

Abstract
Synaptic pathology in Alzheimer's disease brains is thought to involve soluble Aβ42 peptide. Here, sterile incubation in PBS caused small Aβ42 oligomer formation as well as heterogeneous, 6E10-immunopositive aggregates of 80-100 kDa. High molecular weight aggregates (H-agg) formed in a time-dependent manner over an extended 30-day period. Interestingly, an inverse relationship between dimeric and H-agg formation was more evident when incubations were performed at 37°C as compared to 23°C, thus providing an experimental strategy with which to address synaptic compromise produced by the different Aβ aggregates. H-agg species formed faster and to higher levels at 37°C compared to 23°C, and the two aggregate preparations were evaluated in hippocampal slice cultures, a sensitive system for monitoring synaptic integrity. Applied daily at 80-600 nM for 7 days, the Aβ42 preparations caused dose-dependent and aggregation-dependent declines in α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) and N-methyl-D-aspartate (NMDA) receptor subunits as well as in presynaptic components. Unlike the synaptic effects, Aβ42 induced only trace cellular degeneration that was CA1 specific. The 37°C preparation was less effective at decreasing synaptic markers, corresponding with its reduced levels of Aβ42 monomers and dimers. Aβ42 dimers decayed significantly faster at 37°C than 23°C, and more rapidly than monomers at either temperature. These findings indicate that Aβ42 can self-aggregate into potent synaptotoxic oligomers as well as into larger aggregates that may serve to neutralize the toxic formations. These results will add to the growing debate concerning whether high molecular weight Aβ complexes that form amyloid plaques are protective through the sequestration of oligomeric species.
1. Introduction
Alzheimer's disease (AD) is an age-related neurodegenerative disorder characterized by progressive cognitive decline, motor function impairment, and behavioral changes [1]. Multifarious investigations into the causative agent of AD have led to the amyloid cascade hypothesis, implicating the amyloid-β peptide (Aβ) as a major causative factor in AD pathogenesis. Aβ is predominately a 38-, 40-, or 42- amino acid peptide derived through differential cleavage of the amyloid precursor protein (APP) by β- and γ-secretases [2, 3]. Increased amounts of Aβ42 have been associated with AD [4] and this peptide has shown a greater propensity to oligomerize in vitro [5, 6]. Several mutations in the genes encoding APP or subunits of the secretases that promote production of Aβ42 have been linked to familial AD [7-12]. Furthermore, transgenic mice expressing mutated APP and/or secretase proteins display memory deficits associated with increased levels of total Aβ, increased Aβ42/Aβ40 ratio, and intraneuronal Aβ aggregation [13-16]. These discoveries provide strong evidence that Aβ is a critical component of AD-associated brain defects.
Several forms of Aβ have been implicated in AD pathology. Postmortem comparison of brain extracts from AD patients to normal individuals shows a close correlation between increased levels of soluble Aβ and neurodegeneration [17-19]. Additionally, intraneuronal Aβ42 was detected postmortem in patients with impaired cognition [20]. In further studies, attempts were made to isolate the neurotoxic form of Aβ. Extracts from AD brains yielded Aβ42 aggregates with molecular weights from 10 kDa to over 100 kDa [4], whereas another group predominantly found lower molecular weight Aβ oligomers including monomers, dimers, and trimers [17]. In a proof-of-concept study, soluble Aβ dimers isolated from AD brains produced memory impairments in wild-type rats, and also disrupted signaling in vitro [21]. Similarly, Lesné et al. [22] isolated a specific 56 kDa soluble Aβ oligomer that was detected at the onset of memory deficits in a transgenic mouse model of AD and caused spatial memory impairment when inoculated into wild-type rats. These studies reaffirm a pathological role for Aβ oligomers; however, the species and the exact role in neurodegeneration remain elusive.
To circumvent the difficulty of isolating Aβ from in vivo samples, synthetic peptide preparations have become widely used to investigate oligomerization events. Initial studies determined that solutions which are allowed to age for several days become neurotoxic. Analysis revealed that the aged solutions contain high molecular weight aggregates of Aβ as opposed to the predominate monomeric species found in freshly prepared solutions [23]. Several different soluble oligomers of synthetic Aβ have been described, ranging from dimers to 24mers [17, 24], including a soluble dodecamer unique to Aβ42 with a molecular weight of 55.2 kDa [25]. Also, toxicity of the synthetic peptide has been shown as Aβ42 causes neuronal damage and precludes glutamatergic signaling in neuronal cultures [26-28]. Although neuronal cultures have been widely used to study neurotoxicity, organotypic hippocampal slice cultures provide a convenient in vitro model that more closely maintains the structure and neuronal connections of the mature hippocampus [29]. Furthermore, this model is useful in studying Aβ-induced pathology since neurons in the hippocampal slice cultures internalize exogenous Aβ42 leading to decline of the presynaptic marker, synaptophysin [30, 31]. In the present study, we utilized hippocampal slice cultures to determine the effect of pre-aggregated Aβ42 solutions on synaptic integrity. Our data demonstrate that synaptic decline induced by Aβ42 solutions is affected by the conditions under which peptide aggregation occurs.
2. Materials and methods
2.1 Aβ aggregation
Aβ42, a gift from Professor Charles Glabe (University of California, Irvine, CA), and Aβ40 (Bachem, King of Prussia, PA) were reconstituted to 2.22 mM in 0.1 M NaOH and then bath sonicated at room temperature for 1 min. The solutions were diluted to 45 μM in 0.1 M PBS, immediately sterile filtered, and allowed to aggregate in the dark at 37°C or 23°C for 0 - 30 d. Aliquots of the solutions were either prepared for electrophoresis or stored at -80°C. Experiments utilizing Aβ42 obtained from American Peptide Company, Inc. (Sunnyvale, CA) yielded similar results.
2.2 Organotypic Hippocampal Slice Cultures
Brain tissue from postnatal day twelve Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA) was rapidly removed to prepare slices as described [29, 32-35]. Transverse slices of hippocampus (400 μm) were quickly prepared and placed on insert membranes (Millipore Corporation, Billerica, MA) with cultured medium consisting of 50% basal medium Eagle (Sigma, St. Louis, MO), 25% Earle's balanced salts (Sigma), 25% horse serum (Gemini Bio-products, Sacramento, CA), and defined supplements, as described previously [29, 32-35]. Slices were maintained at 37°C in 5% CO2-enriched atmosphere for 14 - 25 d before experimental use and were maintained in culture for the same length of time throughout the experiment by staggering treatments. For Aβ42 treatments, pre-aggregated or fresh solutions were further diluted in serum free media and applied briefly to the surface of the tissue and then placed in the bottom of the well containing the insert for 8-12 h. Then media containing serum was added for 12 h before the application was repeated. For immunoblotting, cultured slices were harvested with a soft brush and sonicated in sets of 6-8 slices using ice-cold homogenization buffer, then protein content was determined using Pierce BCA Protein Assay (Thermo Scientific, Rockford, IL) with similar results obtained with Dc Protein Assay (Bio-Rad, Hercules, CA). All of the studies were carried out in strict accordance with the recommendations from the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health. Animal use was conducted in accordance with an approved protocol from the Institutional Animal Care and Use Committee of the University of North Carolina–Pembroke.
2.3 Immunoblot analysis
Equal protein aliquots (80 μg) of the slice samples were denatured in SDS buffer for 5 min at 100°C, then separated by 4-15% tris-glycine SDS-PAGE (BioRad) and blotted to nitrocellulose (BioRad). Alternatively, aliquots (1 μg) of the Aβ solutions were mixed with SDS buffer, incubated at 40°C for 30 min, cooled to room temperature, separated on 16.5% tris-tricine or 4-20% tris-glycine SDS-PAGE (BioRad), and then transferred to nitrocellulose. For dot blots, samples in 0.15% SDS were treated as above and equal amount protein blotted onto nitrocellulose and allowed to dry. The blots were stained for different markers using the following antibodies: calpain-mediated breakdown product (BDP, as previously described [29, 36]), GluR1 prepared as previously described [37], GluR2 (1:250; Millipore), NR1 (selective for splice variants NR1-1a, NR1-1b, NR1-2a, NR1-2b; 1:100; Millipore), NR2A (1:100; Millipore), NR2B (1:75; Millipore), synaptophysin (1:100; Millipore), synapsin II (1:600; Calbiochem, La Jolla, CA), actin (1:250; Sigma), 6E10 against amino acids 1-16 of Aβ (1:80; Covance, Emeryville, CA), 82E1 against amino acids 1-16 of Aβ (1:100, IBL, Takasake-Shi, Japan), A11 against amyloid oligomers (Professor Charles Glabe, University of California, Irvine, CA). Secondary antibody incubation utilized anti-IgG-alkaline phosphatase conjugates (1:1800, BioRad), and color development used the 5-bromo-4-chloro-3-indolyl phosphate and nitroblue tetrazolium substrate system. Development of immunoreactivity was terminated before maximal intensity was reached on the blots in order to avoid saturation and to ensure a linear relationship with increasing amount of sample protein. Labeled bands were scanned at high resolution to determine integrated optical density with BIOQUANT software (R & M Biometrics, Nashville, Tennessee). The immunostaining of glutamate receptor subunits and other proteins on blots was routinely tested for linearity within the optical density range exhibited in cultured slice samples. Tests were conducted by immunoblotting sets of samples containing 2–100 μg protein from a single tissue preparation. Typical immunoreactivity plots exhibited linear relationships often across >10-fold span of staining intensity and with a high correlation coefficient (r = 0.95–0.99). The specific immunoreactivity of samples across immunoblots was combined by normalizing against the mean measures of common sample groups present in the different blots.
