

Abstract
Identification and quantification of the metabolites of drugs and drug candidates are routinely performed using liquid chromatography-mass spectrometry (LC-MS). The best practice is to generate a standard curve with the metabolite versus the internal standard. However, to avoid the difficulties in metabolite synthesis, standard curves are sometimes prepared using the substrate, assuming that the signal for substrate and the metabolite will be equivalent. We have tested the errors associated with this assumption using a series of very similar compounds that undergo common metabolic reactions using both conventional flow electrospray ionization LC-MS and low-flow captive spray ionization (CSI) LC-MS. The differences in standard curves for four different types of transformations (O-demethylation, N-demethylation, aromatic hydroxylation, and benzylic hydroxylation) are presented. The results demonstrate that the signals of the substrates compared with those of the metabolites are statistically different in 18 of the 20 substrate-metabolite combinations for both methods. The ratio of the slopes of the standard curves varied up to 4-fold but was slightly less for the CSI method.
Introduction
Pharmacokinetic studies are now routinely performed using liquid chromatography-mass spectrometry (LC-MS). When pharmacokinetic studies are required for metabolites, one of the major problems with the use of LC-MS is that the signal varies, depending on the compound. For LC-MS analysis, the best practice is to construct a standard curve using a synthesized metabolite of the substrate and an internal standard. The problem associated with this is the difficulty of metabolite synthesis. Synthesis of the metabolites requires good synthetic skills and, most importantly, time. Furthermore, sometimes the metabolites are too complex to be readily amendable to synthesis. Given these factors, it is not surprising that many studies use alternative methods to determine the amount of a given metabolite produced.
When a synthetic standard of a metabolite is not available, alternative methods can be used to approximately quantify small molecules by mass spectrometry. Most often, a substrate disappearance method is used to determine the stability of a potential drug. With use of this method, the percentage change in substrate over time can be monitored at one or two different concentrations. Low turnover rates or high affinity can lead to large errors in the substrate depletion method because the change in signal is very small. Furthermore, the results depend on the initial substrate concentration used in the analysis. For example, Peng et al. (2010) applied the substrate depletion method at two different concentrations to determine the stability of a series of compounds with different affinities. It was found that at the higher concentration (25 μM), high-affinity/high-clearance compounds gave very low substrate depletion values, whereas at the lower concentration (1 μM), most of the high-affinity compounds were extensively metabolized.
A second method correlates the amount of substrate disappearance with metabolite appearance. This correlation works for single-metabolite quantification but is more problematic when multiple metabolites are formed from a substrate. When multiple metabolites are observed, this method can be used if one assumes that two metabolites give similar signals in the mass spectrometer.
By far, the simplest method is to assume that the substrate gives a signal that is equivalent to that of the product (Koudriakova et al., 1998; Shebley et al., 2009; Liu et al., 2010; Kamdem et al., 2011) and to monitor signal from the product. This assumption makes metabolite quantification using a mass spectrometer convenient, but it is not clear when, or if, it is correct. This study tests the errors associated with this assumption using a series of very similar compounds for which we have synthesized metabolites using both low-flow captive (CSI) and conventional flow (ESI) interfaces. The captive spray ionization technique takes advantage of a commercially available “plug and play” ionization source requiring minimal setup and maintenance. This technique operates at optimal flow rates of 250 nl/min to 50 μl/min, between those of nanospray ionization (<500 nl/min) and conventional flow ESI (50–1500 μl/min). We include the synthesis under the Materials and Methods to illustrate the effort required to make a synthetic metabolic standard, which must be balanced against the errors in assuming that substrate and metabolite give the same MS signal.
Materials and Methods
Solvents and chemicals were purchased from Sigma-Aldrich (St. Louis, MO), Thermo Fisher Scientific (Waltham, MA), EMD Chemicals (Gibbstown, NJ), and Mallinckrodt Baker, Inc. (Phillipsburg, NJ). Solvents and chemicals are used without purification. The 1H NMR spectra were obtained with a 300-MHz spectrometer equipped with a quad-detection probe (1H, 13C, 31P, and 19F). The 1H-decoupled 13C NMR spectra were obtained at 75 MHz. Conventional flow mass spectrometry was performed on a ThermoQuest Surveyor coupled to a Thermo Finnegan LCQ Advantage ESI-MS system. Low-flow mass spectrometry was performed on a Thermo Scientific Accela (pump and autosampler) coupled to a Thermo Scientific LTQ XL system equipped with a captive spray ionization source (Microchrom BioResources, Inc., Auburn, CA).
