

Abstract
Discovering chemosensitivity pathways or nodes is an attractive strategy for formulating new drug combinations for cancer. Microtubules are among the most successful anticancer drug targets. Therefore, we implemented a small interfering RNA (siRNA) synthetic lethal screen targeting 5520 unique druggable genes to identify novel chemosensitivity nodes for vinblastine, a microtubule-destabilizing agent used clinically. We transiently transfected human glioblastoma cells with siRNAs for 48 h and then treated cells with a sublethal concentration of vinblastine. Forty-eight hours later, we analyzed cell viability and, using a series of statistical methods, identified 65 gene products that, when suppressed, sensitized glioblastoma cells to vinblastine. After completion of the secondary assays, we focused on one siRNA, B-cell lymphoma extra large (BCL-xL), because of its role in the intrinsic apoptosis signaling pathway as well as the availability of pharmacological inhibitors. We found that nontoxic concentrations of 4-[4-[[2-(4-chlorophenyl)-5,5-dimethylcyclohexen-1-yl]methyl]piperazin-1-yl]-N-[4-[[(2R)-4-morpholin-4-yl-1-phenylsulfanylbutan-2-yl]amino]-3-(trifluoromethylsulfonyl)phenyl]sulfonylbenzamide (ABT-263), an inhibitor of the BCL-2 family members (BCL-2, BCL-xL, and BCL-w), sensitized glioblastoma and non–small-cell lung cancer cells to vinblastine and induced apoptosis through the intrinsic cell death pathway. These results illustrate the usefulness of unbiased siRNA screens as a method for identifying potential novel anticancer therapeutic combinations.
Introduction
Cancer cells are highly dependent on microtubule dynamics, making microtubules an excellent target for anticancer treatment (Jordan and Wilson, 2004). To date, microtubule-destabilizing agents are among the most successful anticancer therapies, with multiple microtubule-destabilizing agents in clinical use, including the vinca alkaloids, which are used for the treatment of Hodgkin's lymphoma, non-Hodgkin's lymphoma, non–small-cell lung cancer, breast cancer, and glioblastoma multiforme (GBM) (Jordan and Wilson, 2004; Dumontet and Jordan, 2010). Despite the general success of these vinca alkaloids, resistance remains a serious clinical problem in a variety of cancers, including GBM (Feun et al., 1994; Ross et al., 1999; Kavallaris et al., 2001). Therefore, there is a need for the development of novel combination chemotherapeutics to help decrease the clinical resistance associated with microtubule-destabilizing agents (Jordan and Wilson, 2004). Identifying chemosensitivity pathways, or nodes, provides an attractive strategy for enhancing the therapeutic potential of established anticancer agents.
GBM is the most common and aggressive form of glioma; a majority of patients die within a year of being diagnosed (Adamson et al., 2009). These primary brain tumors arise from normal human astrocytes and are especially difficult to treat due to their location, aggressive biological behavior, infiltrating growth, and resistance to current anticancer therapies (Eramo et al., 2006). Although the addition of chemotherapy to resection and radiotherapy has increased the 2-year survival rate from 1 to 20%, a successful treatment, much less a cure, for GBM remains elusive. Patients receiving chemotherapeutic agents, such as vinca alkaloids, still have poor survival rates indicative of a tumor cell population that is resistant to current therapies (Parney and Chang, 2003; Stupp et al., 2005). Thus, there is a desperate need for treatments that can specifically target these chemotherapy-resistant GBM cells and potentially increase the survival rate of what is currently a devastating disease.
Small interfering (siRNA) technology has been used to identify genes that are essential for cancer cell survival (Kaelin, 2005; Iorns et al., 2007). In addition, siRNA synthetic lethal approaches can be used to uncover proteins that suppress the growth inhibitory and cytotoxic effects of anticancer agents (Kaelin, 2005; Whitehurst et al., 2007). Druggable genome siRNA libraries comprising siRNAs against gene products that are theoretical targets for drug development have the potential to streamline the identification of small molecule inhibitors. Because approximately two thirds of the proteins encoded in the human genome are thought to be highly problematic as drug targets, specifically focusing on the druggable genome can eliminate a significant subset of nontherapeutic genes (Hopkins and Groom, 2002).
To help identify novel treatment combinations for GBM, we implemented a siRNA high-throughput screen targeting the druggable genome to identify gene products that sensitized human T98G GBM cells to vinblastine (VBL). In this study, we used a series of statistical methods to identify 65 unique gene products that were toxic to cancer cells in combination with VBL. We found that reduction of the prosurvival protein B-cell lymphoma extra large (BCL-xL) by siRNA sensitized T98G cells to VBL. A sublethal concentration of the B-cell lymphoma 2 (BCL-2) family member inhibitor 4-[4-[[2-(4-chlorophenyl)-5,5-dimethylcyclohexen-1-yl]methyl]piperazin-1-yl]-N-[4-[[(2R)-4-morpholin-4-yl-1-phenylsulfanylbutan-2-yl]amino]-3-(trifluoromethylsulfonyl)phenyl]sulfonylbenzamide (ABT-263) (Tse et al., 2008) mimicked the results seen with BCL-xL siRNA in both human T98G GBM and A549 non–small-cell lung cancer cells. Our study illustrates how siRNA high-throughput screening (HTS) technology can be an efficient, unbiased method to identify novel combination anticancer treatments and thereby potentially increase the efficacy of established anticancer drugs such as VBL.
Materials and Methods
Reagents.