2.4 Histology
Hippocampal slice cultures were maintained until culture day 25 and treated with Aβ42 as described above (2.2). Twenty-four hours after the last Aβ42 treatment, media was replaced with serum-free media containing 10 μg/mL of propidium iodide (PI; Fluka, St. Louis, MO) for 1 h. Excitotoxicity was induced in cultures by maintaining them in media with 100 μM NMDA, 100 μM AMPA, and 2 mM excess CaCl2 for 24 h prior to PI staining. Slices were then fixed in 4% paraformaldehyde for 4 h at 4°C [38], rinsed in phosphate buffer, mounted onto slides, and dried. After imaging for PI, whole mounts were Nissl stained. Alternatively, fixed slices were sectioned to 20 μm using a sliding microtome (Leica Microsystems, Nussloch, Germany) equipped with a freezing stage (Physitemp Instruments, Inc., Clifton, NJ). Immunohistochemistry was performed by BOND-MAX (Leica) using GluR1 primary antibody [37] and Bond Polymer Refine Detection kit (Leica). A Nikon AZ100 Microscope equipped with AZ-FL Epi-fluorescence, Fiber Illuminator, AZ-Plan Fluor 5× lense, and Q-Imaging QI Click camera (Nikon Instruments Inc., Melville, NY) was used for imaging. All images analyzed for PI staining received the same gain, exposure time, intensity threshold, and other measurement parameters that were capsulated within each image file. Analysis was performed with NIS-Elements AR (Nikon): threshold of PI staining was determined in positive controls, and optical density (sum density) at or above threshold, and area of staining at or above threshold (binary area) was determined for all images.
2.5 Statistical Analyses
Integrated optical densities for the various antigens and optical density (sum density) or binary area of threshold PI staining were expressed as mean ± SEM. Statistical significance was determined by unpaired two-tailed t-tests, and one-way and two-way analyses of variance (ANOVA), followed by the Tukey's multiple comparison or Bonferroni's post hoc tests using GraphPad Prism version 3.00 for Windows (GraphPad Software, San Diego, CA). Figures were prepared for publication using Photoshop and Illustrator software (Adobe, San Jose, CA).
3. Results
3.1 Formation of high molecular weight Aβ42 aggregates
In order to examine the extent of Aβ42 self-aggregation, 45 μM Aβ42 in PBS was incubated at either 23°C or 37°C for 7 d, then separated by electrophoresis on a 16.5% tris-tricine gel. Immunoblotting with 6E10 antibody indicated that while a fresh solution of Aβ42 was comprised of monomeric and dimeric species, Aβ42 incubated at 23°C showed these species in addition to higher molecular weight aggregates (H-agg) ranging in size from 80-100 kDa (Fig. 1). Aβ42 incubated at 37°C showed a decrease in the dimeric species and an increase in H-agg species compared to the 23°C solution. Primarily monomeric peptide solubilized in hexafluoroisopropanol was reconstituted in DMSO after speed-vac preparation, and found to exhibit similar H-agg formation after 23°C incubation in PBS (data not shown). To obtain more information on the heterogeneity of the H-agg species, Aβ42 solutions pre-aggregated at 37°C were analyzed using a 4-20% tris-glycine gel, followed by 82E1 immunoblotting (Fig. 2A). The heterogeneous H-agg spans 40 to 200 kDa in this matrix and, as in Figure 1, the molecular weight and band intensity increases over time. Relative integrated optical density of the immunostaining showed a dramatic increase in H-agg during the first three days followed by a slower rate of increase, corresponding with a steady decline in the oligomer of approximately 8 kDa (Fig. 2B). Also in Figure 2, Aβ40 does not aggregate into soluble high molecular weight oligomers even after 14 days of incubation at 37°C. This isoform of Aβ forms low molecular weight oligomers; however some of this material remained at the top of the gel.
Figure 1.
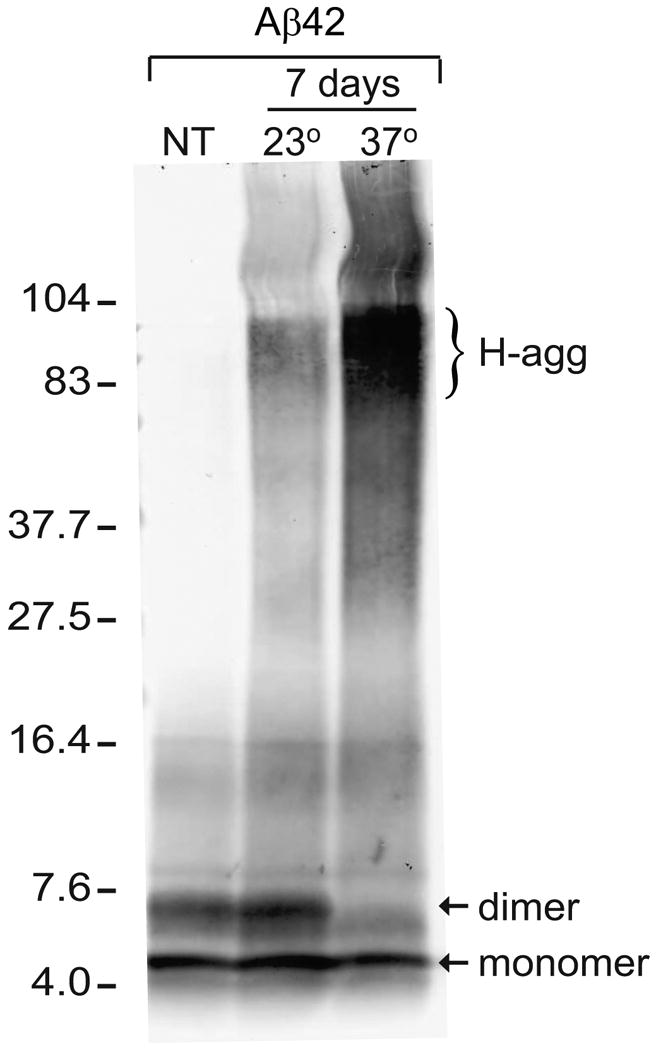
Aβ42 forms high molecular weight aggregates during long-term incubation in PBS. Aliquots of Aβ42 (45 μM in PBS) were incubated in the dark for 7 days at 23°C or 37°C. Loading buffer was then added to equal peptide amounts (1 μg), incubated at 40°C for 30 min, and separated by 16.5% tris-tricine SDS-PAGE alongside an untreated control of non-aggregated Aβ42 (NT). Immunoblotting with 6E10 antibody labeled Aβ42 monomer and dimer (arrows), as well as high molecular weight aggregates (H-agg). The positions of 4- to 104-kDa standards are shown.
Figure 2.
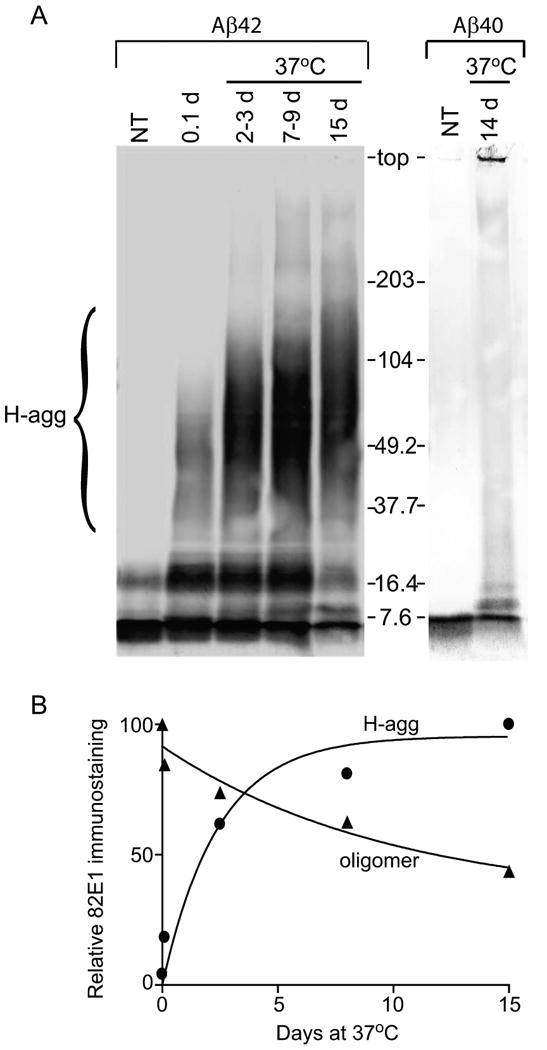
Aβ42, but not Aβ40, forms high molecular weight aggregates when incubated at 37°C. A. Aβ42 and Aβ40 (45 μM in PBS) were incubated for the indicated time (2-3 d samples were pooled from 2 d and 3 d samples; 7-9 d were pooled from 7 d and 9 d samples) or prepared from non-aggregated control (NT). Equal amounts (1 μg) were incubated in loading buffer at 40°C for 30 min, separated by a 4-20% tris-glycine SDS-PAGE, and immunoblotted with 82E1 antibody. The position of 7.6- to 203-kDa standards is shown, and a bracket indicates the heterogeneous high molecular weight aggregates (H-agg). B. Relative integrated optical density of H-agg (circles) and ∼8 kDa (oligomer; triangles) bands were quantified and graphed. Best fit non-linear regression curves are one-phase exponential association and one-phase exponential decay, respectively.
