 2–3 mg/dl higher than in normal persons excreting comparable amounts of uric acid [10]. Renal hyperuricemia may be primary (idiopathic) and unassociated with an identifiable disorder or drug regimen, or it may be secondary to any of the broad array of disorders, treatments, or toxic states that disrupt the mechanisms involved in renal uric acid excretion [2]. Although some patients with primary hyperuricemia and gout may have impaired tubular urate secretory capacity [11], it seems unlikely that there is a unitary mechanism for hyperuricemia due to decreased renal uric acid excretion. In fact, the weight of evidence supports the view that, as in secondary gout, multiple renal processes for handling uric acid can be targets for disruption in patients with primary hyperuricemia.
Despite nearly complete filtration of urate at the glomerulus, uric acid clearance averages only 8–12% that of inulin or creatinine, reflecting the net reabsorption of about 90% of filtered urate. Over several decades, the multiple processes that mediate proximal convoluted tubule handling of filtered urate were operationally defined by studying the effects of inherited mutations and pharmacologic agents on renal uric acid excretion and by applying classic methods of renal physiology [12].
A multicompartmental model for renal uric acid handling, generated using the above approaches, included glomerular filtration; proximal tubule (S1 segment) reabsorption of virtually all filtered urate; secretion of 45–50% of reabsorbed urate (S1 and S2 segments of the proximal tubule); and postsecretory reabsorption of secreted urate (S3 segment of the proximal tubule). Thus, net excretion of uric acid is composed almost entirely of secreted rather than filtered urate. This model did not achieve consensus [12], however; in part this was because the compounds used to evaluate the components were not entirely specific in their sites of action, especially the use of drugs such as pyrazinamide and probenecid. Fortunately, the application of newer methodologies has begun to clarify the dynamics and molecular bases of renal uric acid excretion.
Central role of URAT1 in renal urate reabsorption and the target of uricosuric drugs
Recent molecular biological approaches have resulted in the cloning of an organic anion transporter (OAT) family member called urate transporter (URAT)1 (Fig. 1) [13]. URAT1 has highly specific urate transport activity, exchanging this anion with others including most of the endogenous organic anions (e.g. lactate) and drug anions (e.g. the monocarboxylate metabolite of pyrazinamide) that are known to affect renal uric acid transport. URAT1 localizes selectively to the luminal surface of the renal proximal tubular epithelial cell and mediates a non-voltage-dependent exchange, with properties compatible with a role for URAT1 as a major determinant of urate reabsorption [13,14]. Interaction of URAT1 with the PDZ domain-containing protein PDZK1 is essential for URAT1 to maintain a conformation for the urate exchange function [15]. Of clinical importance, URAT1-mediated urate reabsorption in the proximal tubule is potently suppressed by the uricosuric drugs probenecid, benzbromarone, sulfinpyrazone, and losartan [13] (Fig. 1).
Figure 1.
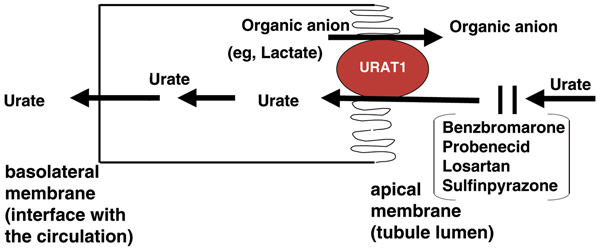
Urate anion transport function of URAT1 in renal proximal tubule epithelial cells. Schematic representation of urate reabsorption in the proximal tubule. Urate reabsorption at the apical (luminal) membrane is critically regulated by URAT1. The organic anion transporter URAT1 exchanges tubular lumen urate with anions inside proximal tubular epithelial cells. Potent stimulators of the exchange process include the intracellular organic ions lactate, nicotinate, and the monocarboxylate metabolite of the anti-tuberculous drug pyrazinimde. Certain intracellular inorganic ions, including chloride, also stimulate urate exchange by URAT1 but with lesser potency. As depicted, urate reabsorption is suppressed at the luminal membrane by some drugs, including benzbromarone, probenecid, losartan, and sulfinpyrazone, consistent with their urisouric properties. Other agents. that affect urate reabsorption in the proximal tubule, including furosemide and salicylates, may act partly by regulating URAT1 function.
That affected individuals in families with 'loss of function' mutations in the gene encoding URAT1 (SLC22A12) demonstrate hypouricemia and hyperuricosuria as well as functional renal impairment supports the suggestion that URAT1 is the most potent regulator of serum urate levels [13,14]. Additional studies support a role of URAT1 and possibly other urate transport/exchange molecules in hyperuricemia and gout. A specific URAT1 gene sequence variant is associated, when carried in the heterozygous state, with less frequent gout and substantially lower serum uric acid levels among Japanese individuals [16]. Pyrazinamide, benzbromarone, and probenecid do not significantly affect urinary uric acid clearance in subjects with defective URAT1 transport function [14]. Finally, sex, sex hormones, aging, diuretic therapy, and experimentally induced hyperuricemia (in rats) have been observed to modulate the intrarenal expression patterns of certain OAT family members [17-20]. For example, estrogen suppresses proximal tubule epithelial cell OAT expression [17], a finding that may explain the higher renal urate clearances, lower serum urate levels, and much reduced incidence of gout in premenopausal women as opposed to men and postmenopausal women.
UAT-1 and UMOD in renal uric acid disposition
A second candidate renal urate transporter, urate transporter/channel 1 (UAT-1), mediates voltage-dependent urate channeling in transfected cells and artificial membranes [21]. UAT-1 is more widely distributed in tissues than is URAT1, and it has been identified as galectin-9, one of a family of proteins with a number of nontransport functions including modulation of cell adhesion and differentiation [21,22]. The role of UAT-1 in the physiologic renal tubular transport or intestinal transport (specifically secretion) of urate remains to be established [13]. A number of other organic acid transporters and additional candidate exchangers may also participate in urate handling, particularly at the basolateral membrane of proximal tubule epithelial cells [23-25].
