

Abstract
Mass spectrometric based methods for absolute quantification of proteins, such as QconCAT, rely on internal standards of stable-isotope labeled reference peptides, or “Q-peptides,” to act as surrogates. Key to the success of this and related methods for absolute protein quantification (such as AQUA) is selection of the Q-peptide. Here we describe a novel method, CONSeQuence (consensus predictor for Q-peptide sequence), based on four different machine learning approaches for Q-peptide selection. CONSeQuence demonstrates improved performance over existing methods for optimal Q-peptide selection in the absence of prior experimental information, as validated using two independent test sets derived from yeast. Furthermore, we examine the physicochemical parameters associated with good peptide surrogates, and demonstrate that in addition to charge and hydrophobicity, peptide secondary structure plays a significant role in determining peptide “detectability” in liquid chromatography-electrospray ionization experiments. We relate peptide properties to protein tertiary structure, demonstrating a counterintuitive preference for buried status for frequently detected peptides. Finally, we demonstrate the improved efficacy of the general approach by applying a predictor trained on yeast data to sets of proteotypic peptides from two additional species taken from an existing peptide identification repository.
The study of cellular systems via identification of their protein components is becoming almost commonplace with the continuing advances in mass spectrometric hardware and associated analytical software. The current drive is now to assign accurate quantitative information to these protein components, such that the data can be used in systems modeling studies. For these models to be effective and the dynamics of these biochemical systems simulated during activation or perturbation, absolute rather than relative quantitative information must be provided (1). Even in the absence of sophisticated modeling studies, evaluating both the qualitative and quantitative aspects of biological networks can permit understanding of the complex interplay of system components, as well as the identification of disease biomarkers (2–4).
Given the dependence of mass spectrometric signal intensity on the nature of the analyte, methods for absolute protein quantification primarily rely on standardization of signal intensity with known quantities of isotope-labeled references that are identical in primary structure to the analyte (5–7). In a typical such proteomics experiment, quantification is performed at the peptide level, often by virtue of selected reaction monitoring experiments, using defined amounts of pure isotope-labeled tryptic peptide as reference for unknown quantities of the tryptic hydrolysate of the protein of interest. Protein amount is subsequently inferred. This can also be combined with label-free approaches to infer absolute peptide quantifications using a subset of peptides as standards (8).
Generation of these isotope-labeled quantification peptides, which we refer to as Q-peptides (fully proteolysed proteotypic peptides (2)), can be achieved either by chemical synthesis, purification, and quantification of each peptide individually, described by Gygi and colleagues as the AQUA strategy (7), or en masse using the QconCAT technique where designer recombinant proteins containing the selected Q-peptides are expressed in labeled form using a bacterial expression system (6, 9–11). A further exciting alternative exploits synthetic peptide libraries to find suitable peptides and reactions (12). Irrespective of the method used for Q-peptide production, selection of the optimal reference peptide is critical for accurate calculation of absolute protein levels in cell extracts (13). An average protein will generate in the region of 30–50 tryptic peptides. However, not all tryptic peptides are consistently observed during mass spectrometric analysis (14) and only a few of these will be suitable Q-peptide candidates. Hydrophilic peptides will not bind to the reversed-phase column used for peptide separation prior to mass spectrometric analysis, and peptides that are extremely hydrophobic are unlikely to be eluted from the stationary phase. The propensity of a peptide to ionize, a prerequisite for detection, is dependent on both its physicochemical properties and the chemical composition of its environment during ionization (15). Moreover, the ionized peptides also need to be stable within the mass analyzer in order to be detected. Most quantitative proteomics experiments rely on peptide analysis using either a transmission quadrupole (QqQ or hybrid Q-ToF) for precursor ion selection (optimally followed by selective transmission of one or more product ions), or a high resolution ion trap (orbitrap). Representative tryptic peptides should therefore have a mass between 600–6000 Da, resulting in charged (2+, 3+) species between m/z 300 and 2000. The Q-peptide selected for use as a reference quantification analyte must also be amenable to detection under the conditions chosen for analysis, these being reversed-phase liquid chromatography (LC)1 and electrospray ionization (ESI) MS in most proteomics experiments. The native peptide should ideally also be in a region of the protein that undergoes complete proteolysis, as the consistent production of limit peptides is essential for accurate quantification. Generally the peptide should also be unique to the protein of interest and not involved in alternate splicing, subject to covalent modifications and/or polymorphisms, all of which could potentially confound quantification.