In order to further characterize the H-agg, unaggregated (0 d) and pre-aggregated Aβ42 (30 d) preparations were subjected to low-speed centrifugation in an attempt to clarify the solution. Previous studies have shown that similar centrifugation will pellet Aβ42 fibrils [39, 40]. Samples taken before centrifugation (pre-spin) showed no difference in H-agg staining as compared to supernatant (super) samples obtained after centrifugation (Fig. 3A). Dot blots indicated that solutions pre-aggregated for 7 d and 30 d contained Aβ42 oligomers whereas unaggregated samples (0 d) did not (Fig. 3B). Interestingly, the 6E10 antibody immunostained pre-aggregated Aβ42 solutions darker than unaggregated samples which may indicate conformation-dependent antibody recognition.
Figure 3.
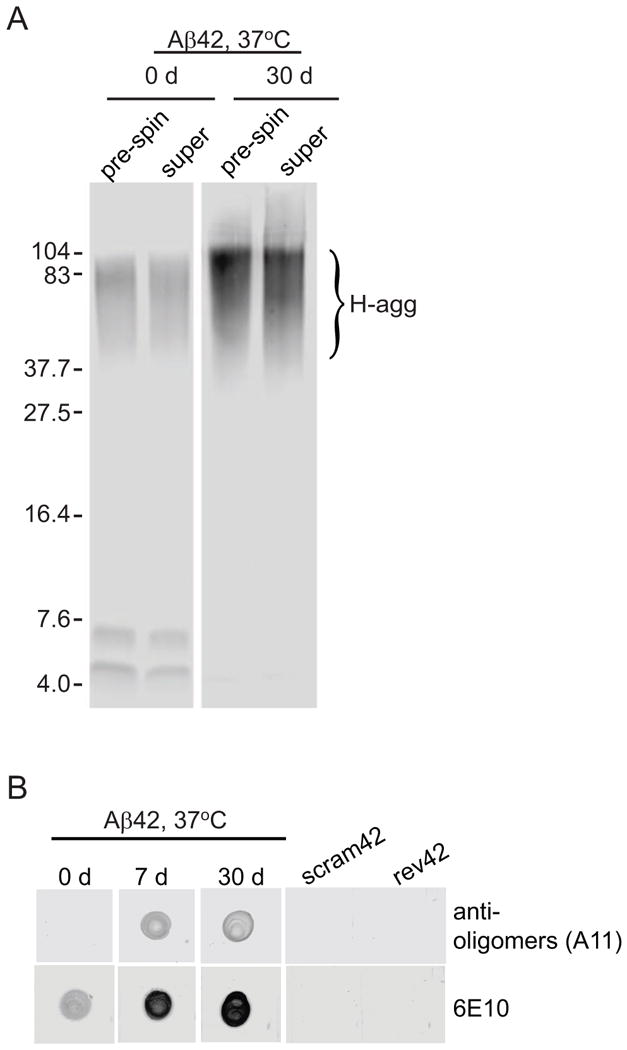
Pre-aggregated Aβ42 solutions are not clarified by low speed centrifugation and contain oligomers. Aβ42 (45 μM in PBS) was allowed to aggregate for indicated time at 37°C. A. Solutions were then centrifuged at 16060 × g for 25 min. Samples taken before centrifugation (pre-spin) were compared with supernatant after centrifugation (super) on a 16.5% tris-tricine SDS-PAGE, blotted onto nitrocellulose, and detected with 82E1 antibody. B. Equal volume and amount of peptide (0.67 μg) was spotted onto nitrocellulose, allowed to air dry, and detected with 6E10 antibody or A11 antibody. Peptides composed of a scrambled sequence of amino acids comprising Aβ42 (scram42) or the reverse sequence of Aβ42 (rev42) were allowed to aggregate for 7 d and also tested by dot blot as negative controls.
3.2 Assessment of Aβ42-induced synaptic decline in organotypic hippocampal slice cultures
Organotypic hippocampal slice cultures were used for the sensitive assessment of synaptic decline because they maintain native neuronal organization and connections throughout the course of long-term experiments [29]. Aβ42 was allowed to pre-aggregate for 5 - 7 d, at the two different temperatures, diluted to 300 - 500 nM in serum free media, and applied daily to slice cultures for 7 consecutive days. Slices were harvested and protein aliquots were assessed by immunoblot for different glutamate receptor subunits. Aβ42 pre-aggregated at 23°C was more effective at compromising synaptic marker expression, causing a decrease in AMPA receptor subunits GluR1 and GluR2 and NMDA receptor subunits NR1, NR2A and NR2B (Fig. 4A). Compared to slices treated with vehicle, Aβ42 pre-aggregated at 23°C reduced GluR1 by 69.8% (p<0.0001), whereas Aβ42 pre-aggregated at 37°C reduced GluR1 by only 22.7% (p=0.01; Fig. 4B). Aβ42 pre-aggregated at 23°C also decreased NR1 levels by 40.8% (p<0.01) however the 37°C solution had no affect on NR1 levels compared to vehicle control (Fig. 4C). Immunohistochemical comparison of slices treated with vehicle vs. Aβ42 pre-aggregated at 23°C revealed reduced GluR1 density in dendritic fields, especially in the s. oriens, corresponding with a striking accumulation of GluR1 immunoreactivity in pyramidal neurons (Fig. 4D, F). This could be due to transport blockage known to be produced by Aβ [41, 42] and the resultant buildup of the synaptic marker in the cell bodies causing downregulation of its expression as found in the immunoblots. The 37°C pre-aggregated Aβ42 solution also caused cellular GluR1 accumulation and reduced dendritic staining (Fig. 4E), yet to a lesser extent than the 23°C solution.
Figure 4.
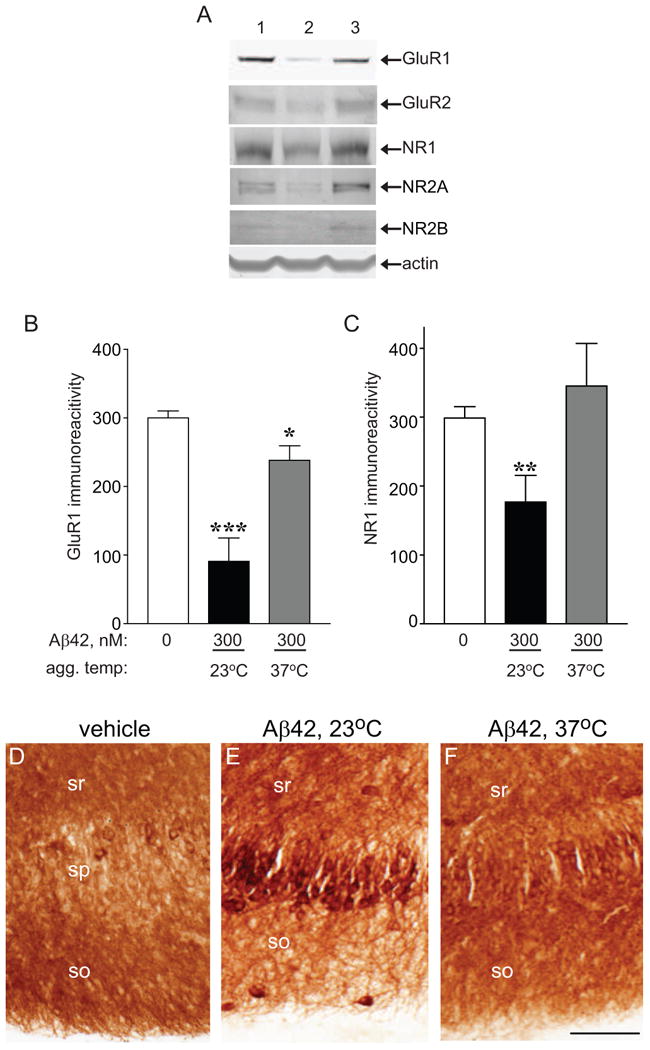
Effects of pre-aggregated Aβ42 on glutamate receptor subunit levels in hippocampal slice cultures. Organotypic hippocampal slices were prepared and maintained in culture for 21 d. Aβ42 at 45 μM in PBS was pre-aggregated at the indicated temperature, diluted with serum free media to 300 nM, and added to the hippocampal slices. A. After 7 daily applications of vehicle control (lane 1), Aβ42 pre-aggregated at 23°C (lane 2), or Aβ42 pre-aggregated at 37°C (lane 3), hippocampal slices were harvested, sonicated, and equal protein immunoblotted for GluR1, GluR2, NR-1, NR2A, NR2B, and actin as a load control. B, C. Mean integrated optical densities ± SEM for GluR1 (B) and NR-1 (C) immunoreactivities were plotted. Unpaired, two-tailed t-test: *p = 0.01, **p<0.01, *** p<0.0001; n=6-12. D-F. After 7 d of indicated treatment, slices were fixed, sectioned to 20 μm, and immunostained with GluR1 primary antibody which was visualized by 3,3-diaminobenzidine. Images were captured on a Nikon AZ100 microscope. so, stratum oriens; sp, stratum pyramidale; sr, stratum radiatum; size bar: 100 μm
Next, the effect of pre-aggregated Aβ42 on presynaptic markers was assessed as studies have shown a decrease in synaptophysin associated with AD [43, 44]. Furthermore, a recent report found an NMDA receptor-dependent decrease in synaptophysin by Aβ42, linking pre- and postsynaptic markers through caspase cascades [45]. In the present study, immunoblots showed a 58.8% decline in synaptophysin by Aβ42 pre-aggregated at 23°C (p<0.01) whereas Aβ42 pre-aggregated at 37°C produced only a 24.2% decline compared to vehicle (Fig. 5B). Other presynaptic markers synapsin IIA and IIB also exhibited marginal declines in response to pre-aggregated Aβ42 (Fig. 5A).