Synthesis of Substrates.
The substrates were synthesized using the method described by Peng et al. (2008), with modifications as described previously. The general synthetic scheme for the synthesis is presented in Scheme 1. Potassium hydroxide (842 mg, 15 mmol) and ethanol (5 ml) were placed in a 100-ml round-bottom flask equipped with a water condenser. The mixture was stirred at 80°C to dissolve the potassium hydroxide. Isatin (5 mmol) was added to the solution followed by dropwise addition of the appropriate ketone (5.5 mmol). The mixture was refluxed at 80°C for 2 days. Then the solvent was evaporated using the rotary evaporator, and the residue was dissolved in 50 ml of water. The aqueous phase was neutralized by dropwise addition of 1 N HCl to pH ∼6. The resulting solid was collected by vacuum filtration to get crude quinoline carboxylic acid. The crude carboxylic acid was used directly to get acyl chloride. The crude carboxylic acid was added to a 50-ml round-bottom flask equipped with a water condenser, and 5 ml of undiluted thionyl chloride was added, followed by reflux at 80°C. After 2 h, the excess thionyl chloride was evaporated using a stream of argon to yield quinoline-4-carbonyl chloride. The resulting solid was dissolved in a 5:1 mixture of dichloromethane-triethylamine (10 ml) in a 50-ml round-bottom flask and appropriate amine (5.5 mmol) was added. The reaction mixture was stirred at room temperature. After 2 h, the solvent was evaporated by rotary evaporation and the product was separated by flash chromatography using a hexanes-dichloromethane-methanol solvent system. The product was further purified by crystallization in hexanes-dichloromethane. The NMR and mass spectral data for characterization of substrates are presented in the supplemental data. All compounds were pure on the basis of NMR spectroscopy and TLC of the product after purification.
Scheme 1.

Synthesis of the quinoline carboxamide analogs as substrate for CYP 3A4. EtOH, ethanol.
1-(Pyrimidin-4-yl)ethanone or 4-Acetylpyrimidine.
1-(Pyrimidin-4-yl) ethanone was prepared as described by Easmon et al. (1992). Dropwise addition of an aqueous solution of 70% tert-butylhydroperoxide (180 mmol) and an aqueous solution (150 ml) of iron(II) sulfate heptahydrate (50 g, 180 mmol), in separate addition funnels, over 30 min to a vigorously stirred ice-cold (10–20°C) solution of sulfuric acid (4 M), acetaldehyde (8 g, 180 mmol), pyrimidine (2.4 g, 30 mmol), and CH2Cl2 (180 ml) yielded crude product. After 1.5 h, the organic phase was separated and extracted with dichloromethane (three 100-ml extractions). The organic phase was then pooled, and the solvent was evaporated in a rotary evaporator to obtain an oil. Flash chromatography with dichloromethane as an eluent was used to obtain the purified product in 49% yield. 1H NMR (CDCl3): δ 2.67 (s, 3H), 7.83 (dd, J = 5.0, 1.5, 1H), 8.91 (d, J = 5.0 Hz, 1H), 9.31 (d, J = 1.5 Hz, 1H); ESI-MS: [M + H]+ = 123.2.
O-Demethylation.
The O-demethylation of the substrate was performed as described previously (McOmie et al., 1968; Gao and Portoghese, 1996) with the modifications as needed (Scheme 2a). To the 10-ml solution of substrate (0.36 mmol) in dichloromethane, boron tribromide (1 M in CH2Cl2, 1.1 mmol) was added over 2 min with stirring. The reaction mixture was stirred for 45 min at room temperature. The reaction was quenched by saturated sodium carbonate to adjust to pH 8 and was extracted with dichloromethane (three 30-ml extractions). The combined organic phase was washed with water. The solvent was removed under reduced pressure, and the residue was partially purified by flash chromatography using the hexanes-dichloromethane-methanol solvent system. The product was further purified by crystallization in hexanes-dichloromethane. The NMR and mass spectral data for characterization of metabolites are presented in the supplemental data.