Further insight into the mechanisms that affect renal uric acid excretion has been provided by studies of familial juvenile hyperuricemic nephropathy (FJHN), an autosomal dominantly inherited disorder that is characterized by early onset of hyperuricemia (with or without gout), hypertension, and progressive renal failure culminating in end-stage renal disease by age 40 years [26]. Severely impaired renal uric acid clearance is present early, and the ensuing progressive nephropathy does not appear to be dependent on urate/uric acid crystal deposition [27]. Most affected families harbor a mutation in the chromosome 16p-linked UMOD gene, usually in a base that is involved in specifying a cysteine residue [28,29]. This gene encodes uromodulin (the Tamm–Horsfall glycoprotein), a disulfide bond-rich molecule that is normally the most abundant protein in human urine [30]. Among several mechanisms proposed to explain the relationship of altered uromodulin expression and the FJHN phenotype [31] is the hypothesis that uromodulin promotes maintenance of the structural and functional integrity of the ascending limb of the loop of Henle by providing a gel-like protective lattice. Compromise of uromodulin structure and function is believed to impair distal nephron sodium chloride reabsorption, with an ensuing fluid volume depletion that activates voltage-dependent sodium/urate cotransport at the proximal tubule. Mutations in UMOD also characterize variant forms of medullary cystic kidney disease (MCKD type 2) and glomerulocystic kidney disease (GCKD) as well as FJHN [32], which are thus allelic disorders with FJHN. These hyperuricemia-associated disorders are characterized by tubulointerstitial nephropathy and fibrosis, although glomerulocystic kidney disease is also associated with dilation of Bowman's space and glomerular tuft collapse. The possibility that other more subtle defects in uromodulin structure and expression may also explain gout in additional patients is under investigation.
Current and future roles of uricosuric agents in urate-lowering strategies
Uricosuric agents have long been used in the treatment of hyperuricemia associated with gout [33]. Probenecid, the only uricosuric agent currently used for this purpose in the USA, remains useful in patients with primary gout who have decreased renal uric acid excretion in the setting of well preserved renal function (creatinine clearance ≥60 ml/min) [1,34]. Unfortunately, all uricosuric agents currently available in the USA are generally ineffective and potentially nephrotoxic when used to treat hyperuricemia associated with moderate chronic kidney disease (creatinine clearance 30–60 ml/min) [33,34]. Given the marked rise in prevalence of both renal insufficiency and gout during the past 2 decades [35], therapy for hyperuricemia in complicated gout is likely to require approaches other than uricosuric drugs, such as safer and more effective xanthine oxidase inhibitors or modified recombinant uricase preparations.
Xanthine oxidase in the generation of uric acid and as a drug target for urate-lowering agents
Degradation of purine nucleotides and nucleosides to purine bases yields the key urate precursors hypoxanthine and guanine (Fig. 2). Ordinarily, most of the hypoxanthine and guanine produced is reutilized in a salvage reaction with 5-phosphoribosyl 1-pyrophosphate (PRPP), which is catalyzed by the enzyme hypoxanthine-guanine phosphoribosyltransferase (HPRT). The remaining guanine is deaminated to xanthine, and unsalvaged hypoxanthine is oxidized to xanthine, which undergoes further oxidation to uric acid. The enzyme xanthine oxidase catalyzes both the conversion of hypoxanthine to xanthine and xanthine to urate (Fig. 3). Xanthine oxidase is a molybdenum-pterin and iron sulfide cluster-containing flavoprotein that exists in two inter-convertible forms: an oxidase form that utilizes oxygen to convert hypoxanthine to xanthine and xanthine to urate, and a nicotinamide adenine dinucleotide (NAD+)-utilizing dehydrogenase form [36,37]. The catalytic events in purine base oxidation involve binding of the purine base substrate to the active site of the oxidase form, with reduction in the molybdenum moiety (valence +6) to reduced molybdenum (+4) by donation of electrons and oxidation of the substrate by water. Electrons are then transferred to the iron-sulfide cluster and then to the flavin center. Reoxidation of the flavin moiety by oxygen is accompanied by release of the product as well as generation of hydrogen peroxide (H2O2) and superoxide (O2-). Inhibition of xanthine oxidase activity is the major action of the most commonly employed class of agents used to lower urate levels in patients with gout.
Figure 2.
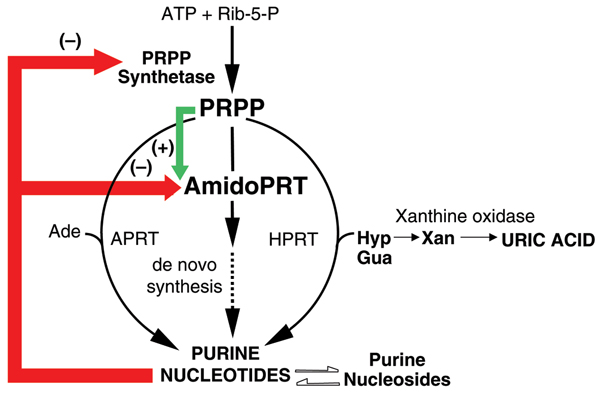
Xanthine oxidase in the context of purine metabolism. Schematic representation of human purine metabolic pathways, culminating in the production of uric acid. Purine nucleotides are synthesized by alternative pathways, each requiring the key regulatory intermediate 5-phosphoribosyl 1-pyrophosphate (PRPP), which is synthesized from ATP and ribose-5-P in a reaction catalyzed by PRPP synthetase. The pathway of purine synthesis de novo involves a sequence of 10 reactions by means of which a purine ring is synthesized on a ribose-phosphate backbone donated by PRPP. The first reaction in the pathway is the rate-limiting step and is catalyzed by the enzyme amidophopshoribosyltransferase (AmidoPRT). The subsequent 9 reactions in the de novo pathway are represented by the dashed arrow. The alternative pathways of purine nucleotide synthesis are single step processes by which preformed purine bases (adenine, Ade; hypoxanthine (Hyp); guanine, Gua) are salvaged in reactions catalyzed by the phosphoribosyltransferase (PRT) enzymes adeninePRT (APRT) and hypoxanthine-guanine (HPRT), respectively. Regulation of nucleotide synthesis is effected mainly at the AmidoPRT step, by means of antagonistic allosteric regulation of the activity of AmidoPRT by inhibitory (-) purine nucleotide products and PRPP activation. Purine nucleotide products also inhibit PRPP synthetase activity. Purine nucleotides and nucleosides are readily interconverted by means of an extensive and complex series of enzyme-catalyzed reactions that provide the cellular requirements for balanced availability of adenine and guanine nucleotides and nucleosides. Phosphorolysis of the nucleosides inosine and guanosine result in production of Hyp and Gua, which are either salvaged (in the HPRT reaction) or are ultimately and irreversibly oxidized through the base xanthine (Xan) to the end product, uric acid, in reactions catalyzed by xanthine oxidase.