To date within our own laboratories, Q-peptide selection for QconCATs has relied exclusively either on the use of defined physicochemical criteria based on manual analysis of proteotypic peptide properties or on the evaluation of experimental data from the proteins of interest. However, the former is limited in its analytical capabilities and the latter requires prior generation of the necessary experimental data and is thus not feasible for large scale quantitative projects, or where no previous analysis has been performed. Computational approaches to the prediction of peptides likely to be observed are clearly desirable. Early studies have characterized properties that contribute to peptide detection after matrix-assisted laser desorption/ionization (MALDI) (16, 17), and several groups have recently developed tools to predict proteotypic peptides for protein quantification under a variety of conditions including LC-ESI (2, 18–20). Although these approaches report success and are of considerable value, no consensus predictors have been developed to date and a complete understanding of the features which promote ionization and detectability is lacking. Moreover, the prediction of proteotypic peptides, i.e. those peptides that are likely to be observed under a given set of conditions, does not necessarily mean that they are suitable for use as reference peptides for protein quantification, given the requirements mentioned previously.
To address these points, we have generated two complementary data sets of proteotypic peptides and developed CONSeQuence, a consensus prediction system built around four independent machine learning algorithms to better understand peptide “detectability.” One data set of tryptic peptides was hand-curated from 13 proteins in equimolar concentration, and a second constructed from the yeast build of the PeptideAtlas database (21). Over 1000 features including a missed cleavage predictor (22) were assessed and used to define those physicochemical characteristics important for the detection of peptides after LC-ESI MS. The resulting feature vectors were then used to train four different algorithms: a Random Forest, a Genetic Program, an artificial neural network and a support vector machine, all using cross-validation. The hand-curated data set is the first data set of this type to be used in the training of machine learning algorithms with a view to predicting proteotypic peptides.
CONSeQuence demonstrates improved performance compared with existing algorithms, exceeding 80% PPV at high prediction sensitivity (50%) on our yeast test sets, thus yielding high numbers of candidate Q-peptides for QconCAT design across the whole proteome. We also demonstrate that the prediction approach enriches for observed peptides when applied to two large metazoan proteome data sets, by a factor of up to 17 times over random, and that the yeast-trained classifiers also show equivalent superior prediction performance on these unseen proteomes. Finally, we demonstrate that in addition to the known roles of peptide size, charge and hydrophobicity, peptide secondary structure is also an important factor in determining “detectability” by electrospray ionization mass spectrometry. CONSeQuence can be applied to lists of peptides or FASTA files containing up to 1000 proteins via a web interface at http://king.smith.man.ac.uk/CONSeQuence. Predictions for larger data sets are available to members of the scientific community on request.
EXPERIMENTAL PROCEDURES
Data Set 1—
Preparation of Samples for Manual Analysis
Protein stocks (Sigma) were resuspended in 25 mm NH4HCO3 to 2–5 mg/ml. The proteins (Table I) were then individually reduced with 4 mm dithiothreitol (10 min, 60 °C) and alkylated with 14 mm iodoacetamide (45 min, RT). Further alkylation was quenched by addition of dithiothreitol to a final concentration of 7 mm, prior to digestion with 2% (w/w) trypsin (Sigma) overnight at 37 °C. Digested proteins were analyzed individually by LC/MS/MS using an Ultimate/Switchos/Famos nanoflow HPLC system (Dionex), arranged in-line with a Q-ToF1 mass spectrometer (Micromass) in positive ion mode. The cone voltage was fixed at 50V and the collision energy was set to change according to precursor m/z, with three MS/MS spectra being acquired per MS survey scan. Peptides were chromatographed at 200 nl/min using a 15 cm × 75 mm i.d. PepMap C18 reversed-phase column (Dionex), with bound peptides being eluted over a 50 min gradient to 65% B (A = 5% MeCN, 0.06% formic acid; B = 95% MeCN, 0.05% formic acid). Processed data for individual proteins were searched against the NCBInr database (release 45) using an in-house Mascot server (Matrix Science) to confirm identity and LC elution time. The retention times of all theoretical tryptic peptides that did not yield tandem MS data were defined by generating an extracted ion chromatogram (XIC) and confirming mass accuracy (≤ 100 ppm) and charge state. For the purposes of this study, the peptides analyzed included all possible charge states of those tryptic peptides generated with either 0 or 1 missed cleavage, where 300 ≤ m/z ≤ 1800 and the peptides contained at least 5 amino acids. The digested proteins were then combined in an equimolar ratio to generate the test set.