Scheme 2.

Synthesis of the metabolites. RT, room temperature; MeOH, methanol; THF, tetrahydrofuran; TBDMS, tert-butyldimethylsilyl; TBAF, tetrabutylammonium fluoride.
N-Demethylation.
The N-demethylation method developed by Acosta et al. (1994) was used to obtain mono N-demethylation directly from the N-dimethylated substrates (Scheme 2b). Iodine (0.27 mmol) was added to the ice-chilled mixture of substrate (0.14 mmol), CaO (1.1 mmol) in tetrahydrofuran (1.6 ml), and methanol (1.2 ml). The mixture was stirred at 0°C and monitored by TLC. After 4 h, all of the reactant was consumed. The reaction mixture was filtered, and filtrate was sequentially washed with 15% sodium thiosulfate solution, water, and brine solution. The organic phase was dried over magnesium sulfate, and solvent was evaporated in a rotary evaporator. The product was separated by flash chromatography using a hexanes-dichloromethane-methanol solvent system. The product was further purified by crystallization in hexanes-dichloromethane. The NMR and mass spectral data for characterization of metabolites are presented in the supplemental data.
4-((tert-Butyldimethylsilyloxy)methyl)aniline.
tert-Butyldimethylsilylchloride (12.2 mol) and imidazole (13.4 mmol) in dichloromethane (20 ml) were added to a solution of 4-aminobenzyl alcohol (12.2 mmol) in 20 ml of dichloromethane. After stirring for 1 h at room temperature, the reaction mixture was diluted with brine solution and then extracted with ethyl acetate. The organic phase was dried over sodium sulfate and filtered. The solvent was evaporated in a rotary evaporator to get the oil with 97% yield. 1H NMR (CDCl3): δ 0.01 (s, 6H), 0.85 (s, 9H), 3.58 (s, 2H), 4.55 (s, 2H), 6.56 (d, J = 8.6 Hz, 2H), 7.03 (d, J = 8.5 Hz, 2H).
Benzylic Hydroxylation.
Protected 4-aminobenzyl alcohol [4-((tert-butyldimethylsilyloxy)methyl)aniline] was reacted with appropriate acid chlorides (the acid chlorides were prepared using the same method described under Synthesis of Substrates) to get the protected benzylic alcohols, which, on deprotection, yielded the desired products (Scheme 2c). The acid chloride (2 mmol) was dissolved in a 5:1 mixture of dichloromethane-triethylamine (5 ml), and 4-((tert-butyldimethylsilyloxy)methyl)aniline (2 mmol) in 1 ml of dichloromethane was added. The reaction mixture was stirred at room temperature for 2 h. After 2 h, the solvent was removed by rotary evaporation, and crude product was purified by flash chromatography (30 g of Silica Gel 60, 0.063–0.200 mm) to get the protected benzylic alcohol using a hexanes-dichloromethane-methanol solvent system. The protected alcohol was dissolved in 20 ml of tetrahydrofuran, and 2 ml of 1 M tetrabutylammonium fluoride solution in tetrahydrofuran. The reaction mixture was stirred, and the progress of the reaction was monitored by TLC. The reaction was completed in 4 h, the solvent was evaporated, and the product was purified by flash chromatography (30 g of Silica Gel 60, 0.063–0.200 mm) using hexanes, dichloromethane, and methanol in 9:10:1 ratio. The product was further purified by crystallization using hexanes-dichloromethane. The NMR and mass spectral data for characterization of metabolites are presented in the supplemental data.
Standard Curves.