Figure 3.

Reactions catalyzed by xanthine oxidase (also known as xanthine oxidoreductase). The enzyme exists in dehydrogenase and oxidase forms, accepting NAD+ in the former conformation and O2 in the latter. Reversible interconversion of the forms involves disulfide bond formation and disruption.
Xanthine oxidase inhibitors
Agents that function through direct inhibition of xanthine oxidase activity include allopurinol and oxypurinol, which are hydroxypyrazolopyrimidine analogs of hypoxanthine and xanthine, respectively; and febuxostat, which is a thiazolecarboxylic acid derivative that is not a purine analog (Fig. 4).
Figure 4.
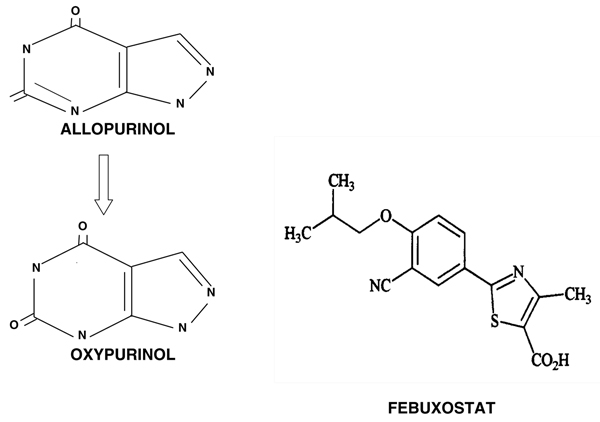
Comparison of structures of allopurinol, oxypurinol, and febuxostat. Clinically utilized xanthine oxidase inhibitors. Allopurinol and its oxidation product oxypurinol are hydroxypyrazolopyrimidine analogues, respectively, of hypoxanthine and xanthine. As such, each can affect the activities of enzymes of purine and pyrimidine metabolism other than xanthine oxidase. Febuxostat is a thiazolecarboxylic acid derivative that does not resemble a purine or pyrimidine and has shown substantial specificity as a xanthine oxidase inhibitor [50].
Allopurinol
Allopurinol is the most commonly used antihyperuricemic agent and is the only xanthine oxidase inhibitor approved to date by the US Food and Drug Administration. This drug inhibits xanthine oxidase by a competitive mechanism with respect to the natural purine base substrates [3,37,38]. Allopurinol binds to the oxidized form of xanthine oxidase at the active site Mo6+-pterin coenzyme, and, in the process of reducing it, generates the oxidized form of allopurinol, namely oxypurinol. Advantages of allopurinol are a once-daily dosing regimen and urate-lowering efficacy regardless of the cause of hyperuricemia.
Few studies comparing the relative efficacy of xanthine oxidase inhibitors and uricosuric agents have been reported. In one prospective study [39], drug efficacy in reducing serum urate concentrations in primary chronic gout was tested in 86 male patients. Of these, 49 patients (including 26 who excreted normal amounts of urinary uric acid daily and 23 with under-excretion) received allopurinol 300 mg/day. A total of 37 under-excretors received benzbromarone (a potent uricosuric drug) at 100 mg/day. Treatment was adjusted to achieve plasma urate concentrations under 6.0 mg/dl. Among patients who received allopurinol, mean plasma urate reductions of 2.75 mg/dl and 3.34 mg/dl were achieved in normal excretors and under-excretors, respectively; a mean reduction of 5.04 mg/dl was observed among those who received benzbromarone (Fig. 5). Only 53% of patients receiving allopurinol at the commonly used dose of 300 mg/day achieved the target urate level as compared with 100% of benzbromarone-treated patients. Increasing the allo-purinol dose improved the urate-lowering efficacy achieved with this drug.
Figure 5.
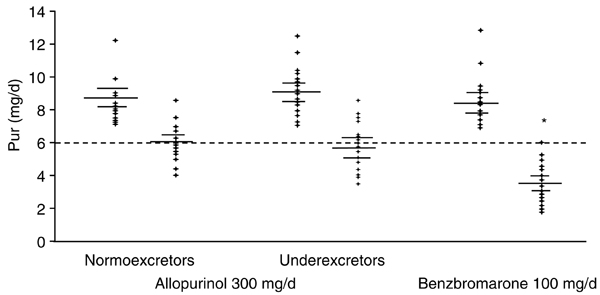
Comparative effects of allopurinol and benzbromarone on plasma urate levels. Initial and final plasma urate (Pur) after standard doses of urate-lowering drugs allopurinol (300 mg/day) or benzbromarone (100 mg/day). *For final versus initial plasma urate, P < 0.01 for allopurinol groups and P < 0.001 for benzbromarone group. Adapted from [39]. Copyright 1998 with permission from BMJ Publishing Group.
Major limitations of allopurinol are summarized in Table 1. The most common undesired effect of xanthine oxidase inhibition is precipitation of acute attacks of gout, particularly among patients in whom colchicine or nonsteroidal anti-inflammatory drug prophylaxis is not utilized. Because sudden increases or decreases in serum urate concentration can trigger acute gouty attacks, xanthine oxidase inhibitor therapy is generally not initiated during an acute attack of gout. A major side effect of allopurinol is hypersensitivity. Rash develops in about 2% of patients receiving allopurinol and is generally reversible on discontinuation of the drug [40]. Severe allopurinol hypersensitivity [41], which can include not only exfoliative dermatitis but also vasculitis and multiorgan system failure, occurs in under 1/1000 patients and is more likely to occur in patients with renal disease or those receiving thiazide diuretics and standard doses of allopurinol. In Han Chinese, severe allopurinol cutaneous reactions were recently linked to the HLA-B*5801 haplotype [42], a finding that has not yet been confirmed in other populations. Current urate-lowering options in allopurinol-hypersensitive gout patients who have had mild-to-moderate but not severe adverse reactions and who are not candidates for uricosuric therapy include: oral allopurinol desensitization, which is efficacious in about 50% of subjects [43]; and the use of oxypurinol (the active metabolite of allopurinol; described below). Cross-sensitivity of allopurinol and oxypurinol limits effective use of oxypurinol in allopurinol-hypersensitive patients.