Figure 5.
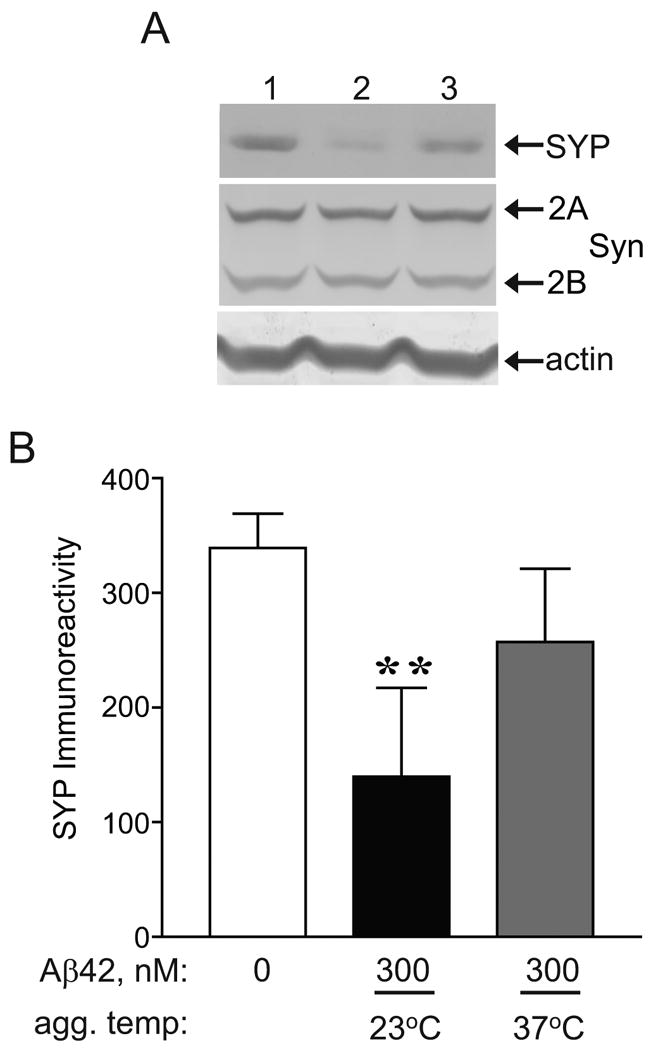
Effects of pre-aggregated Aβ42 on presynaptic markers in hippocampal slice cultures. Organotypic hippocampal slices were prepared and maintained in culture for 21 d. A. Aβ42 at 45 μM in PBS was pre-aggregated at the indicated temperature, diluted with serum free media to 300 nM, and added to the hippocampal slices for 7 d. After daily application of vehicle control (lane 1), Aβ42 pre-aggregated at 23°C (lane 2), or Aβ42 pre-aggregated at 37°C (lane 3), hippocampal slices were harvested, sonicated, and equal protein immunoblotted for synaptophysin (SYP), synapsin 2 (Syn 2A and 2B), and actin as a load control. B. Mean integrated optical densities ± SEM of synaptophysin immunoreactivity were plotted. Unpaired, two-tailed t-test: **p <0.01; n=8-11.
To test whether Aβ42-induced synaptic decline is aggregation-dependent, hippocampal slices were treated with non-aggregated or pre-aggregated Aβ42 solutions (Fig. 6A). As the immunoblot illustrates, non-aggregated Aβ42 does not affect glutamate receptor subunit levels, however, pre-aggregated Aβ42 significantly reduces GluR1 with respect to vehicle (p<0.0001) and reduces GluR1 by 65.6% as compared to non-aggregated Aβ42 (p<0.01; Fig. 6B). Similarly, NR1 is significantly reduced by pre-aggregated Aβ42 (p<0.05) but not by non-aggregated Aβ42 (Fig. 6C). Thus, Aβ42 affects synapses in an aggregation-dependent manner.
Figure 6.

Effect of non-aggregated vs. pre-aggregated Aβ42 on glutamate receptor subunits in hippocampal slice cultures. A. Hippocampal slice cultures were treated with vehicle control (lane 1), 300 nM non-aggregated Aβ42 (lane 2), or 300 nM Aβ42 that was pre-aggregated in PBS at 23°C for 7 d (lane 3). After daily applications, hippocampal slices were harvested, sonicated, and equal protein immunoblotted for GluR1, NR1, and actin levels. B, C. Mean integrated optical densities ± SEM of GluR1 (B) and NR1 (C) immunoreactivities were plotted. One-way ANOVA: p<0.0001(GluR1; n=6-12) and p=0.035 (NR1; n=5-12). Tukey's multiple comparison post-hoc tests: *p<0.05, **p<0.01, ***p<0.001.
Since pre-aggregated Aβ42 solutions significantly decreased glutamate receptor subunit and presynaptic markers, Aβ42-induced cell death was assessed in hippocampal slice cultures. Cultured slices were treated for 7 d with vehicle or Aβ42 pre-aggregated at 23°C, or treated for 24 h with NMDA and AMPA as a positive control for excitotoxicity. Propidium iodide (PI) was used to stain cells with compromised plasma membranes which is indicative of cell death (Figs. 7A, C, E). The threshold of PI staining was determined using NMDA/AMPA treated slices, and the area of PI staining in the CA1, CA3 and dentate gyrus above threshold was measured (Fig. 7G, H; one-way ANOVA; p<0.0001) as well as the fluorescent optical density (Table 1). PI positive cells were observed in only 3 of the 10 Aβ42-treated slices and were localized to the CA1 subfield (Fig. 7C). Nissl stains were performed to assess morphological changes and while pyknotic nuclei were prevalent in the excitotoxic slices (Fig. 7F), little difference was found between vehicle and Aβ42-treated slices (Figs. 7B, D).
Figure 7.
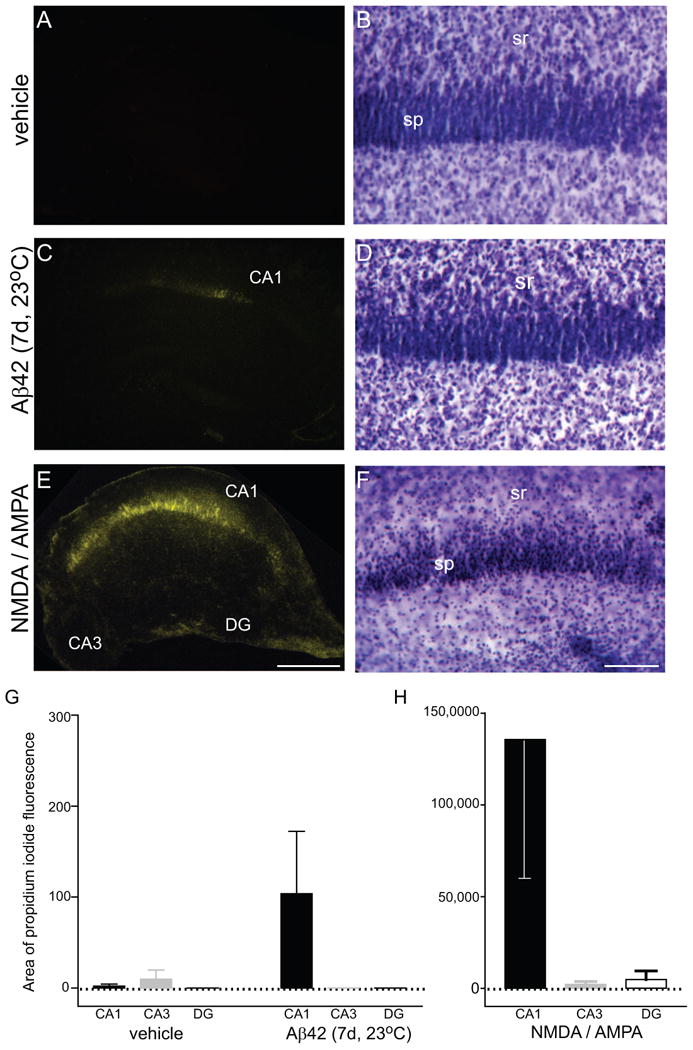
Assessment of neurodegeneration induced by pre-aggregated Aβ42. Organotypic hippocampal slices were prepared and maintained in culture for 25 d. Aβ42 was pre-aggregated at 23°C for 5 d and diluted in serum free media to 500 nM. Hippocampal slices were treated with vehicle (A, B), or Aβ42 solution (C, D) daily for 7 d, and positive controls were incubated with 100 μM NMDA and 100 μM AMPA for 24 h (E, F). Cultures were then incubated with media containing propidium iodide (PI), fixed, mounted, and subfields imaged using epi-fluorescence (A, C, E), followed by Nissl staining and imaging of the CA1 subregion under brightfield (B, D, F). Means ± SEM of image area positive for PI staining are shown (G, H). Note the different scale for the NMDA/AMPA slices that were used as a positive control for neurodegeneration. Non-parametric one-way ANOVA (Kruskal-Wallis test) indicated a significant difference of PI-positive area across the hippocampal subfields of Aβ42-treated slices (p<0.05). DG, dentate gyrus; so, stratum oriens; sp, stratum pyramidale; sr, stratum radiatum; size bar: 500 μm (A, C, E), 100 μm (B, D, F)
Table 1.