Table I. Proteins used to generate the hand-curated data set. In silico tryptic digestion was performed on 13 proteins, resulting in 931 unique peptides.
| Protein name | Accession number | Mr (Da) | # Tryptic peptidesa |
|---|---|---|---|
| Actin | P68135 | 42,051 | 55 |
| Alcohol dehydrogenase I | P00330 | 36,823 | 50 |
| α-Casein | P02662 | 24,529 | 28 |
| Fructose-bisphosphate aldolase A | P00883 | 39,343 | 58 |
| Bovine serum albumin | P02769 | 69,293 | 115 |
| Chick albumin | P01012 | 42,881 | 50 |
| Cytochrome C | P00004 | 11,833 | 27 |
| Enolase | P00924 | 46,802 | 78 |
| Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | P46406 | 35,780 | 55 |
| Human serum albumin | P02768 | 69,367 | 103 |
| Lysozyme | P00698 | 16,239 | 25 |
| Glycogen phosphorylase | P00489 | 97,289 | 160 |
| Transferrin | P02787 | 77,050 | 127 |
| 931 |
a Number of tryptic peptides is defined as those tryptic peptides with ≥5 amino acids, where 300 ≤ m/z ≤ 1800, given the potential for a single missed cleavage, excluding common peptides and those known to be modified.
Analysis of LC-MS Data
To obtain detailed information on detectability (i.e. the ability of a peptide to be generated after proteolysis, separated by LC, ionized by electrospray, analyzed and detected in a mass spectrometer), the test set (1 pmol) (Table I) was diluted in 0.1% (v/v) formic acid and the peptides separated by reversed-phase chromatography as above. MS data only were acquired (Supplemental Fig. 1). Extracted ion chromatograms were generated for each of the peptide ions comprising the test set.
Data Set 2
A flowchart depicting the procedure for generating data set 2 is shown in Fig. 1. A total of 24 non-ICAT yeast experiments were obtained from PeptideAtlas representing a total of 2733 yeast proteins, with all peptides passing a minimum threshold of p > = 0.9 as defined using PeptideProphet (23). All experiments used were Multidimensional Protein Identification Technology type studies where LC separation prior to electrospray ionization had been used to generate tandem mass spectra for identification. Proteins were retained if they were observed in at least two experiments (1788 proteins) and had at least 40% sequence coverage across the experiments (629 proteins). Peptides from the 629 proteins were classed as “flyers” if they were observed at least 50% of the time in these proteins across the 24 experiments. This produced a total of 2121 peptides representative of 536 proteins, with 93 proteins that did not have peptides which passed this criterion. The 536 proteins were then subjected to in silico trypsin digestion, with the resulting peptides limited by length (6–42 residues) and number of internal missed cleavages (< = 2). If a resulting peptide and all of its sub- and super-sequences failed to be observed in any of the 24 experiments (at p > = 0.9) it was taken to be a “nonflyer,” resulting in a total of 19394 “nonflying” peptides. Again, we retained the length thresholds and upper limit on missed cleavages to prevent our negative sets from containing obvious biases on which the machine learning algorithms would be over-trained. All peptides from both data sets can be found in supplemental data 1.
Fig. 1.

Schematic overview of the procedure for obtaining “flyers” and “nonflyers” for data set 2 from PeptideAtlas. Peptide “flyers” are obtained from the high quality PeptideAtlas yeast build (p > 0.9) from commonly observed proteins. Peptides must be detected at least 50% of the time that their parent protein is observed, where the protein itself has been observed in at least two of the 24 non-ICAT experiments and has at least 40% sequence coverage from all peptides in the build. The “nonflyers” are obtained by an in silico digestion of the protein set from which the “flyers” are selected, where the peptide (or any overlapping missed cleaved peptide) is not observed across any of the 24 experiments.
Mapping Peptide Sequences to Feature Vectors
In order to apply machine learning algorithms to build predictive models, the peptide sequences from both the positive “flyers” and negative “nonflyers” were mapped to numerical feature vectors, where each element of the vector represented some characteristic of the corresponding peptide. A total of 1186 features were derived from the peptide sequence. The majority of these were obtained from AAindex1 (24) and were included as both summed and average values for each peptide sequence. The remainder included features calculated from external algorithms including Helix-Coil theory (25), isoelectric point (generated using an in-house perl script), our own in-house missed cleavage predictor (22), and normalized BLAST bit scores obtained from searching peptides sequences against the positive and negative components of data set 2, and ignoring any “self” hits (where the peptide matches its own sequence). A complete list of features can be found in supplemental data 2.