The Silencer Druggable Genome siRNA Library (version 1.1), Silencer Select secondary library, Silencer Select Negative Control 1, and BCL-xL Silencer Select siRNAs (siRNA identification s1920, s1921, and s1922) were purchased from Ambion (Austin, TX). AllStars Hs Cell Death Control siRNA was purchased from QIAGEN (Valencia, CA). DharmaFECT 2 transfection reagent, 5x siRNA resuspension buffers, and the siGENOME Non-Targeting siRNA #1 were purchased from Dharmacon RNA Technologies (Lafayette, CO). Tissue culture-treated 384-well microtiter plates were from Greiner Bio-One (Freckenhausen, Germany). CellTiter-Blue Cell Viability Assay and Caspase-Glo 3/7 Assay were purchased from Promega (Madison, WI). BD BioCoat Collagen I 384-well microplates and BD Falcon 384-well white/clear bottom plates were purchased from BD Biosciences (San Jose, CA). Eagle's minimum essential medium, Opti-MEM, Eagle's basal medium, phosphate-buffered saline (PBS), l-glutamine, penicillin/streptomycin, Hoechst 33342, and 1.0-mm Novex 4 to 20% Tris-glycine gels were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum was purchased from Cellgro (Manassas, VA). ECL Western blotting substrate was from Thermo Fisher Scientific (Waltham, MA). The T98G, A549, U87, U3T3, LNZ 428, MDA-MB-231, and HeLa cell lines and human astrocytes were purchased from the American Type Culture Collection (ATCC; Manassas, VA). VBL and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO). ABT-263 was obtained from ChemieTek (Indianapolis, IN). Glyceraldehyde-3-phosphate dehydrogenase rabbit monoclonal, BCL-xL rabbit polyclonal, and BAX rabbit monoclonal antibodies were purchased from Cell Signaling Technology (Danvers, MA). BCL-xL rabbit monoclonal [E18], cytochrome c mouse monoclonal, and ERAB [5F3] mitochondrial marker mouse monoclonal were purchased from Abcam Inc. (Cambridge, MA). Rabbit and mouse peroxidase-conjugated secondary antibodies were purchased from Jackson ImmunoResearch Laboratories Inc. (West Grove, PA). Mini EDTA-free Protease Inhibitor Cocktail Tablets were purchased from Roche (Nutley, NJ).
Cell Culture and Compounds.
T98G glioblastoma cells were maintained in Eagle's minimum essential medium supplemented with Earle's basic salt solution, nonessential amino acids, sodium pyruvate, 1% l-glutamine, 1% penicillin/streptomycin, and 10% fetal bovine serum. The non–small-cell lung cancer cell line A549 was maintained in Eagle's basal medium supplemented with 1% l-glutamine, 1% penicillin/streptomycin, and 10% fetal bovine serum. Cells were grown in a humidified incubator at 37°C with 5% CO2. T98G and A549 cell lines were validated during the study by the Research Animal Diagnostic Laboratory (University of Missouri at Columbia, Columbia, MO).
VBL and ABT-263 were dissolved in 100% DMSO. Compound treatments were added 48 h after initial cell seeding for both synthetic lethal screens and drug combination studies. Compound treatment and DMSO vehicle controls were diluted in medium to a final DMSO concentration of 0.5%. Cells were incubated for an additional 48 h in the presence of compounds or vehicle controls.
siRNA Synthetic Lethal Screen.
T98G cells were wet reverse-transfected with the Silencer Druggable Genome siRNA Library at a final concentration of 20 nM per target in a one gene per well format as reported previously (Thaker et al., 2010). Each gene was targeted with three pooled siRNAs to ensure efficient protein suppression. The transfected cells were incubated for 48 h in a humidified incubator at 37°C with 5% CO2. After 48 h, the medium was removed and replaced with medium containing either 1.2 nM VBL or 0.5% DMSO vehicle control. Cells were incubated for an additional 48 h in the presence of compounds, and cell viability then was measured by incubating the cells with the CellTiter-Blue Cell Viability Assay (1:5 ratio of CellTiter-Blue to medium) for 3 h, according to the manufacturer's protocol. Plates were read on a SpectraMax M5 (Molecular Devices, Sunnyvale, CA).
Data Analysis for the siRNA Synthetic Lethal Screen.
The siRNA synthetic lethal screens were performed in three independent replicates to uncover sensitizers of T98G cells to VBL. Relative fluorescence units (RFU) from each targeted siRNA well were normalized to in-plate scrambled (SCR) negative control values treated with DMSO (DMSO/SCR siRNA), which allowed for plate-to-plate comparisons. The percentage cell viability was determined using the following equation:
where xRFU is the RFUs for each sample and yRFU is the average RFUs for the DMSO/SCR siRNA control within each plate.
We used median absolute deviation (MAD) analysis, an outlier detection method, which is resistant to the presence of outliers within the samples as reported previously (Thaker et al., 2010). Samples with a MAD score >3.5 were defined as outliers and were discarded. The viability of cells targeted by the siRNA was calculated by averaging the remaining values from the screen.
The samples were ranked according to their viability ratio, VR = (x̄CV/¯yCV), where x̄CV is the average cell viability of the siRNA plus compound treatment and ¯yCV is the average cell viability of the siRNA plus vehicle control. The targeted genes were ranked from lowest to highest according to their viability ratio, and the top 2.5% of genes (138 genes) were selected. Student's t test was performed for these 138 targeted genes to determine the significant difference between cells treated with siRNA plus vehicle control and cells treated with siRNA plus VBL as described previously (Whitehurst et al., 2007). Genes with a p value ≤0.01 were selected, yielding 65 siRNAs that sensitized T98G cells to VBL. The Benjamini-Hochberg false discovery rate (FDR) was used to decrease the possibility of false positives due to multiple comparisons (Benjamini and Hochberg, 2000). The FDR ranks the samples according to their p values, where the lower rank corresponds to less strict criteria (Devlin et al., 2003). By ranking the p values of the Student's t test for each sample from smallest to largest, the FDR can be calculated using the following equation:
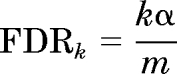 |
where k is the rank of the sample, α is the confidence interval, and m is the total number of t tests performed. Each ordered p value (pk) is compared with the corresponding FDRk, where any test with a pk less than the FDRk is declared significant.