Mean PI fluorescent optical densities (sum density) ± SEM after vehicle, Aβ42, or 100 μM each NMDA/ AMPA treatment in hippocampal slice cultures.
| Vehicle, 7d | Aβ42, 7d | NMDA / AMPA, 24h | |
|---|---|---|---|
| CA1 | 1.42 ± 1.41 | 24.3 ± 16.1 | 21122 ± 15622 |
| CA3 | 3.30 ± 3.30 | 0 ± 0 | 370 ± 308 |
| DG | 0 ± 0 | 0 ± 0 | 504 ± 500 |
Reduced vulnerability of synaptic markers corresponding with the increased H-agg observed with the 37°C pre-aggregated Aβ42 solutions incited an investigation into the potency of 37°C vs. 23°C pre-aggregated Aβ42 solutions. Hippocampal slice cultures were treated daily for 7 consecutive days with indicated concentrations of Aβ42 aggregated at either 37°C or 23°C. Slices were harvested and total protein analyzed by immunoblot, assessing GluR1 levels as a sensitive marker for synaptic decline (Fig. 8A). One-way analysis of variance indicated that Aβ42 pre-aggregated at 23°C caused a dose-dependent decrease in GluR1 levels (p<0.0001). Two-way analysis of variance indicated that both concentration and temperature of aggregation significantly affected GluR1 levels (p<0.0001). Treatment of hippocampal slices with 80 nM Aβ42 aggregated at 37°C showed little change of GluR1 levels compared to control (0 nM); however, 80 nM Aβ42 aggregated at 23°C caused a 38.2 % decrease in GluR1 levels. At 300 nM, the 37°C solution induced only a 23.0% decrease in GluR1; however Aβ42 aggregated at 23°C reduced GluR1 levels by 69.8%. This trend was further accentuated at higher doses as 600 nM of 37°C pre-aggregated solution decreased GluR1 by 42.1% whereas 23°C pre-aggregated solutions caused a 90% reduction (Fig. 8B). In addition to synaptic decline, Aβ42 pre-aggregated at 23°C was found to induce cytoskeletal compromise as indicated by detection of calpain-mediated spectrin breakdown product (BDP; Fig 8A). Allowing Aβ42 to oligomerize at 23°C results in significant synaptic decline in hippocampal slice cultures; whereas pre-aggregation at 37°C abates this effect.
Figure 8.
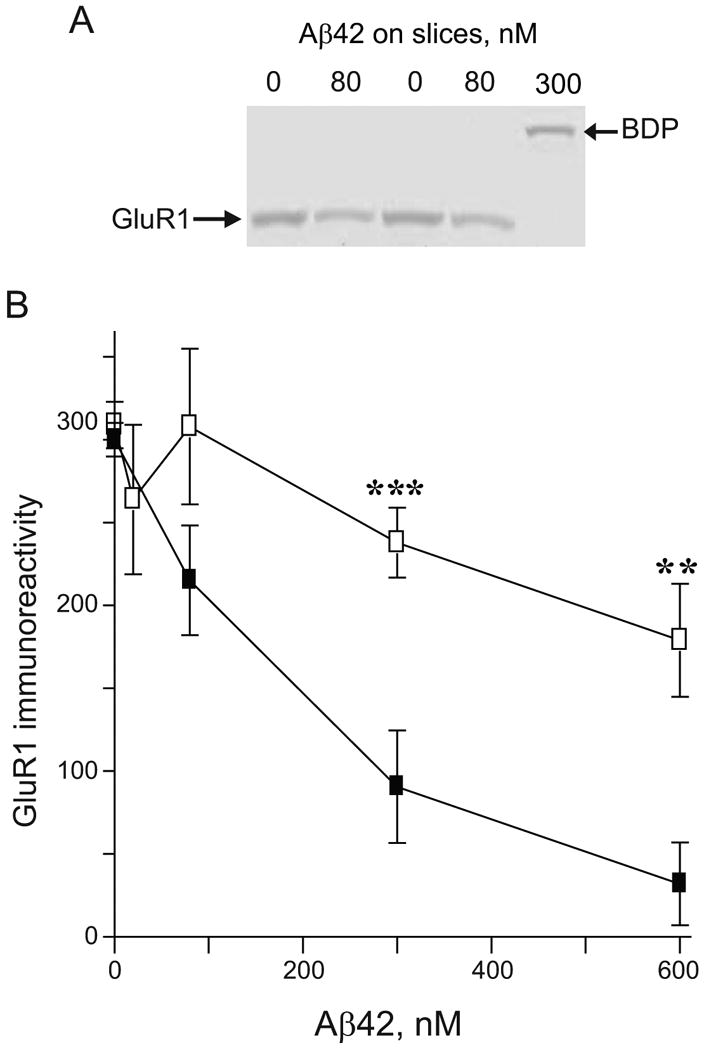
Synaptic compromise potency profile for the different Aβ42 preparations in hippocampal slice cultures. Aβ42 was pre-aggregated in PBS, diluted to the indicated concentration with serum free media, and applied to hippocampal slices. After 7 daily treatments with vehicle control or Aβ42 solutions, the cultured slices were harvested, sonicated, and equal protein immunoblotted for GluR1 and calpain-mediated spectrin breakdown product (BDP). A. Representative immunoblot from hippocampal slice samples treated with vehicle control or different concentrations of Aβ42 pre-aggregated at 23°C. B. Mean integrated optical densities ± SEM of GluR1 immunoreactivity from hippocampal slices treated with Aβ42 pre-aggregated at 37°C (open squares) or 23°C (filled squares) are plotted. One-way ANOVA: p<0.0001, Aβ42 pre-aggregated at 23°C; N.S., Aβ42 pre-aggregated at 37°C. Two-way ANOVA: p<0.0001, n=4-12. Bonferroni's post-tests: **p<0.01 and ***p<0.001.
3.3 Kinetics of Aβ42 aggregation
Since Aβ42 aggregate formation was shown to produce differential synaptic decline, we sought to better characterize the kinetics of peptide species development in the two solutions. A 45 μM solution of Aβ42 was incubated at 37°C for 30 d, and aliquots were removed over time, which were assessed by immunoblotting with 6E10. Formation of H-agg began at 3-4 h after the start of incubation and increased in a time-dependent manner (Fig. 9A). In addition to the increase in concentration of the H-agg species, as indicated by the density of the band, the molecular weight exhibited an apparent increase from 80 kDa at 0.1 d to 100 kDa by 7 d. From the 0.1 d time point, the dimeric species rapidly declined, whereas the monomeric species showed a more gradual decline throughout the time course. Relative integrated optical band densities of the different species were plotted and fit to nonlinear regressions. The monomer and dimer were found to model one-phase exponential decay curves whereas H-agg modeled a one-phase exponential association curve (Fig. 9B). Using this model, the half-life was estimated to be 1.54 d for the dimer and 4.72 d for the monomer, and comparison of their respective decay rates (K) indicated a significant difference in the rate of decay between the two species (p<0.0001; Table 2).
Figure 9.