The conventional flow standard curves (analyte area/internal standard area versus analyte concentration/internal standard concentration) were constructed for the metabolites and the substrates against the internal standard phenacetin using freshly prepared solutions. The assay solution contains 7.14 μM phenacetin and at least four different concentrations of the substrate or metabolite. The substrate and metabolite concentrations were within the linear detection limit of the LC-MS instrument. Data for the standard curve were generated by LC-MS/MS analysis using a ThermoQuest high-performance liquid chromatograph coupled with a Thermo Finnigan LCQ Advantage system. The ions were generated by electrospray ionization and were monitored in positive ion mode. The high-performance liquid chromatography system used reverse-phase chromatography (Eclipse Plus C18 5-μm, 150-mm length, 3.2-mm i.d. column; Agilent Technologies, Santa Clara, CA) beginning with 85% mobile phase A (0.1% acetic acid in water) and 15% mobile phase B (0.1% acetic acid in acetonitrile) with a linear gradient to 85% of mobile phase B over 25 min. After 25 min, mobile phase B was reduced to 15% and run isocratically for 5 min.
The CSI standard curves used 12 concentration points (5–0.002441 μM) and were constructed for the metabolites and the substrates in a 50:50 (v/v) solution of acetonitrile and water with 0.1% formic acid containing 1 μM testosterone as internal standard. At least nine different concentrations of the substrate or metabolite were used for the curves to obtain a linear MS response. Data for the standard curves were generated by LC-MS/MS analysis using a Thermo Scientific Accela high-performance liquid chromatograph coupled with a Thermo Scientific LTQ XL mass spectrometer. Positive ions were generated by the low-flow electrospray ionization captive source. The high-performance liquid chromatography system delivered a mobile phase using an isocratic mixture of 50% mobile phase A (0.1% formic acid in water) and 50% mobile phase B (0.1% formic acid in acetonitrile) at a flow rate of 100 μl/min. The flow was reduced to 5 μl/min using a 1/20 splitter (Accurate Splitter, LC Packings; Dionex, Amsterdam, The Netherlands) before introduction into the source. The following source settings were optimized as generic settings using compound 1 and were used for all subsequent data collection: sheath gas, 0 arbitrary units; auxiliary gas, 0 arbitrary units; spray voltage, 1 kV; capillary temperature, 175°C; capillary voltage, 43 V; and tube lens voltage, 80 V. MS/MS conditions were optimized for each compound to determine the appropriate relative collision energy and product ion to use for selected reaction monitoring analysis. Sample was delivered by flow injections (1 μl) directly into the source, and data were collected and analyzed using Thermo Scientific Xcalibur software (version 2.06). Linear regression analysis was performed using the analyte/internal standard area ratio versus analyte concentration/internal standard concentration.
Results
To assess the equivalency of the signals between the substrate and the metabolite, we synthesized 20 substrates for cytochrome P450 3A4 and the corresponding metabolite for each substrate. Cytochrome P450 3A4 is a major drug-metabolizing enzyme that metabolizes approximately 50% of clinically used drugs (Ortiz de Montellano, 2005). The substrates are quinoline carboxamide analogs that have been used as model compounds for cytochrome P450 kinetic assays (Peng et al., 2008, 2010; Pearson et al., 2011). The metabolites represent common transformations mediated by cytochrome P450 enzymes including O-demethylation, N-demethylation, aromatic hydroxylation, and benzylic hydroxylation. The structures of the synthesized substrates and the metabolites are presented in Table 1. The synthesized metabolites were confirmed to be the cytochrome P450 3A4 metabolites by comparing the retention time and fragmentation patterns in LC-MS with those of the enzymatically formed metabolites (data not shown).
TABLE 1.