Western Blot Analyses and Lysate Preparation.
Western blot analyses were performed as described previously (Thaker et al., 2010). Membranes were probed with antibodies at various dilutions targeting BCL-xL (1:500), β-tubulin (1:10,000), glyceraldehyde-3-phosphate dehydrogenase (1:1000), BAX (1:200), cytochrome c (1:200), and ERAB (1:200). Positive antibody reactions were visualized using either rabbit or mouse peroxidase-conjugated secondary antibodies (1:1000) and chemiluminescence by ECL Western Blotting Substrate (GE Healthcare, Piscataway, NJ) according to the manufacturer's protocol. Membranes were imaged using a FujiFILM LAS-3000 imager (R&D Systems, Minneapolis, MN).
Concentration-Response Curves.
For VBL sensitization assays using BCL-xL siRNAs, cells were transfected in collagen-coated 384-well plates with increasing concentrations of BCL-xL siRNA (0.63–10 nM), as described in the siRNA screen. Forty-eight hours post-transfection, the cells were treated with VBL (25 pM to 50 nM) with a final DMSO concentration of 0.5%. The percentage cell viability of each sample was determined by CellTiter-Blue as described above. All of the samples were normalized to the DMSO/SCR siRNA negative control, which was termed 100% cell viability. Toxicity of siRNA alone was determined by comparing the DMSO/siRNA percentage cell viability to that of the DMSO/SCR siRNA negative control. The “cell death” siRNA (AllStars Hs Cell Death Control siRNA) was used as a positive control for transfection efficiency and for determining cytotoxicity. Only experiments with >90% cell death with the cell death positive control samples were considered for analysis.
For drug combination studies with both T98G and A549 cells, we first determined their sensitivities to VBL (25 pM to 50 nM) and ABT-263 exposure (0.8–50 μM). On the basis of these data, the two drugs were titrated in a “checkerboard” format on a 384-well plate, where the concentration range of VBL was arranged in the columns and the concentration range of ABT-263 was arranged in the rows in the plate, with DMSO in the last rows and columns providing a standard concentration response curve for each drug. Cells were plated at a density of 500 cells per well. We treated plates after 48 h with VBL and ABT-263, and after an additional 48-h cell viability was determined using the CellTiter-Blue assay. All of the wells were normalized to the DMSO control, and the percentage cell viability of each sample was determined as described above.
Mitochondrial Fractionation.
Mitochondrial isolation by differential centrifugation was adapted from methods described previously (Shiva et al., 2001). In brief, T98G cells were plated and treated in 100-mm dishes. Samples were placed on ice and washed with ice-cold PBS to inhibit cellular activity. Samples were scraped on ice with 500 μl of ice-cold PBS into 1.5-ml Eppendorf tubes. The samples were centrifuged at 1500g for 5 min at 4°C to generate a cellular pellet. The supernatant was discarded, and the pellet was homogenized in 500 μl of STE [250 mM sucrose, 10 mM Tris, and 1 mM EGTA, (pH 7.4)] at 4°C on ice. Samples were centrifuged at 1000g for 5 min to remove cellular debris. To separate the mitochondrial fraction from the cytosolic fraction, we centrifuged samples at 10,000g for 10 min. The supernatant fraction was collected for the cytosolic fraction, and the pellet was collected for the mitochondrial fraction. The mitochondrial pellet was homogenized in 100 μl of STE and centrifuged at 10,000g for 10 min, the supernatant was discarded, and the pellet was collected for the mitochondrial fraction. The mitochondrial pellet was resuspended in 50 μl of STE, and the cytosolic and mitochondrial fractions were frozen at −80°C overnight. Samples were prepared for Western blot analysis as described above.
Caspase-Glo 3/7 Assay.
The Caspase-Glo 3/7 Assay was performed according to the manufacturer's protocol. In brief, T98G cells were plated as described previously in the drug combination studies. Cells (500 cells per well) were plated in BD Falcon 384-well white/clear bottom plates. Forty-eight hours after cell seeding, plates were treated with the VBL/ABT-263 combination. After an additional 48 h, the medium containing drugs was removed, and the Caspase-Glo 3/7 reagent was added to wells in a 1:1 ratio of reagent to medium (25 μl of reagent and 25 μl of medium per well). Plates were incubated in the dark at room temperature for 1 h. The plates were read using an EnVision multiplate plate reader (PerkinElmer Life and Analytical Sciences, Waltham, MA). Levels of caspase-3/7 were normalized to those of the DMSO controls.
siRNA Sequences.
The Silencer Druggable Genome siRNA Library consists of 16,560 Silencer siRNA duplexes with three siRNAs targeting each of 5520 gene products. For the initial screen, we pooled the three individual siRNAs at a 1:1:1 ratio. The sequences for BCL-xL siRNAs are available in Supplemental Table 2.
Results
Identification of Gene Products That Sensitize T98G Cells to VBL Using siRNA HTS Methodology.
We performed siRNA HTS, as described previously (Thaker et al., 2010), to identify gene products that sensitize T98G GBM cells to VBL. Three pooled siRNAs were plated in 384-well plates in a one gene per well format with SCR and cell death controls on each plate, which allowed for plate-to-plate comparisons. We wet reverse-transfected T98G cells with three pooled siRNAs against each target in two identical plates and determined cell viability. We applied a series of statistical methods outlined in Supplemental Fig. 1 to determine the gene products that sensitized T98G cells to VBL. In brief, MAD analysis was performed to determine outliers with 95% confidence within the screen. After all of the outliers were removed, we determined the viability ratios of each gene product and averaged all three screens. The targeted genes were ranked according to their viability ratios, and the top 2.5% of gene products was selected (138 genes). We performed Student's t test on the 138 genes, comparing the siRNA to siRNA plus VBL treatments, and determined the gene products that, when suppressed, sensitized T98G cells to VBL with 99% confidence (p ≤ 0.01). This process yielded 65 high-confidence gene products that sensitized cells to VBL (Fig. 1). To control for false positives due to multiple comparisons, we used the Benjamini-Hochberg FDR procedure, which decreased the probability of at least one false positive due to multiple comparisons when performing t tests on numerous samples (Benjamini and Hochberg, 1995). Applying an α of 0.02, we found that all 65 gene products identified by the viability ratios and Student's t test were declared significant according to the FDR (Supplemental Table 1).