Table 1.
Major limitations of allopurinol therapy
| Rash in about 2% of treated patients |
| Additional intolerance in up to 10% of patients (includes hepatic enzyme, gastrointestinal, and central nervous system effects) |
| Allopurinol hypersensitivity syndrome: <1/1000 treated patients, but 20% fatality rate |
| Prolonged renal elimination of active oxypurinol metabolite necessitates reduced dose in renal disease with impaired function |
| Contraindicated in patients receiving azathioprine or 6-mercaptopurine |
Other allopurinol side effects, including hepatic toxicity with serum transaminase elevation, render approximately 10% of treated persons unable to tolerate this agent. Furthermore, for patients with chronic kidney disease [40] the prolonged half-life of renal elimination of oxypurinol limits the recommended maximum allopurinol dosage because of concerns about an increased incidence of hypersensitivity reactions. A major issue currently limiting effective allopurinol use is patient compliance. In one study [44], for example, compliance with allopurinol over 2 years of treatment was only about 20%. It seems likely that insufficient patient education regarding the long-term aims of urate-lowering therapy contributes to this problem, which is shared by therapies for other treatable disorders that may be asymptomatic for substantial periods of time.
Oxypurinol
Oxypurinol [45] is currently only available on a compassionate-use basis in the USA and Canada, and may be useful and effective in most (but not all) patients with mild-to-moderate allopurinol intolerance.
Like allopurinol, oxypurinol is a competitive inhibitor of xanthine oxidase. In contrast to allopurinol, oxypurinol binds to the reduced form of the enzyme with very high affinity but is released from xanthine oxidase when the enzyme is reoxidized. Oral absorption of oxypurinol is poor relative to allopurinol, and oxypurinol doses may require extended titration to achieve satisfactory lowering of serum urate.
Febuxostat
Febuxostat is an orally administered selective inhibitor of xanthine oxidase that is not a purine analog. The mechanism of febuxostat inhibition of the enzyme is mixed inhibition, contrasting with the competitive kinetics exhibited by allopurinol. This difference in kinetic mechanism is consistent with the findings that febuxostat blocks substrate access to the molybdenum-pterin moiety of xanthine oxidase by occupying a channel in the enzyme leading to the active site (Fig. 6), and that febuxostat inhibits both the oxidized and reduced forms of xanthine oxidase [46,47]. In animal studies [47-49] febuxostat was shown to provide more potent and longer lasting hypouricemic activity than allopurinol, and has minimal effects on other enzymes that are involved in purine and pyrimidine metabolism (Fig. 7) [50]. Unlike currently available xanthine oxidase inhibitors, febuxostat is metabolized primarily by hepatic glucuronide formation and oxidation and is excreted in about equal proportions in stool and urine [51].
Figure 6.
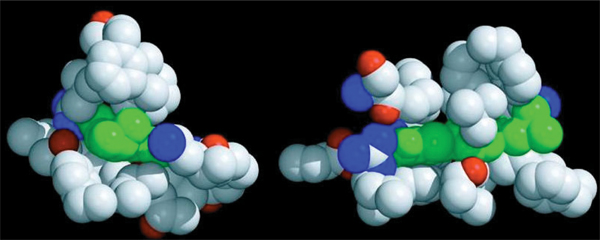
Febuxostat blockade of substrate access to the active site of both reduced and oxidized xanthine oxidase. Space-taking representation of the interaction of febuxostat and amino acid residues of xanthine oxidase. Febuxostat, represented in green, occupies the channel accessing the Mo-pterin moiety in the active site of xanthine oxidase. In contrast to allopurinol and oxypurinol, which bind directly to the active site and result in competitive inhibition of enzyme activity, febuxostat blocks substrate access to the channel resulting in a pattern of mixed inhibition of enzyme activity. Reprinted with permission from [46]. © 2006 American Society for Biochemistry and Molecular Biology.
Figure 7.
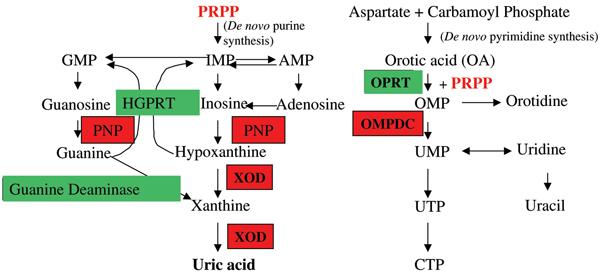
Multiple sites of allopurinol action on pathways of purine and pyrimidine metabolism. Reactions inhibited by allopurinol are shown in red; reactions unaffected are shown in green. Allopurinol or its metabolites also result in depletion of intracellular 5-phosphoribosyl 1-pyrophosphate (PRPP) concentrations. AMP, adenosine monophosphate; CTP, cytosine triphosphate; GMP, guanosine monophosphate; HGPRT, hypoxanthine-guanine phosphoribosyltransferase; IMP, inosine monophosphate; OMPDC, orotidine-5'-monophosphate decarboxylase; OMP, orotidine monophosphate; OPRT, orotate phosphoribosyltransferase; PNP, purine nucleoside phosphorylase; PRPP, 5-phosphoribosyl 1-pyrophosphate; UMP, uridine monophosphate; UTP, uridine triphosphate; XOD, xanthine oxidase.
The efficacy of febuxostat was examined in a phase II study [52] in which 153 patients with gout and hyperuricemia (baseline serum urate levels ≥ 8.0 mg/dl) were randomly assigned to therapy with febuxostat (40, 80, or 120 mg/day) or to placebo once daily for 28 days. Patients received colchicine prophylaxis during a 2-week washout period and for the first 2 weeks of double-blind treatment. The primary end-point was the percentage of patients with serum urate levels below 6.0 mg/dl after 28 days of therapy. The primary end-point was achieved in 0% of patients in the placebo group and, in a dose-related manner, in between 56% and 94% of patients in the febuxostat-treated groups. Mean serum urate reduction from baseline was 2% in the placebo group; among patients in the febuxostat groups the reductions ranged from 37% in the 40 mg/day group to 59% in the 120 mg/day group. The incidence of treatment-related adverse events was similar in the placebo and 40 mg/day febuxostat groups. Gout flares occurred in a similar percentage of patients in the febuxostat 40 mg/day and placebo groups (37% and 35%, respectively), with higher frequency among patients who received the 80 mg/day dose (43%) and 120 mg/day dose (55%) of febuxostat. The proportions of patients in the intent-to-treat population who achieved the primary end-point are shown in Fig. 8.