Structures of the cytochrome P450 3A4 substrates and metabolites used for the study and the standard curve slope ratio of metabolite to substrate
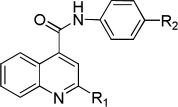
| R1 | Substrate R2 | Substrate | Metabolite R2 | Metabolite | Slope Ratio |
|
|---|---|---|---|---|---|---|
| ESIa | CSIb | |||||
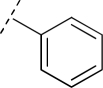 |
OCH3 | 1 | OH | 1M | 0.7 | 1.3 |
| N(CH3)2 | 2 | NH(CH3) | 2M | 0.9 | 0.4 | |
| H | 3 | OH | 3M | 1.5 | 0.9 | |
| CH3 | 4 | CH2OH | 4M | 2.6 | 0.5 | |
 |
OCH3 | 5 | OH | 5M | 1.7 | 3.6 |
| N(CH3)2 | 6 | NH(CH3) | 6M | 2.5 | 0.7 | |
| H | 7 | OH | 7M | 4.0 | 1.2 | |
| CH3 | 8 | CH2OH | 8M | 2.5 | 0.5 | |
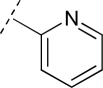 |
OCH3 | 9 | OH | 9M | 2.4 | 1.9 |
| N(CH3)2 | 10 | NH(CH3) | 10M | 1.3 | 0.5 | |
| H | 11 | OH | 11M | 2.1 | 0.9 | |
| CH3 | 12 | CH2OH | 12M | 2.2 | 0.7 | |
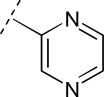 |
OCH3 | 13 | OH | 13M | 2.3 | 1.6 |
| N(CH3)2 | 14 | NH(CH3) | 14M | 2.7 | 3.0 | |
| H | 15 | OH | 15M | 3.1 | 0.8 | |
| CH3 | 16 | CH2OH | 16M | 2.4 | 1.2 | |
 |
OCH3 | 17 | OH | 17M | 3.0 | 1.2 |
| N(CH3)2 | 18 | NH(CH3) | 18M | 0.6 | N.D. | |
| H | 19 | OH | 19M | 2.7 | 0.7 | |
| CH3 | 20 | CH2OH | 20M | 0.9 | 2.8 | |
N.D., not determined.
Slope (average) of the standard curve for metabolite versus internal standard divided by slope (average) of the standard curve for substrate versus internal standard using ESI.
Slope (average) of the standard curve for metabolite versus internal standard divided by slope (average) of the standard curve for substrate versus internal standard using nanospray ionization. Slopes are presented in the supplemental data.
The synthesis of a metabolite can be challenging. Synthesis of the mono N-demethylated metabolite failed using several literature procedures. For example, N-demethylation using N-iodosuccinamide (Stenmark et al., 2000) and using the method developed by Rosenau et al. (2004) did not work in our hands, although N-demethylation was eventually successfully performed for all the aniline substrates using the method of Acosta et al. (1994), which uses calcium oxide and iodine in the presence of methanol (Scheme 2b). Attempts to perform benzylic hydroxylation directly on the substrates were also unsuccessful. Therefore, the protected 4-aminobenzyl alcohol was reacted with appropriate acid chloride to get the protected benzylic alcohol, which, upon deprotection, yielded the desired product (Scheme 2c).
Having the substrates and the metabolites in hand, we constructed the standard curves for the substrates or the metabolites versus the internal standard using conventional flow LC-MS methodology, and CSI LC-MS. As an example, a representative pair of standard curves for a substrate (compound 14) and a metabolite (compound 14M) are presented in Fig. 1 using each type of methodology. The slopes vary by a ratio of 2.7 for conventional flow and 3.0 for CSI. A graphical representation of the slopes associated with the standard curves for all 20 of the substrates versus metabolites is presented in Fig. 2 for both methods. To assess whether the difference in signal associated with the substrate versus that of the metabolite was statistically significant, we performed Student's t tests for the slopes of the substrates and those of the metabolites. The calculated independent, two-sample t values and the slopes for substrate and metabolite pairs are presented in Supplemental Tables S1 and S2. For 18 of the 20 pairs of substrates and metabolites, the slopes are significantly different at the 95% confidence level. Two substrate/metabolite pairs (compound 10 and compound 20) have insignificant differences in their slopes using ESI, whereas compounds 3 and 11 show insignificant differences using CSI.
Fig. 1.

Representative standard curves. The curves were constructed by plotting the ratio of area of analyte to internal standard (IS) against the ratio of concentration of analyte to IS. A, standard curve of substrate 14 versus IS using electrospray ionization. B, standard curve of metabolite 14M versus IS using electrospray ionization. C, standard curve of substrate 14 versus IS using CSI. D, standard curve of metabolite 14M versus IS using CSI.
Fig. 2.

Slopes of the standard curves for the substrate and metabolite pairs. The standard curves (analyte area/internal standard area versus analyte concentration/internal standard concentration) were prepared from the LC-MS analysis using at least four concentrations of substrate and metabolite. The error bar represents the S.D. All slopes except those for compounds 10 and 20 are significantly different at the 95% confidence limit. A, results for ESI. B, results for CSI.