Fig. 1.

Reduction in cell viability with the top 65 gene products from the primary siRNA HTS. Percentage cell viabilities were determined for the siRNA plus DMSO (white bars) and siRNA plus VBL (black bars), as described under Materials and Methods, for all of the genes in the druggable genome library. The top hits from the screen, according to the viability ratios and p values (65 genes), are listed on the abscissa. In most cases, the siRNAs were not toxic to the cells when used alone; however, upon the addition of a nontoxic concentration of VBL, the combination was toxic. Each value is the mean of three independent experiments. Bars, S.D.
To confirm positive siRNAs from the primary HTS, we performed a secondary assay using two unique siRNAs rather than the pooled siRNAs from the primary high-throughput screen. We found 40 siRNAs targeting 29 different gene products that sensitized T98G cells to VBL (Table 1). For nine of the 29 gene products, both unique siRNAs sensitized cells (Table 1).
TABLE 1.
Confirmation of VBL sensitization with individual siRNAs and T98G cells
| Gene Symbol |
p Value |
Gene Symbol |
p Value |
Gene Symbol |
p Value |
|||
|---|---|---|---|---|---|---|---|---|
| siRNA A | siRNA B | siRNA A | siRNA B | siRNA A | siRNA B | |||
| ACOT11 | 0.023 | <0.001 | ADSL | NS | 0.003 | POLR1A | NS | 0.041 |
| AKT3 | 0.033 | 0.002 | ANKRD30A | NS | 0.042 | PRDX6 | 0.014 | NS |
| BCL-xL | 0.035 | 0.041 | BTN3A2 | NS | 0.041 | PROCR | 0.002 | NS |
| FUK | 0.006 | 0.003 | CDC42 | NS | 0.002 | RAB9A | NS | 0.031 |
| ITPR3 | 0.003 | 0.010 | DHFRL1 | 0.046 | NS | SDHA | NS | <0.001 |
| KDELR1 | 0.029 | 0.030 | GSTA1 | 0.027 | NS | SERPINA10 | NS | 0.024 |
| NOS1 | 0.008 | 0.003 | HSD11B1L | 0.012 | NS | SERPINB13 | NS | 0.012 |
| NRAS | 0.003 | 0.001 | LOC136242 | 0.030 | NS | SPINK1 | 0.004 | NS |
| SERPINA3 | 0.004 | 0.008 | NEIL3 | <0.001 | NS | UGT1A9 | 0.046 | NS |
| NIT1 | NS | 0.045 | UGT2B17 | NS | 0.004 | |||
| PMM2 | 0.023 | NS | WARS2 | 0.012 | NS | |||
NS, not significant; siRNAs in the secondary assay with p > 0.05.
BCL-xL siRNA Sensitizes T98G Cells to VBL at Cytostatic Concentrations.
Due to the availability and the biological relevance of inhibitors targeting the prosurvival protein BCL-xL (Tse et al., 2008; Kang and Reynolds, 2009), we focused further on BCL-xL-specific siRNAs to determine whether they could sensitize T98G cells to increasing concentrations of VBL as indicated by the primary and secondary assays (Fig. 2). We treated T98G cells with increasing concentrations of VBL in the presence and absence of decreasing concentrations of pooled BCL-xL siRNAs (sequences of siRNAs in Supplemental Table 2). We found that cells were sensitized to the higher VBL concentrations (3.13–25 nM) when treated in combination with nontoxic concentrations of BCL-xL siRNAs (0.63–10 nM; Fig. 2). We performed Western blot analyses to determine whether the sensitization of VBL to T98G cells was due to specific knockdown of BCL-xL protein. At both 48- (time of drug addition) and 98-h time points (end point of the assay), BCL-xL protein levels were undetectable at all of the concentrations tested relative to those of SCR siRNA controls (Fig. 3).
Fig. 2.

BCL-xL siRNA sensitization of T98G cells to VBL. Three siRNAs targeting BCL-xL were pooled and tested as sensitizers of T98G cells to VBL. A–E, cells were transfected with negative control siRNAs (SCR), positive control siRNAs (cell death), and BCL-xL siRNAs at decreasing concentrations: (A) 10 nM, (B) 5 nM, (C) 2.5 nM, (D) 1.25 nM, and (E) 0.63 nM. Forty-eight hours post-transfection, the cells were treated with increasing concentrations of VBL ranging from 25 pM to 25 nM. After 48 h, cell viability was determined by CellTiter-Blue fluorometric assay as described under Materials and Methods. All of the data points were normalized to a SCR/DMSO control to allow for well to-well comparisons. The effect of different concentrations of VBL on cell viability in cells treated with SCR or BCL-xL siRNA was determined. ○, T98G cells treated with VBL in combination with siRNA negative control (SCR). □, T98G cells treated with VBL in combination with pooled BCL-xL siRNAs. ▴, T98G cells transfected with cell death negative control siRNA. F, SCR and BCL-xL siRNAs were nontoxic to cells. Each value is the mean of four independent experiments. Bars represent S.E.M. *, p ≤ 0.05.
Fig. 3.

Reduction in BCL-xL protein levels in T98G cells after siRNA treatment. Cells were transfected with either negative control (SCR) siRNA or BCL-xL pooled siRNA at various concentrations (2.5, 1.25, 0.63, and 0.31 nM), and protein levels of BCL-xL were measured at 48 and 96 h by Western blot analyses. BCL-xL siRNA decreased protein levels at all of the concentrations tested relative to the SCR control, at both 48 and 96 h. The Western blot is representative of three independent experiments.