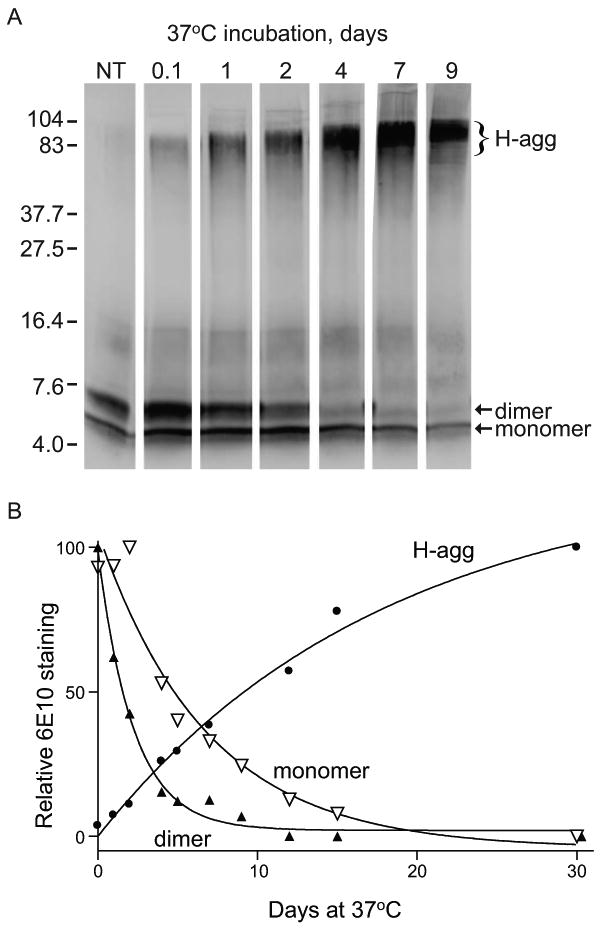
Time-dependent formation of Aβ42 high molecular weight aggregates at 37°C. Aβ42 at 45 μM in PBS was aggregated for the indicated number of days at 37°C or prepared from fresh stock (NT). Aliquots of peptide (1 μg) were mixed with loading buffer, incubated 30 min at 40°C, run on 16.5% tris-tricine SDS-PAGE, and immunoblotted for Aβ. A. The 6E10 antibody labeled monomer and dimer species (arrows), as well as high molecular weight aggregates (H-agg). Positions of 4.0- to 104-kDa standards are shown. B. Relative integrated optical densities of bands corresponding to monomer (inverted open triangle), dimer (filled triangle), and high molecular weight aggregates (filled circle) were plotted. The best fit nonlinear regressions were curves for one-phase exponential decay (monomer and dimer) and one-phase exponential association (H-agg).
Table 2.
Decay rate (K) ± SEM of Aβ42 species.
| Incubation temperature | 23°C | 37°C | 23°C vs. 37°C |
|---|---|---|---|
| Monomer | K=0.047 ± 0.052 | K=0.147 ± 0.038 | N.S. |
| Dimer | K=0.082 ± 0.016 | K=0.451 ± 0.037 | p<0.0001 |
| Monomer vs. Dimer | N.S. | p<0.0001 |
Given the increased potency of the Aβ42 solutions pre-aggregated at 23°C (Fig. 8), a comparison was made between Aβ42 solutions incubated at 23°C and 37°C. Here, the same procedure as the previous experiment was completed using aliquots of Aβ42 at each temperature. The immunoblot shows that Aβ42 incubated at either temperature begins to form H-agg within a few hours, and both samples show an increase in H-agg band density and molecular weight over the time course (Fig. 10). However, Aβ42 incubated at 37°C shows a much higher concentration of H-agg (compare band densities at 5 d and 9 d) than Aβ incubated at 23°C. Also, while the concentration of the dimeric and monomeric species decreased slightly over the time course in the 23°C solution, the dimeric species is barely visible by 9 d in the 37°C solution. Band intensities were plotted over time and as in Figure 9B, one-phase exponential decay curves were used as a model for the monomer (Fig. 10B) and dimer (Fig. 10C). The calculated half-life for the monomeric species and dimeric species at 23°C was 14.7 d and 8.45 d, respectively. There was no difference in the exponential decay rates between the monomer and dimer in the 23°C solutions; however, there was a significant difference between the exponential decay rate of the dimeric species upon comparison of the 23°C and 37°C solutions (p<0.0001; Table 2). Also, a comparison of the monomer/dimer ratio revealed a significant difference between the two solutions from 0.1 - 12 d (p<0.01; paired t-test; data not shown).
Figure 10.
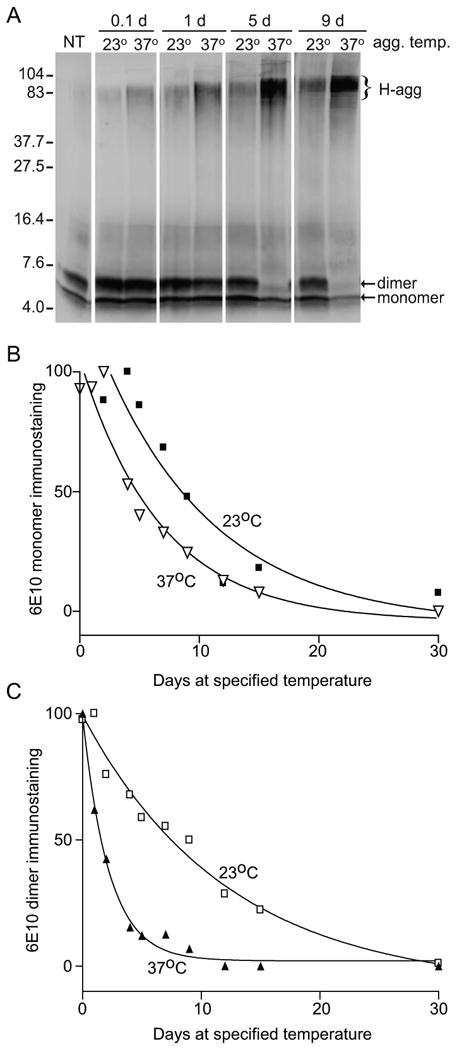
Comparison of time-dependent Aβ42 aggregation at 23°C vs. 37°C. Aβ42 in PBS was aggregated at 23°C or 37°C for the indicated times or prepared from fresh stock (NT). Aliquots of 1 μg peptide were mixed with loading buffer, incubated 30 min at 40°C, separated on 16.5% tris-tricine SDS-PAGE, and immunoblotted with the 6E10 antibody. A. Molecular weight standards from 4.0 to 104 kDa are indicated; arrows show monomeric and dimeric species and bracket indicates high molecular weight aggregates. B-C. Relative integrated optical density of bands corresponding to monomer (B) and dimer (C) were compared for solutions incubated at 23°C (squares) and 37°C (triangles). The best fit nonlinear regression curve (one-phase exponential decay) for each is illustrated.
4. Discussion
This study explores the propensity of soluble Aβ42 to aggregate, and the extent to which aggregation is time and temperature dependent. The heterogeneous Aβ42 aggregate solutions were evaluated for their ability to reduce the expression of glutamate receptor subunits and presynaptic markers in hippocampal slice cultures. Incubating Aβ42 solutions at 23°C or 37°C promoted the formation of high molecular weight aggregates at the expense of dimeric and monomeric species. However, formation of high molecular weight aggregates occurs more quickly and to a greater extent at 37°C compared to solutions incubated at 23°C. The most significant rate of decay was exhibited by the dimer at 37°C; the temperature that promotes high molecular weight aggregate formation. Thus, dimers are likely to play a significant role in the formation of high molecular weight aggregates. Related to our findings is the fact that previous work in which modified Aβ40 dimers seeded higher order aggregates much more readily than Aβ40 monomers [46]. This potentially explains the significantly faster decay rate of the dimeric species at 37°C.
Incubating long-term hippocampal slice cultures with the pre-aggregated Aβ42 solutions was found to cause sequelae of synaptic compromise, whereas non-aggregated Aβ42 solutions at the submicromolar concentrations tested had no effect on synaptic markers. GluR1 and NR1 levels decreased in response to Aβ42 pre-aggregated at 23°C, with GluR1 declining by concentrations as low as 80 nM. Other subunits of the excitatory amino acid receptors, GluR2, NR2A, and NR2B, followed the same trend, declining in response to Aβ2 pre-aggregated at 23°C. Perhaps overactivation of the AMPA and NMDA receptors by Aβ42 leads to downregulated expression of their subunits and disruption of synaptic signaling. In recent studies, nanomolar levels of Aβ oligomers present in the AD brain were reported to increase NMDA receptor activation and thereby impair synaptic plasticity [47, 48]. NR2B-containing NMDA receptors were the focus of the two studies and such receptors have been proposed to play a particularly important role in excitotoxicity. Evidence of overactivated excitatory receptors in the current study is indicated by the detection of calpain-mediated spectrin breakdown product, a sensitive marker of excitotoxic events and early stage neuronal degeneration [29, 49]
Both AMPA and NMDA signaling affect long-term potentiation (LTP), which is widely viewed as a model for memory formation and a measure of synaptic plasticity (review in [50]). Glutamatergic signaling through both the NMDA and AMPA receptors is significantly affected in the AD brain [51], and the degree of cognitive impairment demonstrated by patients with AD strongly correlates with synaptic decline [52-54]. The data presented here corroborate previous work that has shown a decrease in both GluR1 levels and surface expression of NMDA receptors in cultured neurons isolated from transgenic mouse models of AD [27, 55]. Also, aggregated Aβ42, but not Aβ40, inhibits the ability of CA1 hippocampal pyramidal neurons to signal through AMPA receptors [26], and it induces endocytosis of NMDA receptors in cortical neuronal cultures [27]. The multiple roles of NMDA receptors is of continued interest due to recent findings indicating that not only does Aβ influence NMDA receptors, but NMDA responses can both elevate synaptic Aβ generation or reduce APP processing into Aβ depending on the level of signaling intensity [56]. Physiological concentrations of naturally-secreted Aβ dimers and trimers have also been shown to require the activity of NMDA receptors to mediate spine loss in organotypic cultures [57]. In addition to glutamate receptor subunit decline, Aβ42 pre-aggregated at 23°C induced a significant decline in synaptophysin, further confirming the early synaptotoxic effects of this preparation. Collectively, these results indicate that pre-aggregated Aβ42 has the ability to induce the synaptic decline that is characteristic of AD-type pathogenesis. Despite the detrimental effect on synaptic integrity, aggregated Aβ42 induced limited cellular degeneration that was localized to CA1. This may be related to the fact that exogenous Aβ42 uptake is specific to pyramidal neurons in CA1 [30], and this study confirms the susceptibility of this region to aggregated Aβ42. We believe that the 7 d treatment with submicromolar concentration of Aβ42 that resulted in synaptic decline may not be long enough for overt neuronal degeneration to be observed in the hippocampal slice model.