The ratio of the slope of the metabolite to that of the substrate is presented in Table 1. The largest slope difference was 4.0 for compounds 7 and 7M using ESI. Whereas some of the metabolites gave a higher signal relative to that of the substrate for a given concentration, this was not consistently true. For example, the slope of the metabolite 18M was almost half that of the substrate 18 for ESI, whereas this is true for the majority of the CSI results. These results illustrate that the determination of the metabolite concentration using substrate to construct the standard curve can yield a predicted concentration of the metabolite up to 4-fold lower than the actual concentration to approximately 2-fold higher than the actually metabolite concentration.
Discussion
It has been proposed that low-flow LC-MS techniques such as nanospray may provide less variability in ionization (Benetton et al., 2003; Hop et al., 2005; Ramanathan et al., 2011) (see below) than regular LC-MS. To test this hypothesis we also compared standard curves generated using CSI LC-MS. The results are shown in Table 1, and the overall variability is less than that for normal-flow LC-MS. To look at the absolute deviations associated with the difference in signals from parent and metabolite, we calculated the average of the absolute differences of the ratio of the metabolite and parent signals from 1 (unsigned average difference). If the ratio was a fraction, it was inverted and subtracted from 1. For example, the slope ratio for CSI of compound 5 is 0.3; inverting this number gives 3.3, which has an absolute error from perfect agreement of 2.3. The unsigned average difference of the substrate/metabolite ratios from 1 indicates that for normal LC-MS the error is 1.2, whereas for captive spray LC-MS it is 0.8, the largest error being 3.0 for compound 7 for LC-MS and 2.3 for compounds 5 and 14 for captive spray LC-MS.
The metabolic pathways for this series of compounds cover benzylic hydroxylation, aromatic hydroxylation, N-dealkylation, and O-dealkylation, which are some of the most commonly observed metabolic transformations. It is of interest to determine whether some metabolic transformations give more similar metabolite/substrate slope ratios than others. Although we expected to see consistent deviation for each metabolic pathway as a result of pKa differences or changes in lipophilicity in the parent versus metabolite, this was not the case. The results did not show a consistent trend for signal differences between substrate and metabolite with respect to different metabolic transformations using either ESI or CSI. Using ESI as an example, the ratio of the slopes of the metabolite to that of the substrate for O-demethylation for five different substrates, 1, 5, 9, 13, and 17, are 0.7, 1.7, 2.4, 2.3, and 3, respectively, with the unsigned average difference in the slopes being 1.2. Likewise, for N-demethylation of substrates 2, 6, 10, 14, and 18, the slope ratios are 0.9, 2.5, 1.3, 2.7, and 0.6, respectively, with the unsigned average difference in the slopes being 0.85. For aromatic oxidation, all the slopes are positive with the average difference in slopes from 1 being 1.7, whereas for benzylic hydroxylation, the unsigned average difference in the slopes from 1 is 1.2. These observations indicate that the signal of a metabolite can be higher or lower than that of the substrate and that none of the different metabolic transformations give a consistently better agreement in the slopes. This result does not appear to be the case for carboxylic acid metabolites (Hop et al., 2005; Valaskovic et al., 2006; Ramanathan et al., 2007) or for the loss of an amine by N-dealkylation (Benetton et al., 2003), both of which consistently show a lower signal for the metabolite. Indeed, the fact that all the compounds tested have multiple nitrogen atoms and no other ionizable group would lead one to conclude that this is a best case scenario and that one might expect larger deviations for metabolites that introduce a negative charge or for the loss of nitrogen.
Thus, it appears that the use of substrate to estimate the LC-MS signal, although convenient, leads to significant errors. In this regard, other methodologies show promise. For example, a number of investigators have used nanospray LC-MS to decrease the differences between the signal from parent drug and metabolites (Benetton et al., 2003; Hop et al., 2005; Valaskovic et al., 2006; Ramanathan et al., 2007). Furthermore, a number of other methods to estimate exposure to a metabolite using corrected responses in LC-MS/MS (Vishwanathan et al., 2009; Gao et al., 2010; Ma et al., 2010; Tong et al., 2010; Mutlib et al., 2011; Walker et al., 2011) or NMR based methods (Vishwanathan et al., 2009; Mutlib et al., 2011; Walker et al., 2011) have been developed.