Figure 8.
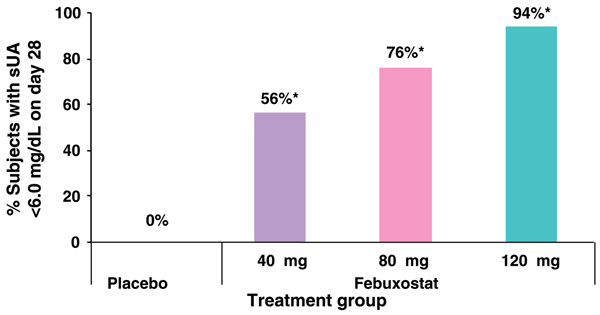
Serum urate-lowering efficacy of febuxostat compared with placebo. Subjects with gout and baseline serum urate levels > 8.0 mg/dl received once daily doses of placebo or febuxostat for 28 days. Modified from [52].
In a recently reported 1-year phase III randomized controlled trial comparing 80 mg/day and 120 mg/day doses of febuxostat with allopurinol at 300 mg/day in patients with gout and serum urate levels of 8.0 mg/dl or greater [53], both doses of febuxostat resulted in sustained and superior (to allopurinol) urate-lowering efficacy. Nevertheless, after 1 year of treatment, the secondary outcomes of reduction in gout flares and tophus area were not different in the febuxostat and allopurinol group, and the overall incidence of treatment-related adverse events-most of which were mild and moderate in severity- was similar for all treatment groups. However, a significantly greater number of patients in the febuxostat 120/day group discontinued therapy (P = 0.003).
Uricase
The importance of the hepatic enzyme uricase in urate balance has been underlined by the marked hyperuricemia and renal tubulopathy induced by engineered uricase gene inactivation in the mouse. Serum urate levels in uricase knockout mice are about 10-fold higher than in wild-type mice (1 mg/dl) [54]. Recombinant Aspergillus flavus uricase (Rasburicase™; Sanofi-Synthelabo, Malvern, PA, USA), administered in a single short-term course, is US Food and Drug Administration approved for prevention of tumor lysis syndrome [55]. Recombinant uricase therapy profoundly lowers serum urate levels. Unlike currently available urate-lowering therapies, uricase can promote accelerated tophus dissolution in a therapeutic course limited to 3 months in duration [56]. However, unmodified uricase is highly immunogenic and can trigger severe and potentially lethal side effects (e.g. anaphylaxis) [57-59]. Moreover, hydrogen peroxide, a product of the uricase reaction (Fig. 9), stimulates cell transformation in vitro [60], raising concern with respect to long-term therapy with uricase. Uricase-induced redox stress can trigger hemolysis and methemoglobinemia in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency [57-59], precluding uricase treatment of patients with this disorder. This also prompts the question of whether concurrently clearing the serum of the major circulating antioxidant (urate) and generating hydrogen peroxide without providing for alternative antioxidants will compromise the unstable lesions (e.g. arteriosclerotic plaques or other sites of chronic redox stress) in which urate can accumulate [61,62].
Figure 9.
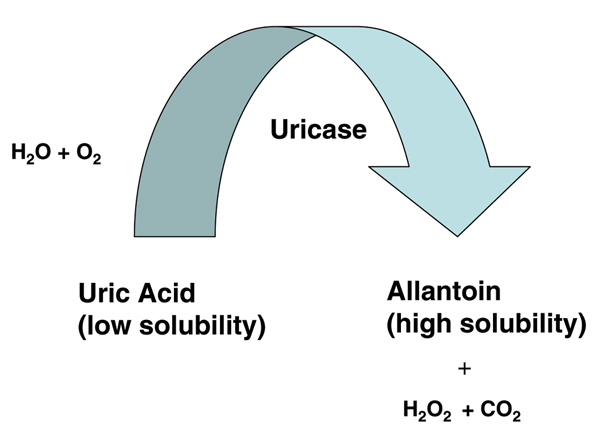
Action of uricase to catabolize uric acid to allantoin. Uricase (urate oxidase) gene expression and the depicted uricase reaction are lacking in humans, as discussed in the text. PEGylated recombinant uricase preparations are now under investigation for treatment of refractory hyperuricemia in gout, and have the potential to rapidly reduce ("de-bulk") tophus burden.
Neutralizing antibodies frequently develop in the course of treatment with purified native or unmodified recombinant uricase [55,57-59]. In order to reduce antigenicity and prolong the half-life of uricase activity, polyethyleneglycol modified forms of recombinant uricase have been developed, and two such biologic agents are currently in clinical trials [63,64]. It is likely that the therapeutic niches for modified uricases in the treatment of gout will be for short-term acceleration of tophus dissolution ('debulking') in carefully selected patients with large tophus burdens and perhaps for more long-term use in patients intolerant of or unresponsive to all other forms of urate-lowering therapy.
Conclusion
Gout is among the oldest recognized rheumatic diseases. Current research into the physiologic mechanisms and molecular processes that underlie hyperuricemia and gout has identified a range of targets for pharmacotherapy and is fostering the development of new drugs and biologic agents for reversing hyperuricemia. The future promises more effective treatments for patients with difficult-to-manage gout, including those with renal insufficiency, allopurinol intolerance, and extensive tophi.
Abbreviations
FJHN = familial juvenile hyperuricemic nephropathy; MSU = monosodium urate; OAT = organic anion transporter; UAT = urate transporter/channel 1; URAT = urate transporter.
Competing interests
RT and MAB serve as paid consultants for TAP Pharmaceutical Products Inc. DAB has declared no relevant financial relationships.