ABT-263 Sensitizes T98G and A549 Cells to VBL.
To determine whether ABT-263, a prosurvival BCL-2 family inhibitor, sensitized cells to VBL, we treated T98G cells with increasing concentrations of VBL in the presence and absence of a nontoxic concentration of ABT-263 (Fig. 4A). As seen in Fig. 4B, the toxicity of VBL at concentrations ≥1.56 nM was increased by the addition of 1.56 μM ABT-263.
Fig. 4.

Sensitization of T98G and A549 cells to VBL by ABT-263. A, T98G cells were treated with increasing concentrations of VBL (25 pM to 25 nM) in the presence and absence of a nontoxic concentration of ABT-263 (1.56 μM). B, A549 cells were treated with increasing concentrations of VBL (25 pM to 25 nM) in the presence and absence of a nontoxic concentration of ABT-263 (12.5 μM). At higher concentrations of VBL (1.56–25 nM for T98G and 3.13–25 nM for A549), ABT-263 significantly decreases the cell viability relative to VBL alone in both T98G and A549 cancer cell lines. Cell viability was determined using a CellTiter-Blue assay. All of the values were normalized to the DMSO control (no ABT-263). ○, Cells treated with increasing concentrations of VBL. ■, Cells treated with increasing concentrations of VBL in the presence of nontoxic ABT-263 concentrations. Each value is the mean of four independent experiments. Bars represent S.E.M. *, p ≤ 0.05.
We compared expression levels of BCL-xL protein in T98G cells, other human glioblastoma cells (U87, U373 and LNZ 428), and other human cancer cells (MDA-MB-231 breast, HeLa cervical, and A549 lung cancer cells) to those of normal human astrocytes by Western blot analyses (Supplemental Fig. 2). We found that, relative to the normal human astrocytes, BCL-xL protein was overexpressed in the majority of the cancer cell lines (MDA-MB-231, A549, T98G, U87, and U3T3). To determine whether BCL-xL sensitization of VBL was unique to T98G cells, we treated A549 non-small lung cancer cells with increasing concentrations of VBL in the presence and absence of nontoxic concentrations of ABT-263. We found that, in the presence of 12.5 μM ABT-263, A549 cells also were sensitized to VBL at concentrations >3.13 nM (Fig. 4B).
It is notable that a 48-h exposure of either T98G or A549 cells to VBL alone did not reduce cell viability markedly below 50%, whereas the addition of ABT-263 greatly reduced cell viability (Fig. 4). Therefore, we performed growth inhibition studies using cell death siRNAs, which were known to induce >90% cell death in these cells (Thaker et al., 2010). By comparing the cell viability at the time of VBL addition and the 48-h end point of the assay, we could distinguish between no growth inhibition, which was defined as having the same cell viability as the negative control, and complete growth inhibition, which was defined as having the cell viability of the negative control at the time of plating. We defined cytotoxicity in terms of added loss of cell viability and normalized cell viability to that of the cell death control after subtracting the cytostatic effect. In all of the conditions tested (BCL-xL siRNA in T98G cells and ABT-263 in T98G and A549 cells), VBL induced a cytostatic effect at concentrations of ≥1.56 nM (Supplemental Fig. 3). In the presence of BCL-xL siRNA in T98G cells, however, the loss of BCL-xL protein sensitized the cells to VBL, and we propose that the cytostatic concentrations became cytotoxic (Supplemental Fig. 3A). In the T98G and A549 cell lines, the addition of a nontoxic concentration of ABT-263 also sensitized the cells to VBL, inducing a cytotoxic effect at cytostatic concentrations of VBL alone (Supplemental Fig. 3, B and C, respectively).
ABT-263 and VBL Induce Cell Death through Mitochondria-Dependent Apoptosis.
We examined caspase-3/7 activation to assess the possible role of mitochondria-dependent apoptosis (Lakhani et al., 2006) in the presence of nontoxic concentrations of ABT-263 and VBL in T98G cells (Fig. 5A). From the time of drug addition up to 8 h, we did not observe any significant increase in caspase-3/7 activity by nontoxic concentrations of VBL or ABT-263; however, at 24 h, we noted caspase-3/7 activation at 25 nM VBL and 1.56 μM ABT-263 (Fig. 5A). We then treated cells with increasing concentrations of VBL in the presence and absence of 1.56 μM ABT-263 and measured caspase-3/7 activity (Fig. 5B). At 24 h, there was a VBL concentration-dependent increase in caspase-3/7 activation, which was greatly enhanced after the addition of 1.56 μM ABT-263 (Fig. 5B).
Fig. 5.

Induction of intrinsic apoptosis induced by VBL and ABT-263. A, activation of caspase-3/7 in T98G cells was measured at toxic (25 μM) and nontoxic (1.56 μM) concentrations of ABT-263 and toxic (25 nM) concentrations of VBL over time. Caspase-3/7 activation was measured at 0 (black), 1 (checkered), 8 (diagonal lines), and 24 h (Whitehurst et al., 2007). B, T98G cells were treated with increasing concentrations of VBL (25 pM to 25 nM) in the presence and absence of 1.56 μM ABT-263. Caspase-3/7 activation was measured at 24 h using a Caspase-3/7 Glo Assay kit. Δ, Cells treated with increasing concentrations of VBL. ■, Cells treated with combinations of VBL and ABT-263. Each value is the mean of three independent experiments. Bars represent S.E.M. *, p ≤ 0.05.
We also examined cytochrome c localization to the mitochondria to determine whether the observed cell death was occurring through mitochondria-dependent apoptosis. We initially treated T98G cells with increasing concentrations of ABT-263 and observed a concentration-dependent decrease in cytochrome c localization to the mitochondria (Fig. 6, A and B), which was decreased further in the presence of VBL (Fig. 6, C and D). This is indicative of cytochrome c release into the cytoplasm and ultimately the induction of mitochondria-dependent apoptosis.