In summary, we compared the slopes of the standard curves between substrates and metabolites with a common internal standard to test whether the signal is equivalent and whether the substrates can be used to quantify the metabolites in LC-MS. The results demonstrated that the signals of the substrates compared with those of the metabolites are significantly different statistically for metabolite/substrate pairs for O-demethylation, N-demethylation, aromatic hydroxylation, and benzylic hydroxylation. The ratio of the signal for the metabolite to that of the substrate was found to be up to 4-fold different, which may be an unacceptable error unless you have very low or high amounts of metabolite formed. The results also demonstrate that the signal of the compound is an intrinsic property of the compound and not related to any given metabolic pathway.
Supplementary Material
Acknowledgments
We thank Carolyn Joswig-Jones for help formatting the figures.
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grant GM84546].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/dmd.111.040865.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
- LC
- liquid chromatography
- MS
- mass spectrometry
- CSI
- captive spray ionization
- ESI
- electrospray ionization
- TLC
- thin-layer chromatography.
Authorship Contributions
Participated in research design: Dahal, Jones, Davis, and Rock.
Conducted experiments: Dahal and Davis.
Contributed new reagents or analytic tools: Dahal, Jones, Davis, and Rock.
Performed data analysis: Dahal, Davis, and Jones.
Wrote or contributed to the writing of the manuscript: Dahal, Jones, Davis, and Rock.
References
- Acosta K, Cessac JW, Rao PN, Kim HK. (1994) Oxidative demethylation of 4-substituted N,N-dimethylanilines with iodine and calcium oxide in the presence of methanol. J Chem Soc Chem Commun 1994:1985–1986 [Google Scholar]
- Benetton S, Kameoka J, Tan A, Wachs T, Craighead H, Henion JD. (2003) Chip-based P450 drug metabolism coupled to electrospray ionization-mass spectrometry detection. Anal Chem 75:6430–6436 [DOI] [PubMed] [Google Scholar]
- Easmon J, Heinisch G, Holzer W, Rosenwirth B. (1992) Novel thiosemicarbazones derived from formyl- and acyldiazines: synthesis, effects on cell proliferation, and synergism with antiviral agents. J Med Chem 35:3288–3296 [DOI] [PubMed] [Google Scholar]
- Gao H, Deng S, Obach RS. (2010) A simple liquid chromatography-tandem mass spectrometry method to determine relative plasma exposures of drug metabolites across species for metabolite safety assessments. Drug Metab Dispos 38:2147–2156 [DOI] [PubMed] [Google Scholar]
- Gao P, Portoghese PS. (1996) Boron tribromide-catalyzed rearrangement of 7,7-diphenylhydromorphone to 6,7-diphenylmorphine: a novel conversion of ketones to allylic alcohols. J Org Chem 61:2466–2469 [Google Scholar]
- Hop CE, Chen Y, Yu LJ. (2005) Uniformity of ionization response of structurally diverse analytes using a chip-based nanoelectrospray ionization source. Rapid Commun Mass Spectrom 19:3139–3142 [DOI] [PubMed] [Google Scholar]
- Kamdem LK, Flockhart DA, Desta Z. (2011) In vitro cytochrome P450-mediated metabolism of exemestane. Drug Metab Dispos 39:98–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koudriakova T, Iatsimirskaia E, Utkin I, Gangl E, Vouros P, Storozhuk E, Orza D, Marinina J, Gerber N. (1998) Metabolism of the human immunodeficiency virus protease inhibitors indinavir and ritonavir by human intestinal microsomes and expressed cytochrome P4503A4/3A5: mechanism-based inactivation of cytochrome P4503A by ritonavir. Drug Metab Dispos 26:552–561 [PubMed] [Google Scholar]
- Liu D, Zheng X, Tang Y, Zi J, Nan Y, Wang S, Xiao C, Zhu J, Chen C. (2010) Metabolism of tanshinol borneol ester in rat and human liver microsomes. Drug Metab Dispos 38:1464–1470 [DOI] [PubMed] [Google Scholar]