Interestingly, pre-aggregated Aβ42 solutions containing higher concentrations of H-agg are less capable of reducing synaptic proteins. This indicates that high molecular weight Aβ aggregates may be protective through sequestration of synaptotoxic oligomers. AD-associated cognitive decline was initially attributed to amyloid plaques [58-61]; however, it was later discovered that neurodegeneration more closely correlates with soluble Aβ levels [17, 18]. Much effort has been placed into identifying the form of Aβ that is neurotoxic. Interestingly, the data presented here indicate that an increased rate of decay of Aβ42 dimers in favor of higher molecular weight aggregate formation corresponds with decreased neuronal compromise. This supports the growing number of studies that implicate low molecular weight oligomers, especially Aβ42 dimers and trimers, as the primary neurotoxic species [21, 62-64]. Together, the findings substantiate that high molecular weight Aβ42 complexes have the potential to sequester toxic oligomeric species, thereby lowering their concentration below the threshold for synaptotoxic effects. A recent study in C. elegans showed that experimental induction of amyloid deposits leads to decreased levels of Aβ oligomers and protection against neuromuscular synaptic defects [65]. We also found that the Aβ42 dimer decays faster than the monomeric species in the less toxic solutions pre-aggregated at 37°C. Thus, as an alternative hypothesis, since monomers have been shown to be neuroprotective [66], is that the difference in the monomer/dimer ratio could play a role in promoting or suppressing synaptotoxicity.
Therapeutic strategies have been targeted at disaggregation of Aβ, either extracellularly or intracellularly, to facilitate lysosomal degradation and promote cellular uptake of the peptide for trafficking to lysosomes [67]. The present study suggests extracellular pro-aggregation of Aβ oligomers as a self-repair mechanism in the brain and as a novel therapeutic avenue. We have shown that aggregation of Aβ42 is time- and temperature-dependent, species of Aβ oligomers are differentially affected by temperature, and that this affects the synaptotoxic properties of Aβ. The tendency of Aβ42 to form high molecular weight species and the dimers being the first pool of low molecular weight species to be correspondingly depleted has important implications for the treatment of AD. If the high molecular weight complexes are indeed nontoxic, increasing the rate of high molecular weight aggregate formation may in fact reduce low molecular weight oligomers that are responsible for synaptic degeneration and thus cognitive decline.
Research Highlights.
Formation of high molecular weight Aβ42 aggregates is time-dependent
High molecular weight aggregates appear to influence Aβ42 dimer decay rate
Long term Aβ42 aggregation events lead to reduced synaptotoxicity
Sequestering Aβ42 dimers into large aggregates may be protective
Acknowledgments
This work was supported in part by National Institutes of Health grants R25 GM077634, 1R41 AG031590, and 1G11 HD052381-01A1, Merz Pharmaceuticals GmbH, and the Oliver Smithies Grant from the North Carolina Biotechnology Center (Research Triangle Park, North Carolina). The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors would like to thank Drs. Christopher Parsons, Charles Glabe, and Lee Phillips for helpful discussions, and Shanna Harrelson, Hollie Young-Oxendine, and Johnathan Locklear for their technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Welsh-Bohmer KA, White CL., III Alzheimer disease: What changes in the brain cause dementia? Neurology. 2009;72:354–360. doi: 10.1212/01.wnl.0000343818.11392.d9. [DOI] [PubMed] [Google Scholar]
- 2.Walsh DM, Selkoe DJ. Aβ oligomers - a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 4.Kuo YM, Emmerling MR, Vigo-Pelfrey C, Kasunic TC, Kirkpatrick JB, Murdoch GH, Ball MJ, Roher AE. Water-soluble Aβ (N-40, N-42) oligomers in normal and Alzheimer disease brains. J Biol Chem. 1996;271:4077–4081. doi: 10.1074/jbc.271.8.4077. [DOI] [PubMed] [Google Scholar]
- 5.Burdick D, Soreghan B, Kwon M, Kosmoski J, Knauer M, Henschen A, Yates J, Cotman C, Glabe C. Assembly and aggregation properties of synthetic Alzheimer's A4/βamyloid peptide analogs. J Biol Chem. 1992;267:546–554. [PubMed] [Google Scholar]
- 6.Jarrett JT, Berger EP, Lansbury PT. The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 7.St George-Hyslop PH, Haines J, Rogaev E, Mortilla M, Vaula G, Pericak-Vance M, Foncin JF, Montesi M, Bruni A, Sorbi S, Rainero I, Pinessi L, Pollen D, Polinsky R, Nee L, Kennedy J, Macciardi F, Rogaeva E, Liang Y, Alexandrova N, Lukiw W, Schlumpf K, Tanzi R, Tsuda T, Farrer L, Cantu JM, Duara R, Amaducci L, Bergamini L, Gusella J, Roses A, McLachlan DC. Genetic evidence for a novel familial Alzheimer's disease locus on chromosome 14. Nat Genet. 1992;2:330–334. doi: 10.1038/ng1292-330. [DOI] [PubMed] [Google Scholar]
- 8.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HAR, Haines JL, Pericak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St George-Hyslop PH. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 9.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericak-Vance M, Roses A, Williamson R, Rossor M, Owen M, Hardy J. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 10.Chartier-Harlin MC, Crawford F, Houlden H, Warren A, Hughes D, Fidani L, Goate A, Rossor M, Roques P, Hardy J. Early-onset Alzheimer's disease caused by mutations at codon 717 of the β-amyloid precursor protein gene. Nature. 1991;353:844–846. doi: 10.1038/353844a0. [DOI] [PubMed] [Google Scholar]
- 11.Kumar-Singh S, Theuns J, Van Broeck B, Pirici D, Vennekens K, Corsmit E, Cruts M, Dermaut B, Wang R, Van Broeckhoven C. Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Aβ42 and decreased Aβ40. Hum Mutat. 2006;27:686–695. doi: 10.1002/humu.20336. [DOI] [PubMed] [Google Scholar]
- 12.Suzuki N, Cheung TT, Cai XD, Odaka A, Otvos L, Eckman C, Golde TE, Younkin SG. An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science. 1994;264:1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 13.Kuo YM, Beach TG, Sue LI, Scott S, Layne KJ, Kokjohn TA, Kalback WM, Luehrs DC, Vishnivetskaya TA, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Weller RO, Roher AE. The evolution of Aβ peptide burden in the APP23 transgenic mice: implications for Aβ deposition in Alzheimer disease. Mol Med. 2001;7:609–618. [PMC free article] [PubMed] [Google Scholar]
- 14.Lord A, Kalimo H, Eckman C, Zhang XQ, Lannfelt L, Nilsson LN. The Arctic Alzheimer mutation facilitates early intraneuronal Aβ aggregation and senile plaque formation in transgenic mice. Neurobiol Aging. 2006;27:67–77. doi: 10.1016/j.neurobiolaging.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 15.Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer's disease-linked presenilin 1 variants elevate Aβ1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 16.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–103. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 17.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Konrad V, Bush AI, Masters CL. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 18.Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J, Dickson DW, Trojanowski JQ, Lee VM. The evels of soluble versus insoluble brain Aβ distinguish Alzheimer's disease from normal and pathologic aging. Exp Neurol. 1999;158:328–337. doi: 10.1006/exnr.1999.7085. [DOI] [PubMed] [Google Scholar]
- 20.Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Aβ42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 23.Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW. In vitro aging of β-amyloid protein causes peptide aggregation and neurotoxicity. Brain Res. 1991;563:311–314. doi: 10.1016/0006-8993(91)91553-d. [DOI] [PubMed] [Google Scholar]
- 24.Chromy BA, Nowak RJ, Lambert MP, Viola KL, Chang L, Velasco PT, Jones BW, Fernandez SJ, Lacor PN, Horowitz P, Finch CE, Krafft GA, Klein WL. Self-Assembly of Aβ(1-42) into globular neurotoxins. Biochemistry. 2003;42:12749–12760. doi: 10.1021/bi030029q. [DOI] [PubMed] [Google Scholar]
- 25.Bernstein SL, Dupuis NF, Lazo ND, Wyttenbach T, Condron MM, Bitan G, Teplow DB, Shea JE, Ruotolo BT, Robinson CV, Bowers MT. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer's disease. Nat Chem. 2009;1:326–331. doi: 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parameshwaran K, Sims C, Kanju P, Vaithianathan T, Shonesy BC, Dhanasekaran M, Bahr BA, Suppiramaniam V. Amyloid β-peptide Aβ1–42 but not Aβ1–40 attenuates synaptic AMPA receptor function. Synapse. 2007;61:367–374. doi: 10.1002/syn.20386. [DOI] [PubMed] [Google Scholar]