References
- Terkeltaub RA. Clinical practice. Gout. N Engl J Med. 2003;349:1647–1655. doi: 10.1056/NEJMcp030733. [DOI] [PubMed] [Google Scholar]
- Becker MA, Jolly M. In: Arthritis and Allied Conditions. 15. Koopman WJ, Moreland LW, editor. Philadelphia: Lippincott, Williams & Wilkins; 2005. Clinical gout and the pathogenesis of hyperuricemia; pp. 2303–2339. [Google Scholar]
- Wyngaarden JB, Kelley WN. Gout and Hyperuricemia. New York: Grune & Stratton; 1976. pp. 1–512. [Google Scholar]
- Benedict JD, Forsham PH, Stetten DeW Jr. The metabolism of uric acid in the normal and gouty human studied with the aid of isotopic uric acid. J Biol Chem. 1949;181:183–193. [PubMed] [Google Scholar]
- Sorensen LB. Degradation of uric acid in man. Metabolism. 1959;8:687–703. [PubMed] [Google Scholar]
- Watanabe S, Kang DH, Feng L, Nakagawa T, Kanellis J, Lan H, Mazzali M, Johnson RJ. Uric acid, hominoid evolution, and the pathogenesis of slat sensitivity. Hypertension. 2002;40:355–360. doi: 10.1161/01.HYP.0000028589.66335.AA. [DOI] [PubMed] [Google Scholar]
- Perez-Ruiz F, Calabozo M, Garcia-Erauskin G, Ruibal A, Herrero-Beites AM. Renal underexcretion of uric acid is present in patients with apparent high urinary uric acid output. Arthritis Rheum. 2002;47:610–613. doi: 10.1002/art.10792. [DOI] [PubMed] [Google Scholar]
- Becker MA, Kim M. Regulation of rates of purine synthesis de novo by purine nucleotides and phosphoribosylpyrophosphate. J Biol Chem. 1987;262:14532–14537. [PubMed] [Google Scholar]
- Becker MA. Phosphoribosylpyrophosphate synthetase and the regulation of phosphoribosylpyrophosphate production in human cells. Prog Nucl Acid Res Mol Biol. 2001;69:115–148. doi: 10.1016/s0079-6603(01)69046-9. [DOI] [PubMed] [Google Scholar]
- Simkin PA. Urate excretion in normal and gouty men. Adv Exp Med Biol. 1977;76B:41–45. doi: 10.1007/978-1-4684-3285-5_5. [DOI] [PubMed] [Google Scholar]
- Rieselbach R, Sorensen LB, Shelp WD, Steele TH. Diminished renal urate secretion per nephron as a basis for gout. Ann Intern Med. 1970;70:359–366. doi: 10.7326/0003-4819-73-3-359. [DOI] [PubMed] [Google Scholar]
- Roch-Ramel F, Diezi J. In: Diseases of the Kidney. 6. Schreier RW, Gottschalk CE, editor. Boston: Little Brown; 1996. Renal transport of organic ions and uric acid; pp. 231–249. [Google Scholar]
- Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, Hosoyamada M, Takeda M, Sekine T, Igarashi T. et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature. 2002;417:447–452. doi: 10.1038/nature742. [DOI] [PubMed] [Google Scholar]
- Ichida K, Hosoyamada M, Hisatome I, Enomoto A, Hikita M, Endou H, Hosoya T. Clinical and molecular analysis of patients with renal hypouricemia in Japan: influence of URAT1 gene on urinary urate excretion. J Am Soc Nephrol. 2004;15:164–173. doi: 10.1097/01.ASN.0000105320.04395.D0. [DOI] [PubMed] [Google Scholar]
- Anzai N, Miyazaki H, Noshiro R, Khamdang S, Chairoungdua A, Shin HJ, Enomoto A, Sakamoto S, Hirata T, Tomita K, Kanai Y, Endou H. The multivalent PDZ-domain-containing protein PDZKi1 regulates transport activity of renal urate-anion exchanger urate1 via its C terminus. J Biol Chem. 2004;279:45942–45950. doi: 10.1074/jbc.M406724200. [DOI] [PubMed] [Google Scholar]
- Taniguchi A, Urano W, Yamanaka M, Yamanaka H, Hosoyamada M, Endou H, Kamatani N. A common mutation in an organic ion transporter gene, SLC22A12, is a suppressing factor for the development of gout. Arthritis Rheum. 2005;52:2576–2577. doi: 10.1002/art.21242. [DOI] [PubMed] [Google Scholar]
- Ljubojevic M, Herak-Kramberger CM, Hagos Y, Bahn A, Endou H, Burckhardt G, Sabolic I. Rat renal OAT1 and OAT3 exhibit gender differences determined by both androgen stimulation and estrogen inhibition. Am J Physiol Renal Physiol. 2004;287:F124–F138. doi: 10.1152/ajprenal.00029.2004. [DOI] [PubMed] [Google Scholar]
- Kim GH, Na KY, Kim SY, Joo KW, Oh YK, Chae SW, Endou H, Han JS. Up-regulation of organic anion transporter 1 protein is induced by chronic furosemide or hydrochlorothiazide infusion in rat kidney. Nephrol Dial Transplant. 2003;18:1505–1511. doi: 10.1093/ndt/gfg186. [DOI] [PubMed] [Google Scholar]
- Kojima R, Sekine T, Kawachi M, Cha SH, Suzuki Y, Endou H. Immunolocalization of multispecific organic anion transporters, OAT1, OAT2, and OAT3, in rat kidney. J Am Soc Nephrol. 2002;13:848–857. doi: 10.1681/ASN.V134848. [DOI] [PubMed] [Google Scholar]
- Habu Y, Yano I, Takeuchi A, Saito H, Okuda M, Fukatsu A, Inui K. Decreased activity of basolateral organic ion transports in hyperuricemic rat kidney: roles of organic ion transporters, rOAT1, rOAT3 and rOCT2. Biochem Pharmacol. 2003;66:1107–1114. doi: 10.1016/S0006-2952(03)00466-0. [DOI] [PubMed] [Google Scholar]
- Lipkowitz MS, Leal-Pinto E, Rappoport JZ, Najfeld V, Abramson RG. Functional reconstitution, membrane targeting genomic structure, and chromosomal localization of a human urate transporter. J Clin Invest. 2001;107:1103–1115. doi: 10.1172/JCI12471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkowitz MS, Leal-Pinto E, Cohen BE, Abramson RG. Galectin 9 is the sugar-regulated urate transporter/channel UAT. Glycoconj J. 2004;19:491–498. doi: 10.1023/B:GLYC.0000014078.65610.2f. [DOI] [PubMed] [Google Scholar]