- Ma S, Li Z, Lee KJ, Chowdhury SK. (2010) Determination of exposure multiples of human metabolites for MIST assessment in preclinical safety species without using reference standards or radiolabeled compounds. Chem Res Toxicol 23:1871–1873 [DOI] [PubMed] [Google Scholar]
- McOmie JF, Watts ML, West DE. (1968) Demethylation of aryl methyl ethers by boron tribromide. Tetrahedron 24:2289–2292 [Google Scholar]
- Mutlib A, Espina R, Vishwanathan K, Babalola K, Chen Z, Dehnhardt C, Venkatesan A, Mansour T, Chaudhary I, Talaat R, et al. (2011) Application of quantitative NMR in pharmacological evaluation of biologically generated metabolites: implications in drug discovery. Drug Metab Dispos 39:106–116 [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano PR. (2005) Cytochrome P450: Structure, Mechanism, and Biochemistry, Kluwer Academic/Plenum Publishers, New York [Google Scholar]
- Pearson J, Dahal UP, Rock D, Peng CC, Schenk JO, Joswig-Jones C, Jones JP. (2011) The kinetic mechanism for cytochrome P450 metabolism of type II binding compounds: evidence supporting direct reduction. Arch Biochem Biophys 511:69–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng CC, Cape JL, Rushmore T, Crouch GJ, Jones JP. (2008) Cytochrome P450 2C9 type II binding studies on quinoline-4-carboxamide analogues. J Med Chem 51:8000–8011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng CC, Pearson JT, Rock DA, Joswig-Jones CA, Jones JP. (2010) The effects of type II binding on metabolic stability and binding affinity in cytochrome P450 CYP3A4. Arch Biochem Biophys 497:68–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan R, Raghavan N, Comezoglu SN, Humphreys WG. (2011) A low flow ionization technique to integrate quantitative and qualitative small molecule bioanalysis. Int J Mass Spectrom 301:127–135 [Google Scholar]
- Ramanathan R, Zhong R, Blumenkrantz N, Chowdhury SK, Alton KB. (2007) Response normalized liquid chromatography nanospray ionization mass spectrometry. J Am Soc Mass Spectrom 18:1891–1899 [DOI] [PubMed] [Google Scholar]
- Rosenau T, Hofinger A, Potthast A, Kosma P. (2004) A general, selective, high-yield N-demethylation procedure for tertiary amines by solid reagents in a convenient column chromatography-like setup. Org Lett 6:541–544 [DOI] [PubMed] [Google Scholar]
- Shebley M, Kent UM, Ballou DP, Hollenberg PF. (2009) Mechanistic analysis of the inactivation of cytochrome P450 2B6 by phencyclidine: effects on substrate binding, electron transfer, and uncoupling. Drug Metab Dispos 37:745–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark HG, Brazzale A, Ma Z. (2000) Biomimetic synthesis of macrolide/ketolide metabolites through a selective N-demethylation reaction. J Org Chem 65:3875–3876 [DOI] [PubMed] [Google Scholar]
- Tong W, Chowdhury SK, Su AD, Alton KB. (2010) Quantitation of parent drug and its unstable metabolites by in situ coulometric oxidation and liquid chromatography-tandem mass spectrometry. Anal Chem 82:10251–10257 [DOI] [PubMed] [Google Scholar]
- Valaskovic GA, Utley L, Lee MS, Wu JT. (2006) Ultra-low flow nanospray for the normalization of conventional liquid chromatography/mass spectrometry through equimolar response: standard-free quantitative estimation of metabolite levels in drug discovery. Rapid Commun Mass Spectrom 20:1087–1096 [DOI] [PubMed] [Google Scholar]
- Vishwanathan K, Babalola K, Wang J, Espina R, Yu L, Adedoyin A, Talaat R, Mutlib A, Scatina J. (2009) Obtaining exposures of metabolites in preclinical species through plasma pooling and quantitative NMR: addressing metabolites in safety testing (MIST) guidance without using radiolabeled compounds and chemically synthesized metabolite standards. Chem Res Toxicol 22:311–322 [DOI] [PubMed] [Google Scholar]
- Walker GS, Ryder TF, Sharma R, Smith EB, Freund A. (2011) Validation of isolated metabolites from drug metabolism studies as analytical standards by quantitative NMR. Drug Metab Dispos 39:433–440 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.