Machine Learning Methods—
Feature Selection
Because of the high level of redundancy in the full feature set, a small subset of features was chosen in order to build a robust model. Such a reduction also helps with interpretation of results, and provides a better insight into which features are more important in terms of Q-peptide prediction. The Kullback-Leibler (KL) distance measure was used to estimate differences between the probability distribution of “flyers” and “nonflyers,” with highly discriminatory features having a relatively high KL value. Many of the descriptive features produced similar score distributions across the whole data set. Using the whole of data set 2, for example, the top two features Average Positive Charge and Proportion of Basic Residues (KL = 0.92 and 0.89 respectively, data not shown) had a Pearson correlation co-efficient of 0.997 (p < 0.00001). Including both of these features would add negligible information for separating “flyers” from “nonflyers,” within feature space, than if only one of the two features was taken. The 50 top ranked features were therefore selected with the additional criterion that no two features had a Pearson correlation of >0.7 or < –0.7 (supplemental Table 1). The feature value distributions of the “flyers” and “nonflyers” for the top four ranked and filtered features for data set 2 are shown in Fig. 2.
Fig. 2.

Distribution of feature scores of “flyers” and “nonflyers” for the top four KL ranked features for data set 2. A, Average positive charge of amino acids across each peptide, B, average helix termination parameter at position j+1, C, sum of “buriability” (related to hydrophobicity) and D, average principal property value z3 (the 3rd principal component from a PCA analysis) which is related to electronic properties encompassing pKa and pI (43), across the peptide.
Support Vector Machines
A support vector machine is a type of supervised learning approach for classification (26). Support Vector Machines (SVMs) create a hyperplane between data points that effectively splits the data into two classes, using a kernel function to transform data into higher dimensional space for nonlinear problems. In both linear and nonlinear cases, SVMs attempt to maximize the distance between the hyperplane and its nearest data points. For this reason SVMs have been used to approach a wide variety of biological problems (27). In this study, the SVMlight (28) package was chosen for its ease of use. In this instance the radial basis function kernel was used and the gamma parameter was optimized such that the error function (SN × PPV) was maximized. The default method, grid search, was used for estimation of the cost parameter C, which controls the trade-off between allowing training errors and forcing rigid margins. Additionally, a weighting factor equal to the ratio of “nonflyers” to “flyers” was used to weight the training errors of the unbalanced data sets.
Random Forests
Random Forests is a classifier that consists of set of decision trees (29). A single decision tree structure consists of “branches” and “leaves,” where branches represent combinations of features used to divide and subdivide the data and leaves represent the classifications as a result of the branch descriptions. Random Forests is essentially a collection of decision trees that are each grown using a subset of the input features in the training data. The final classification of the data can then be assigned by the proportion of votes a classification has across the whole forest of decision trees. Random Forests have previously been successfully applied to proteotypic peptide prediction (20).
Here the FORTRAN implementation of Random Forests was obtained from http://www.stat.berkeley.edu/∼breiman/RandomForests/cc_software.htm. Models were trained using class weights reflecting the ratio between negative and positive examples in the training set. The number of features to sample randomly at each branch point in the decision trees was optimized so as to maximize the cost function Sensitivity (SN) × Positive Predictive Value (PPV).
Genetic Programming
Genetic Programming (GP) is a methodology inspired by the process of natural evolution (30). It involves the representation of a solution to a problem as a tree structure. In this case, each solution is a mathematical function created by the combination of simple arithmetic operations (+, -, ×, ÷). A “population” of potential solutions to a problem are randomly generated and in successive “generations” effective solutions are selected and modified, either through random “mutations” of a single solution or “crossover” between two solutions. We have previously demonstrated GP's success in the prediction of peptide detectability (31).