Fig. 6.

ABT-263-dependent caspase-3/7 induction of intrinsic apoptosis. A, T98G cells were treated with decreasing concentrations of ABT-263 (3.13–0.31 μM), and the levels of cytochrome c in the mitochondrial fractions were visualized by Western blot analyses. ERAB, a mitochondria-specific marker, was used as a loading control. B, quantification of cytochrome c localization to the mitochondrial fraction relative to ERAB expression levels. All of the values were normalized to the DMSO control. C, levels of cytochrome c in the mitochondrial fractions in the presence of DMSO, 1.56 μM ABT-263, 12.5 nM VBL, and ABT-263 plus VBL. D, quantification of localization of cytochrome c to the mitochondrial fraction in the presence of ABT-263, VBL, or ABT-263 plus VBL. Expression levels were normalized to ERAB expression levels as well as the DMSO control. Blots are representative of three independent experiments. Bars, S.E.M.
Discussion
siRNA HTS technology has facilitated the identification of gene products that when suppressed result in decreased proliferation or even death of cancerous cells (Willingham et al., 2004; Thaker et al., 2010). To help identify novel combination treatments for cancer, we performed VBL-dependent siRNA HTS targeting the druggable genome. Using this siRNA methodology combined with a series of statistical methods, we identified gene products that sensitized GBM cells to VBL. siRNA HTS is an inherently variable type of cellular assay, where data sets can fluctuate daily due to differences in ambient conditions, cell density, cellular functionality, human or mechanical error, and siRNA transfection efficiency (Birmingham et al., 2009). Even in the most consistent siRNA screens, the transfection efficiency is challenging to regulate and monitor on a well-to-well basis, which increases the possibility of variability among replicates. Thus, complementary but independent statistical methods are necessary to identify high-confidence gene products from the primary screen.
MAD analysis has been used previously as a method for “hit” detection (Chung et al., 2008) but for our purposes served as an unbiased outlier detection methodology. Using the MAD analysis, we were able to distinguish replicates within the individual screens that did not corroborate the overall results. In removing these replicates, we uncovered a subset of possible false negatives that were being biased heavily by the presence of outliers. To determine gene products that sensitized GBM cells to VBL, we used a similar statistical approach to that of Whitehurst et al. (Kaelin, 2005; Whitehurst et al., 2007), using both the Student's t test and the viability ratios as a measure of “hit” determination. In contrast to the strategy used by Whitehurst and colleagues (Kaelin, 2005; Whitehurst et al., 2007), we performed a linear analysis, where we first limited the 5520 gene products of the druggable genome by their viability ratios (top 2.5%), then performed Student's t tests on the remaining 138 genes. By first limiting the number of Student's t tests from 5520 to 138, we decreased the possibility of a false positive due to multiple comparisons from 100 to 75%. As indicated by the FDR with which we used an α of 0.02, we confirmed that all 65 high-confidence gene products identified by performing 138 Student's t tests, versus 5520 tests, were likely true positives. Further evaluation with different siRNAs produced nine prioritized gene products. Assuming that an inhibitor targeting these gene products could have the same molecular effect as decreasing protein expression by siRNA, we were particularly interested in the top gene products with commercially available inhibitors to mimic the effect of the siRNA. Of particular interest was BCL-xL, a member of the BCL-2 prosurvival proteins that plays an essential role in the intrinsic cell death pathway. When the prosurvival proteins BCL-2 and BCL-xL are overexpressed in cancer, the ratio of pro- and antiapoptotic proteins is disturbed, and the intrinsic cell death pathway can be evaded (Trask et al., 2002; Kang and Reynolds, 2009; Li et al., 2009; Hockenbery, 2010). This is significant, because the majority of cancer chemotherapies, including cytotoxic drugs such as VBL, induce cell death through the intrinsic signaling pathway. In some cases, overexpression of BCL-2 prosurvival proteins actually can enhance the resistance of the cells to these anticancer therapies (Tse et al., 2008).
Oblimersen is a BCL-2 antisense oligonucleotide that has been used clinically to specifically decrease the expression of BCL-2 protein in cancer cells. BCL-2 prosurvival protein siRNAs, such as oblimersen, can resensitize chemotherapy-resistant cancer cells overexpressing the BCL-2 prosurvival proteins BCL-2, BCL-xL, and BCL-w (Gazitt et al., 1998; Liu et al., 1999; Castilla et al., 2006). Decreasing the expression of the BCL-2 prosurvival proteins by siRNA predisposes the cells to an apoptotic phenotype that reestablishes the balance between prosurvival and proapoptotic proteins (Supplemental Fig. 4). Although the combination of oblimersen with the microtubule-stabilizing agents paclitaxel and docetaxel has had clinical success in small cell lung cancer, non–small-cell lung cancer, and hormone refractory prostate cancer (Tolcher et al., 2005; Liu et al., 2008; Kang and Reynolds, 2009), oblimersen has not been tested clinically, to our knowledge, in combination with VBL and other microtubule-destabilizing agents.
From the primary and secondary siRNA HTS, we found that, similarly to the combination of oblimersen with microtubule stabilizing agents, upon the addition of BCL-xL siRNA to T98G GBM cells, we could sensitize these cells to the microtubule destabilizing agent VBL. Likewise, when we combined a sublethal concentration of ABT-263, which is a small molecule inhibitor that binds the Bcl-2 homology domain 3 of BCL-2, BCL-xL, and BCL-w, with VBL, we observed a greater than additive toxic effect as indicated by the siRNA HTS. The activation of caspase-3/7 might have resulted from mitochondria-dependent apoptotic pathways that are regulated by BCL-2 family members (Lakhani et al., 2006), although we recognize that effector caspases, such as caspase 3, also are activated by mitochondria-independent mechanisms (Guicciardi and Gores, 2009). Interestingly, the sensitization of T98G and A549 cancer cell lines to VBL by BCL-xL siRNA and ABT-263 only occurred with VBL concentrations (≥1.56 nM) that alone reduced cell viability. This is consistent with the notion that an external stimulus, such as an anticancer agent, is necessary for the activation of intrinsic apoptosis (Ackler et al., 2008, 2010; Tse et al., 2008).