- Anzai N, Enomoto A, Endou H. Renal urate handling: clinical relevance of recent advances. Curr Rheumatol Rep. 2005;7:227–234. doi: 10.1007/s11926-996-0044-0. [DOI] [PubMed] [Google Scholar]
- Rafey MA, Lipkowitz MS, Leal-Pinto E, Abramson RG. Uric acid transport. Curr Opin Nephrol Hypertens. 2003;12:511–516. doi: 10.1097/00041552-200309000-00005. [DOI] [PubMed] [Google Scholar]
- Hediger MA, Johnson RJ, Miyazaki H, Endou H. Molecular physiology of urate transport. Physiology. 2005;20:125–133. doi: 10.1152/physiol.00039.2004. [DOI] [PubMed] [Google Scholar]
- Calabrese G, Simmonds HA, Cameron JS, Davies PM. Precocious familial gout with reduced fractional urate clearance and normal purine enzymes. QJM. 1990;78:441–445. [PubMed] [Google Scholar]
- Puig JG, Miranda ME, Mateos FA, Picazo ML, Jimenez ML, Calvin TS, Gil AA. Hereditary nephropathy associated with hyperuricemia and gout. Arch Intern Med. 1993;153:357–365. doi: 10.1001/archinte.153.3.357. [DOI] [PubMed] [Google Scholar]
- Dahan K, Devuyst O, Smaers M, Vertommen D, Loute G, Poux JM, Viron B, Jacquot C, Gagnadoux MF, Chauveau D. et al. A cluster of mutations in the UMOD gene causes familial juvenile hyperuricemic nephropathy with abnormal expression of uromodulin. J Am Soc Nephrol. 2003;14:2883–2893. doi: 10.1097/01.ASN.0000092147.83480.B5. [DOI] [PubMed] [Google Scholar]
- Kudo E, Kamatani N, Tezuka O, Taniguchi A, Yamanaka H, Yabe S, Osabe D, Shinohara S, Nomura K, Segawa M. et al. Familial juvenile hyperuricemic nephropathy: Detection of mutations in the uromodulin gene in five Japanese families. Kidney Int. 2004;65:1589–1597. doi: 10.1111/j.1523-1755.2004.00559.x. [DOI] [PubMed] [Google Scholar]
- Weichhart T, Zlabinger GJ, Saemann MD. The multiple functions of Tamm-Horsfall protein in human health and disease: a mystery clears up. Wien Klin Wochenschr. 2005;117:316–322. doi: 10.1007/s00508-005-0353-8. [DOI] [PubMed] [Google Scholar]
- Cameron JS, Simmonds HA. Hereditary hyperuricemia and renal disease. Semin Nephrol. 2005;25:9–18. doi: 10.1016/j.semnephrol.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Rampoldi L, Caridi G, Santon D, Boaretto F, Bernascone I, Lamorte G, Tardanico R, Dagnino M, Colussi G, Scolari F. et al. Allelism of MCKD, FJHN, and GCKD caused by impairment of uromoduline export dynamics. Hum Molec Genet. 2003;12:3369–3384. doi: 10.1093/hmg/ddg353. [DOI] [PubMed] [Google Scholar]
- Perez-Ruiz F, Inaki H, Herrero-Beites AM. In: Crystal-induced Arthropathies. Wortmann RL, Schumacher HR Jr, Becker MA, Ryan LM, editor. New York: Elsevier; Uricosuric therapy. in press . [Google Scholar]
- Emmerson BT. The management of gout. N Engl J Med. 1996;334:445–451. doi: 10.1056/NEJM199602153340707. [DOI] [PubMed] [Google Scholar]
- Bieber JD, Terkeltaub RA. Gout: on the brink of novel therapeutic options for an ancient disease. Arthritis Rheum. 2004;50:2400–2414. doi: 10.1002/art.20438. [DOI] [PubMed] [Google Scholar]
- Hille R, Nishino T. Flavoprotein structure and mechanism. 4. Xanthine oxidase and xanthine dehydrogenase. FASEB J. 1995;9:995–1003. [PubMed] [Google Scholar]
- Raivio KO, Saksela M, Lapatto R. In: The Metabolic and Molecular Bases of Inherited Disease. 8. Scriver CR, Beaudet AL, Sly WS, Valle D, editor. New York: McGraw-Hill; 2001. Xanthine oxidoreductase-role in human pathophysiology and in hereditary xanthinuria; pp. 2639–2652. [Google Scholar]
- Berry CE, Hare JM. Xanthine oxidoreductase and cardiovascular disease. Molecular mechanisms and pathophysiological implications. J Physiol. 2004;555:589–606. doi: 10.1113/jphysiol.2003.055913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Ruiz F, Alonso-Ruiz A, Calabozo M, Herrero-Beites A, Garcia-Erauskin G, Ruiz-Lucea E. Efficacy of allopurinol and benzbromarone for the control of hyperuricaemia. A pathogenic approach to the treatment of primary chronic gout. Ann Rheum Dis. 1998;57:545–549. doi: 10.1136/ard.57.9.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fam AG. Difficult gout and new approaches for control of hyperuricemia in the allopurinol-allergic patient. Curr Rheumatol Rep. 2001;3:29–35. doi: 10.1007/s11926-001-0048-8. [DOI] [PubMed] [Google Scholar]
- Arellano F, Sacristan JA. Allopurinol hypersensitivity syndrome: a review. Ann Pharmacother. 1993;27:337–343. doi: 10.1177/106002809302700317. [DOI] [PubMed] [Google Scholar]
- Hung S-I, Chung W-H, Liou L-B, Chu CC, Lin M, Huang HP, Lin YL, Lan JL, Yang LC, Hong HS. et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci USA. 2005;102:4134–4139. doi: 10.1073/pnas.0409500102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fam AG, Dunne SM, Iazetta J, Paton TW. Efficacy and safety of desensitization to allopurinol following cutaneous reactions. Arthritis Rheum. 2001;44:231–238. doi: 10.1002/1529-0131(200101)44:1<231::AID-ANR30>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Riedel AA, Nelson M, Joseph-Ridge N, Wallace K, MacDonald P, Becker M. Compliance with allopurinol therapy among managed care enrollees with gout: a retrospective analysis of administrative claims. J Rheumatol. 2004;31:1575–1581. [PubMed] [Google Scholar]