The parameters used for the GP runs were: population size = 100; maximum tree-size = 250 nodes; maximum tree-depth = 17; crossover rate = 0.6; mutation rate = 0.2; objective function = SN × PPV. Trees were selected for reproduction and for replacement using a 4-way tournament and trees were replaced one at a time, i.e. steady state evolution. Crossover was carried out by randomly selecting a cut-point from each parent and swapping the subtrees below these points. Mutation was performed by randomly selecting a mutation operator from: subtree replacement, node replacement, shrink, swap, constant value mutation and node expansion. These mutation operators and the other GP parameters are described in detail in (32). For each GP run, one-third of the training data was held back for ‘validation’. Evolution was stopped after 500 generations or when the objective function on the validation data had shown no improvement for 50 generations, if this was sooner. Each GP run was performed 10 times and the best solution from the run that yielded the best objective score on the validation data was the model selected.
Artificial Neural Networks
Artificial Neural Networks (ANN) are mathematical models, inspired by the animal brain, used for nonparametric regression and classification using parallel processing (33). Each neuron accepts several inputs, calculates a weighted sum of these inputs and emits the output of a logistic function applied to this sum. Multiple inputs are passed via “hidden layer” neurons to an output neuron, whose output is interpreted as a prediction (in this case “flyer”/”nonflyer”). By repeatedly adjusting the weights between neurons using the method of “back-propagated gradient descent,” the accuracy of ANNs predictions may be improved.
The inputs to the ANNs were normalized to the range [–1, 1]. The ANNs contained a single hidden layer composed of 20 neurons, each using a bipolar logistic transfer function. Gradient descent was implemented using the Levenberg-Marquardt algorithm (34). Networks were trained for a maximum of 200 epochs but training was finished early if no improvement in the mean square error on the training data was observed for five consecutive epochs.
PeptideSieve and ESPPredictor
In order to assess the performance of our machine learning and consensus predictors, they were compared against both PeptideSieve (2) and ESPPredictor (20), two leading published tools for predicting proteotypic peptides. PeptideSieve accepts protein sequences and returns scores for individual tryptic peptides based upon the likelihood of the derived peptides being observed during mass spectrometry. By selecting different threshold values it is possible to produce a series of predictors with varying levels of strictness. A low threshold is likely to result in a large number of predicted “flyers,” but with low confidence. A higher threshold will reduce the number of predicted “flyers,” but result in a higher confidence level.
The Enhanced Signature Peptide Predictor takes a list of peptides as input and returns a probability of high response for each peptide. The probabilities are calculated separately for each protein, with a higher probability indicating a higher likelihood that a peptide is detectable compared with other peptides within the same protein.
Prediction Assessment and Consensus Machine Learning Approaches
Cross-validation was used to assess the prediction performances of the machine learning methods. Each data set was split into ten equal subsets, with each subset containing the same number of “flyers” and “nonflyers.” These subsets were used to create 10 cross-validation sets of training (90%) and testing (10%) peptides, ensuring no overlap of peptide between testing sets. For each cross-validation the feature selection and subsequent machine learning algorithms were performed on the training set alone. The performance was then assessed on the corresponding test set, which was held back from the feature selection and machine learning steps. Performance statistics were averaged across all ten runs.
From the individual machine learning predictors, a consensus predictor was constructed using a voting procedure. In this method, a peptide was predicted to “fly” if it obtained a specific number of “votes” (1, 2, 3, or 4) from the individual predictors, leading to four prediction states labeled CONS1, CONS2, CONS3, and CONS4. This procedure was agnostic as to which particular algorithms predicted a peptide would “fly,” counting only the total number of votes in each case. Increasing the number of votes required makes the consensus predictor more selective and less sensitive, in a similar manner to increasing the PeptideSieve threshold or ESPPredictor associated probability of high response.
In an additional step for data set 2, the raw cross-validated prediction scores from three out of the four machine learning methods (ANN, SVM, and RandomForests) were normalized (unit variance about a mean of zero). The peptide data set and normalized scores were then shuffled to create 10 further cross-validation sets (90% training and 10% testing). SVMlight was then used to train models with the linear kernel function using flyer/nonflyer as the target output on the 10 training sets and tested on the respective 10% test sets. The output distances from the hyperplane were then scaled between 0 and 1, to generate a final prediction score, termed CONSeQuence rank.
Prediction algorithm success was measured using the standard statistical terms, as follows
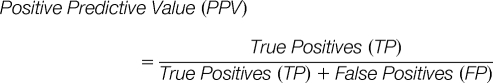 |
Both high SN and high PPV are desirable in a good predictor, i.e. we would like to predict a large number of peptides with a high confidence level. When assessing our predictors, we have therefore considered SN, PPV, and the product SN × PPV in particular, as well as the Area Under the Curve (AUC) Receiver Operator Characteristic statistic.