In our studies, relatively stringent criteria were used in the data analysis. The application of a more lenient viability ratio or p value would have expanded the subset of candidate gene products that sensitize VBL to cells. Indeed, if we had selected a p value of ≤0.05 instead of ≤0.01, then both BCL-2 and BCL-w would have been included in the primary data set (Supplemental Fig. 5). Due to the previous success of VBL as an individual anticancer agent (Giannakakou et al., 2000) and ABT-263 as a sensitizer to other anticancer therapies (Ackler et al., 2008, 2010), we believe that the combination of ABT-263 with VBL is worthy of further investigation, especially with those tumor types, such as GBM, which are dependent on the overexpression of BCL-2 prosurvival proteins.
In summary, we have used an unbiased siRNA HTS to expose potential drug combinations that may be of clinical utility in the treatment of cancer. In addition to BCL-xL, we identified other gene products (Table 1) that function to protect T98G cells from the cytotoxic actions of VBL and have available pharmacological inhibitors or inducers. Clearly, these are worthy of further study. Overall, these studies illustrate the value of unbiased siRNA HTS for detecting novel chemosensitivity nodes and potential new anticancer drug combinations.
Supplementary Material
Acknowledgments
We thank Nikhil Thaker, Fang Zhang, and Crystal Zellefrow as well as the rest of the Lazo laboratory for their intellectual input in the design and experimental methods for the siRNA HTS. We also thank Larry J. Kitchens for his intellectual input in the statistical analysis and hit determination methods for the primary and secondary high-throughput screens.
This work was supported by the National Institutes of Health National Cancer Institute [Grant CA078039]; the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant NS40923]; the National Institutes of Health National Institute of Allergy and Infectious Diseases [Grant AI063021]; and a grant from the Fiske Drug Discovery Fund.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.111.184879.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
- GBM
- glioblastoma multiforme
- ABT-263
- 4-[4-[[2-(4-chlorophenyl)-5,5-dimethylcyclohexen-1-yl]methyl]piperazin-1-yl]-N-[4-[[(2R)-4-morpholin-4-yl-1-phenylsulfanylbutan-2-yl]amino]-3-(trifluoromethylsulfonyl)phenyl]sulfonylbenzamide
- BCL-2
- B-cell lymphoma 2
- BCL-xL
- B-cell lymphoma extra large
- DMSO
- dimethyl sulfoxide
- FDR
- false discovery rate
- MAD
- median absolute deviation
- HTS
- high-throughput screening
- PBS
- phosphate-buffered saline
- RFU
- relative fluorescent unit
- siRNA
- small interfering RNA
- SCR
- scrambled
- VBL
- vinblastine.
Authorship Contributions
Participated in research design: Kitchens, Shun, and Lazo.
Conducted experiments: Kitchens and McDonald.
Contributed new reagents or analytic tools: Pollack.
Performed data analysis: Kitchens and Shun.
Wrote or contributed to the writing of the manuscript: Kitchens, Pollack, and Lazo.
References
- Ackler S, Mitten MJ, Foster K, Oleksijew A, Refici M, Tahir SK, Xiao Y, Tse C, Frost DJ, Fesik SW, et al. (2010) The Bcl-2 inhibitor ABT-263 enhances the response of multiple chemotherapeutic regimens in hematologic tumors in vivo. Cancer Chemother Pharmacol 66:869–880 [DOI] [PubMed] [Google Scholar]
- Ackler S, Xiao Y, Mitten MJ, Foster K, Oleksijew A, Refici M, Schlessinger S, Wang B, Chemburkar SR, Bauch J, et al. (2008) ABT-263 and rapamycin act cooperatively to kill lymphoma cells in vitro and in vivo. Mol Cancer Ther 7:3265–3274 [DOI] [PubMed] [Google Scholar]
- Adamson C, Kanu OO, Mehta AI, Di C, Lin N, Mattox AK, Bigner DD. (2009) Glioblastoma multiforme: a review of where we have been and where we are going. Expert Opin Investig Drugs 18:1061–1083 [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B 57:289–300 [Google Scholar]
- Benjamini Y, Hochberg Y. (2000) On the adaptive control of the false discovery rate in multiple testing with independent statistics. J Educ Behav Statist 25:60–83 [Google Scholar]
- Birmingham A, Selfors LM, Forster T, Wrobel D, Kennedy CJ, Shanks E, Santoyo-Lopez J, Dunican DJ, Long A, Kelleher D, et al. (2009) Statistical methods for analysis of high-throughput RNA interference screens. Nat Methods 6:569–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilla C, Congregado B, Chinchón D, Torrubia FJ, Japón MA, Sáez C. (2006) Bcl-xL is overexpressed in hormone-resistant prostate cancer and promotes survival of LNCaP cells via interaction with proapoptotic Bak. Endocrinology 147:4960–4967 [DOI] [PubMed] [Google Scholar]
- Chung N, Zhang XD, Kreamer A, Locco L, Kuan PF, Bartz S, Linsley PS, Ferrer M, Strulovici B. (2008) Median absolute deviation to improve hit selection for genome-scale RNAi screens. J Biomol Screen 13:149–158 [DOI] [PubMed] [Google Scholar]
- Devlin B, Roeder K, Wasserman L. (2003) False discovery or missed discovery? Heredity 91:537–538 [DOI] [PubMed] [Google Scholar]
- Dumontet C, Jordan MA. (2010) Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov 9:790–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eramo A, Ricci-Vitiani L, Zeuner A, Pallini R, Lotti F, Sette G, Pilozzi E, Larocca LM, Peschle C, De Maria R. (2006) Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ 13:1238–1241 [DOI] [PubMed] [Google Scholar]