- 27.Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-β. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 28.Ditaranto K, Tekirian TL, Yang AJ. Lysosomal membrane damage in soluble Aβ-mediated cell death in Alzheimer's disease. Neurobiol Dis. 2001;8:19–31. doi: 10.1006/nbdi.2000.0364. [DOI] [PubMed] [Google Scholar]
- 29.Bahr BA. Long-term hippocampal slices: A model system for investigating synaptic mechanisms and pathologic processes. J Neurosci Res. 1995;42:294–305. doi: 10.1002/jnr.490420303. [DOI] [PubMed] [Google Scholar]
- 30.Bahr BA, Hoffman KB, Yang AJ, Hess US, Glabe CG, Lynch G. Amyloid β protein is internalized selectively by hippocampal field CA1 and causes neurons to accumulate amyloidogenic carboxyterminal fragments of the amyloid precursor protein. J Comp Neurol. 1998;397:139–147. [PubMed] [Google Scholar]
- 31.Li M, Chen L, Lee DH, Yu LC, Zhang Y. The role of intracellular amyloid β in Alzheimer's disease. Prog Neurobiol. 2007;83:131–139. doi: 10.1016/j.pneurobio.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 32.Bendiske J, Caba E, Brown QB, Bahr BA. Intracellular deposition, microtubule destabilization, and transport failure: an “early” pathogenic cascade leading to synaptic decline. J Neuropathol Exp Neurol. 2002;61:640–650. doi: 10.1093/jnen/61.7.640. [DOI] [PubMed] [Google Scholar]
- 33.Butler D, Brown QB, Chin DJ, Batey L, Karim S, Mutneja MS, Karanian DA, Bahr BA. Cellular responses to protein accumulation involve autophagy and lysosomal enzyme activation. Rejuvenation Res. 2005;8:227–237. doi: 10.1089/rej.2005.8.227. [DOI] [PubMed] [Google Scholar]
- 34.Karanian DA, Brown QB, Makriyannis A, Bahr BA. Blocking cannabinoid activation of FAK and ERK1/2 compromises synaptic integrity in hippocampus. Eur J Pharmacol. 2005;508:47–56. doi: 10.1016/j.ejphar.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 35.Karanian DA, Brown QB, Makriyannis A, Kosten TA, Bahr BA. Dual modulation of endocannabinoid transport and fatty acid amide hydrolase protects against excitotoxicity. J Neurosci. 2005;25:7813–7820. doi: 10.1523/JNEUROSCI.2347-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bahr BA, Bendiske J, Brown QB, Munirathinam S, Caba E, Rudin M, Urwyler S, Sauter A, Rogers G. Survival signaling and selective neuroprotection through glutamatergic transmission. Exp Neurol. 2002;174:37–47. doi: 10.1006/exnr.2001.7852. [DOI] [PubMed] [Google Scholar]
- 37.Bahr BA, Hoffman KB, Kessler M, Hennegriff M, Park GY, Yamamoto RS, Kawasaki BT, Vanderklish PW, Hall RA, Lynch G. Distinct distributions of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptor subunits and a related 53,000 Mr antigen (GR53) in brain tissue. Neuroscience. 1996;74:707–721. doi: 10.1016/0306-4522(96)00133-9. [DOI] [PubMed] [Google Scholar]
- 38.Brana C, Benham C, Sundstrom L. A method for characterising cell death in vitro by combining propidium iodide staining with immunohistochemistry. Brain Res Brain Res Protoc. 2002;10:109–114. doi: 10.1016/s1385-299x(02)00201-5. [DOI] [PubMed] [Google Scholar]
- 39.Hilbich C, Kisters-Woike B, Reed J, Masters CL, Beyreuther K. Aggregation and secondary structure of synthetic amyloid βA4 peptides of Alzheimer's disease. J Mol Biol. 1991;218:149–163. doi: 10.1016/0022-2836(91)90881-6. [DOI] [PubMed] [Google Scholar]
- 40.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 41.Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- 42.Shah SB, Nolan R, Davis E, Stokin GB, Niesman I, Canto I, Glabe C, Goldstein LS. Examination of potential mechanisms of amyloid-induced defects in neuronal transport. Neurobiol Dis. 2009;36:11–25. doi: 10.1016/j.nbd.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 43.Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:933–944. doi: 10.1097/00005072-199708000-00011. [DOI] [PubMed] [Google Scholar]
- 44.Heinonen O, Soininen H, Sorvari H, Kosunen O, Paljärvi L, Koivisto E, Riekkinen PJ. Loss of synaptophysin-like immunoreactivity in the hippocampal formation is an early phenomenon in alzheimer's disease. Neuroscience. 1995;64:375–384. doi: 10.1016/0306-4522(94)00422-2. [DOI] [PubMed] [Google Scholar]
- 45.Liu J, Chang L, Roselli F, Almeida OF, Gao X, Wang X, Yew DT, Wu Y. Amyloid-β induces caspase-dependent loss of PSD-95 and synaptophysin through NMDA receptors. J Alzheimers Dis. 2010;22:541–556. doi: 10.3233/JAD-2010-100948. [DOI] [PubMed] [Google Scholar]
- 46.O'Nuallain B, Freir DB, Nicoll AJ, Risse E, Ferguson N, Herron CE, Collinge J, Walsh DM. Amyloid β-protein dimers rapidly form stable synaptotoxic protofibrils. J Neurosci. 2010;30:14411–14419. doi: 10.1523/JNEUROSCI.3537-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. 2011;31:6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rammes G, Hasenjäger A, Sroka-Saidi K, Deussing JM, Parsons CG. Therapeutic significance of NR2B-containing NMDA receptors and mGluR5 metabotropic glutamate receptors in mediating the synaptotoxic effects of β-amyloid oligomers on long-term potentiation (LTP) in murine hippocampal slices. Neuropharmacology. 2011;60:982–990. doi: 10.1016/j.neuropharm.2011.01.051. [DOI] [PubMed] [Google Scholar]
- 49.Czogalla A, Sikorski AF. Spectrin and calpain: a ‘target’ and a ‘sniper’ in the pathology of neuronal cells. Cell Mol Life Sci. 2005;62:1913–1924. doi: 10.1007/s00018-005-5097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miyamoto E. Molecular mechanism of neuronal plasticity: induction and maintenance of long-term potentiation in the hippocampus. J Pharmacol Sci. 2006;100:433–442. doi: 10.1254/jphs.cpj06007x. [DOI] [PubMed] [Google Scholar]
- 51.Proctor DT, Coulson EJ, Dodd PR. Post-synaptic scaffolding protein interactions with glutamate receptors in synaptic dysfunction and Alzheimer's disease. Prog Neurobiol. 2011;93:509–521. doi: 10.1016/j.pneurobio.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 52.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 53.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 54.DeKosky ST, Scheff SW, Styren SD. Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration. 1996;5:417–421. doi: 10.1006/neur.1996.0056. [DOI] [PubMed] [Google Scholar]
- 55.Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, Gouras GK. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis. 2005;20:187–198. doi: 10.1016/j.nbd.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 56.Verges DK, Restivo JL, Goebel WD, Holtzman DM, Cirrito JR. Opposing synaptic regulation of amyloid-β metabolism by NMDA receptors in vivo. J Neurosci. 2011;31:11328–11337. doi: 10.1523/JNEUROSCI.0607-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 59.Giannakopoulos P, Hof PR, Michel JP, Guimon J, Bouras C. Cerebral cortex pathology in aging and Alzheimer's disease: a quantitative survey of large hospital-based geriatric and psychiatric cohorts. Brain Res Brain Res Rev. 1997;25:217–245. doi: 10.1016/s0165-0173(97)00023-4. [DOI] [PubMed] [Google Scholar]
- 60.Crystal H, Dickson D, Fuld P, Masur D, Scott R, Mehler M, Masdeu J, Kawas C, Aronson M, Wolfson L. Clinico-pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer's disease. Neurology. 1988;38:1682–1687. doi: 10.1212/wnl.38.11.1682. [DOI] [PubMed] [Google Scholar]
- 61.Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, Yen SH, Aronson MK. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging. 1992;13:179–189. doi: 10.1016/0197-4580(92)90027-u. [DOI] [PubMed] [Google Scholar]
- 62.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 63.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lacor PN, Buniel MC, Furlow PW, Sanz Clemente A, Velasco PT, Wood M, Viola KL, Klein WL. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rebolledo DL, Aldunate R, Kohn R, Neira I, Minniti AN, Inestrosa NC. Copper reduces Aβ oligomeric species and ameliorates neuromuscular synaptic defects in a C. elegans model of inclusion body myositis. J Neurosci. 2011;31:10149–10158. doi: 10.1523/JNEUROSCI.0336-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giuffrida ML, Caraci F, Pignataro B, Cataldo S, De Bona P, Bruno V, Molinaro G, Pappalardo G, Messina A, Palmigiano A, Garozzo D, Nicoletti F, Rizzarelli E, Copani A. β-amyloid monomers are neuroprotective. J Neurosci. 2009;29:10582–10587. doi: 10.1523/JNEUROSCI.1736-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Butler D, Hwang J, Estick C, Nishiyama A, Kumar SS, Baveghems C, Young-Oxendine HB, Wisniewski ML, Charalambides A, Bahr BA. Protective effects of positive lysosomal modulation in Alzheimer's disease transgenic mouse models. PloS One. 2011;6:e20501. doi: 10.1371/journal.pone.0020501. [DOI] [PMC free article] [PubMed] [Google Scholar]