- Walter-Sack I, de Vries JX, Kutschker C, Ittensohn A, Voss A. Disposition and uric acid lowering effect of oxipurinol: comparison of different oxipurinol formulations and allopurinol in healthy individuals. Eur J Clin Pharmacol. 1995;49:215–220. doi: 10.1007/BF00192382. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Eger BT, Nishino T, Kondo S, Pai EF, Nishino T. An extremely potent inhibitor of xanthine oxidoreductase. Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J Biol Chem. 2003;278:1848–1855. doi: 10.1074/jbc.M208307200. [DOI] [PubMed] [Google Scholar]
- Horiuchi H, Ota M, Kobayashi M, Kaneko H, Kasahara Y, Nishimura S, Kondo S, Komoriya K. A comparative study on the hypouricemic activity and potency in renal xanthine calculus formation of two xanthine oxidase/xanthine dehydrogenase inhibitors: TEI-6720 and allopurinol in rats. Res Commun Mol Pathol Pharmacol. 1999;104:307–319. [PubMed] [Google Scholar]
- Osada Y, Tsuchimoto M, Fukushima H, Takahashi K, Kondo S, Hasegawa M, Komoriya K. Hypouricemic effect of the novel xanthine oxidase inhibitor, TEI-6720, in rodents. Eur J Pharmacol. 1993;241:183–188. doi: 10.1016/0014-2999(93)90201-R. [DOI] [PubMed] [Google Scholar]
- Komoriya K, Osada Y, Hasegawa M, Horiuchi H, Kondo S, Couch RC, Griffin TB. Hypouricemic effect of allopurinol and the novel xanthine oxidase inhibitor TEI-6720 in chimpanzees. Eur J Pharmacol. 1993;250:455–460. doi: 10.1016/0014-2999(93)90033-E. [DOI] [PubMed] [Google Scholar]
- Takano Y, Hase-Aoki K, Horiuchi H, Zhao L, Kasahara Y, Kondo S, Becker MA. Selectivity of febuxostat, a novel non-purine inhibitor of xanthine oxidase/xanthine dehydrogenase. Life Sci. 2005;76:1835–1847. doi: 10.1016/j.lfs.2004.10.031. [DOI] [PubMed] [Google Scholar]
- Hoshide S, Nishimura S, Ishii S, Matsuzawa K, Saito N, Tanaka T. Metabolites of TMX-67, a new pharmaceutical entity for the treatment of gout or hyperuricemia, and their pharmaco-kinetic profiles in humans. Drug Metab Rev. 2000. p. 269.
- Becker MA, Schumacher HR Jr, Wortmann RL, MacDonald PA, Palo WA, Eustace D, Vernillet L, Joseph-Ridge N. Febuxostat, a novel nonpurine selective inhibitor of xanthine oxidase: a twenty-eight-day, multicenter, phase II, randomized, double-blind, placebo-controlled, dose-response clinical trial examining safety and efficacy in patients with gout. Arthritis Rheum. 2005;52:916–923. doi: 10.1002/art.20935. [DOI] [PubMed] [Google Scholar]
- Becker MA, Schumacher HR Jr, Wortmann RL, MacDonald PA, Eustace D, Palo WA, Streit J, Joseph-Ridge N. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med. 2005;353:2450–2461. doi: 10.1056/NEJMoa050373. [DOI] [PubMed] [Google Scholar]
- Kelly SJ, Delnomdedieu M, Oliverio MI. Diabetes insipidus in uricase-deficient mice: a model for evaluating therapy with poly(ethylene glycol)-modified uricase. J Am Soc Nephrol. 2001;12:1001–1009. doi: 10.1681/ASN.V1251001. [DOI] [PubMed] [Google Scholar]
- Pui C-H, Jeha S, Carnitta B. Recombinant urate oxidase (ras-buricase) in the prevention and treatment of malignancy-associated hyperuricemia in pediatric and adult patients: results of a compassionate-use trial. Leukemia. 2001;15:1505–1509. doi: 10.1038/sj.leu.2402235. [DOI] [PubMed] [Google Scholar]
- Baraf HSB, Kim S, Matsumoto AK. et al. Resolution of tophi with intravenous Peg-uricase in refractory gout. Arthritis Rheum. 2005;52:S105. doi: 10.1002/art.20724. [DOI] [Google Scholar]
- Pui CH. Urate oxidase in the prophylaxis or treatment of hyperuricemia: the United States experience. Semin Hematol. 2001. pp. 13–21. [DOI] [PubMed]
- Goldman SC. Rasburicase: potential role in managing tumor lysis in patients with hematological malignancies. Expert Rev Anticancer Ther. 2003;3:429–433. doi: 10.1586/14737140.3.4.429. [DOI] [PubMed] [Google Scholar]
- Yim BT, Sims-McCallum RP, Chong PH. Rasburicase for the treatment and prevention of hyperuricemia. Ann Pharmacother. 2003;37:1047–1054. doi: 10.1345/aph.1C336. [DOI] [PubMed] [Google Scholar]
- Chu R, Lin Y, Reddy KC, Pan J, Rao MS, Reddy JK, Yeldandi AV. Transformation of epithelial cells stably transfected with H2O2-generating peroxisomal urate oxidase. Cancer Res. 1996;56:4846–4852. [PubMed] [Google Scholar]
- Patetsios P, Rodino W, Wisselink W, Bryan D, Kirwin JD, Panetta TF. Identification of uric acid in aortic aneurysms and athero-sclerotic artery. Ann N Y Acad Sci. 1996;800:243–245. doi: 10.1111/j.1749-6632.1996.tb33318.x. [DOI] [PubMed] [Google Scholar]
- Langemann H, Feuerstein T, Mendelowitsch A, Gratzl O. Microdialytical monitoring of uric and ascorbic acids in the brains of patients after severe brain injury and during neurovascular surgery. J Neurol Neurosurg Psychiatry. 2001;71:169–174. doi: 10.1136/jnnp.71.2.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundy JS, Becker MA, Baraf HSB, Barkhuizen A, Moreland L, Huang B, Alton M. A phase 2 study of multiple doses of intravenous polyethylene glycol (PEG)-uricase in patients with hyperuricemia and refractory gout. Arthritis Rheum. 2005;52:S679. doi: 10.1002/art.20959. [DOI] [Google Scholar]
- Bomalaski JS, Goddard DH, Grezlak D. Phase I study of uricase formulated with polyethylene glycol (Uricase-PEG 20) Arthritis Rheum. 2002;46:S141. [PubMed] [Google Scholar]