- Feun LG, Savaraj N, Landy HJ. (1994) Drug resistance in brain tumors. J Neurooncol 20:165–176 [DOI] [PubMed] [Google Scholar]
- Gazitt Y, Rothenberg ML, Hilsenbeck SG, Fey V, Thomas C, Montegomrey W. (1998) Bcl-2 overexpression is associated with resistance to paclitaxel, but not gemcitabine, in multiple myeloma cells. Int J Oncol 13:839–848 [DOI] [PubMed] [Google Scholar]
- Giannakakou P, Sackett D, Fojo T. (2000) Tubulin/microtubules: still a promising target for new chemotherapeutic agents. J Natl Cancer Inst 92:182–183 [DOI] [PubMed] [Google Scholar]
- Guicciardi ME, Gores GJ. (2009) Life and death by death receptors. FASEB J 23:1625–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockenbery DM. (2010) Targeting mitochondria for cancer therapy. Environ Mol Mutagen 51:476–489 [DOI] [PubMed] [Google Scholar]
- Hopkins AL, Groom CR. (2002) The druggable genome. Nat Rev Drug Discov 1:727–730 [DOI] [PubMed] [Google Scholar]
- Iorns E, Lord CJ, Turner N, Ashworth A. (2007) Utilizing RNA interference to enhance cancer drug discovery. Nat Rev Drug Discov 6:556–568 [DOI] [PubMed] [Google Scholar]
- Jordan MA, Wilson L. (2004) Microtubules as a target for anticancer drugs. Nat Rev Cancer 4:253–265 [DOI] [PubMed] [Google Scholar]
- Kaelin WG., Jr (2005) The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 5:689–698 [DOI] [PubMed] [Google Scholar]
- Kang MH, Reynolds CP. (2009) Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res 15:1126–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavallaris M, Tait AS, Walsh BJ, He L, Horwitz SB, Norris MD, Haber M. (2001) Multiple microtubule alterations are associated with Vinca alkaloid resistance in human leukemia cells. Cancer Res 61:5803–5809 [PubMed] [Google Scholar]
- Lakhani SA, Masud A, Kuida K, Porter GA, Jr, Booth CJ, Mehal WZ, Inayat I, Flavell RA. (2006) Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science 311:847–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Zang Y, Li C, Patel NS, Grandis JR, Johnson DE. (2009) ABT-737 synergizes with chemotherapy to kill head and neck squamous cell carcinoma cells via a Noxa-mediated pathway. Mol Pharmacol 75:1231–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Kolesar J, McNeel DG, Leith C, Schell K, Eickhoff J, Lee F, Traynor A, Marnocha R, Alberti D, et al. (2008) A phase I pharmacokinetic and pharmacodynamic correlative study of the antisense Bcl-2 oligonucleotide g3139, in combination with carboplatin and paclitaxel, in patients with advanced solid tumors. Clin Cancer Res 14:2732–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Page C, Beidler DR, Wicha MS, Núñez G. (1999) Overexpression of Bcl-x(L) promotes chemotherapy resistance of mammary tumors in a syngeneic mouse model. Am J Pathol 155:1861–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parney IF, Chang SM. (2003) Current chemotherapy for glioblastoma. Cancer J 9:149–156 [DOI] [PubMed] [Google Scholar]
- Ross DD, Yang W, Abruzzo LV, Dalton WS, Schneider E, Lage H, Dietel M, Greenberger L, Cole SP, Doyle LA. (1999) Atypical multidrug resistance: breast cancer resistance protein messenger RNA expression in mitoxantrone-selected cell lines. J Natl Cancer Inst 91:429–433 [DOI] [PubMed] [Google Scholar]
- Shiva S, Brookes PS, Patel RP, Anderson PG, Darley-Usmar VM. (2001) Nitric oxide partitioning into mitochondrial membranes and the control of respiration at cytochrome c oxidase. Proc Natl Acad Sci USA 98:7212–7217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996 [DOI] [PubMed] [Google Scholar]
- Thaker NG, McDonald PR, Zhang F, Kitchens CA, Shun TY, Pollack IF, Lazo JS. (2010) Designing, optimizing, and implementing high-throughput siRNA genomic screening with glioma cells for the discovery of survival genes and novel drug targets. J Neurosci Methods 185:204–212 [DOI] [PubMed] [Google Scholar]
- Tolcher AW, Chi K, Kuhn J, Gleave M, Patnaik A, Takimoto C, Schwartz G, Thompson I, Berg K, D'Aloisio S, et al. (2005) A phase II, pharmacokinetic, and biological correlative study of oblimersen sodium and docetaxel in patients with hormone-refractory prostate cancer. Clin Cancer Res 11:3854–3861 [DOI] [PubMed] [Google Scholar]
- Trask DK, Wolf GT, Bradford CR, Fisher SG, Devaney K, Johnson M, Singleton T, Wicha M. (2002) Expression of Bcl-2 family proteins in advanced laryngeal squamous cell carcinoma: correlation with response to chemotherapy and organ preservation. Laryngoscope 112:638–644 [DOI] [PubMed] [Google Scholar]
- Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, et al. (2008) ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 68:3421–3428 [DOI] [PubMed] [Google Scholar]
- Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L, Peyton M, Minna JD, Michnoff C, Hao W, Roth MG, et al. (2007) Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature 446:815–819 [DOI] [PubMed] [Google Scholar]
- Willingham AT, Deveraux QL, Hampton GM, Aza-Blanc P. (2004) RNAi and HTS: exploring cancer by systematic loss-of-function. Oncogene 23:8392–8400 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.