RESULTS
Cross-validated Prediction Performance Results
Table II shows the SN, PPV, and SN × PPV performances on data sets 1 and 2, for both the individual predictors and the CONSeQuence binary predictor. Based on SN×PPV, a comparison of the individual predictors shows that SVM results in the best performance for Data set 1 and the ANN is the best performer for Data set 2. We note that predictive performance of the CONS binary classifiers is comparable to the best of the individual classifiers, and that superior PPV is achieved in particular for CONS3,4. However, our principal aim is to achieve the maximal PPV at an acceptable SN, because we require high confidence that our predicted peptides are indeed “flyers,” while ensuring enough candidate Q-peptides are predicted per protein to support QconCAT design or for stable-isotope based peptide synthesis. Fig. 3 shows precision-recall curves plotting PPV against SN for our consensus predictor and for PeptideSieve and ESPPredictor for data set 1 and data set 2. The predictors become more selective from right to left, as the number of votes required by the CONSeQuence binary predictor increases from 1 to 4 and the CONSeQuence rank predictor scores increase. Similarly, the thresholds used for PeptideSieve and ESPPredictor increase from 0.1 to 0.9, in steps of 0.1, from right to left. From Fig. 3, it is clear that both CONSeQuence methods outperform both the other algorithms on data set 2. Although ESPPredictor can achieve a PPV of 0.82, this is only at an extremely low and impractical level of sensitivity (0.03) and is therefore of limited utility.
Table II. Performance statistics for individual machine learning methods and for CONSeQuence on Dataset 1 and Dataset 2.
| SVMlight | Random forests | Genetic programming | Neural networks | CONS1 | CONS2 | CONS3 | CONS4 | |
|---|---|---|---|---|---|---|---|---|
| Dataset 1 | ||||||||
| Sensitivity | 0.95 | 0.72 | 0.94 | 0.74 | 0.99 | 0.95 | 0.80 | 0.61 |
| PPV | 0.59 | 0.71 | 0.58 | 0.67 | 0.56 | 0.60 | 0.67 | 0.74 |
| Sensitivity x PPV | 0.56 | 0.51 | 0.54 | 0.50 | 0.56 | 0.57 | 0.54 | 0.45 |
| Dataset 2 | ||||||||
| Sensitivity | 0.62 | 0.91 | 0.75 | 0.60 | 0.94 | 0.87 | 0.75 | 0.54 |
| PPV | 0.56 | 0.41 | 0.47 | 0.72 | 0.35 | 0.47 | 0.60 | 0.75 |
| Sensitivity x PPV | 0.35 | 0.38 | 0.35 | 0.43 | 0.33 | 0.41 | 0.45 | 0.40 |
Fig. 3.

Precision-recall curves are shown, plotting Sensitivity (SN) against Positive Predictive Value (PPV) for CONSeQuence, PeptideSieve and ESPPredictor for A, Data set 1 (experimentally derived) and B, Data set 2 (empirically derived). Depicted are the Sensitivity and PPV for increasing PeptideSieve and ESPPredictor score cut-offs (0.1 to 0.9) and the increasing “number of votes required” (1 to 4) of CONSeQuence binary and increasing PPV (0.1 to 0.9) for CONSeQuence rank. Comparative performance of the individual ML algorithms are shown (C) against CONSeQuence binary and CONSeQuence rank.
Although PeptideSieve was, like CONSeQuence, trained on yeast data, we believe that superior performance in maximizing PPV is achieved through more stringent selection criteria for the negative data set and, at least in part, the use of consensus prediction approaches. The CONS1–4 points and CONSeQuence rank curve dominates all PeptideSieve and ESPPredictor points in Fig. 3. For example, the CONS3 and CONS4 binary predictors dominate the precision-recall points for the SVM and Random Forest methods, when applied independently (Fig. 3C). Similarly, the ranking predictor dominates SVM, Random Forest and shows modest improvement above ANN for sensitivity over 0.3, although we note this is only a small improvement. Nevertheless, the AUC coefficients for the CONSeQuence rank predictor are superior to all individual machine learning methods, when Receiver Operator Characteristic curves are considered (see supplementary Fig. S2). CONSeQuence remains uniformly superior to Peptide Sieve and ESP Predictor, which have AUC scores of 0.86 and 0.65 compared with equivalent AUC scores of 0.937, 0.945, and 0.948 for the SVM, Random Forests and Neural Networks respectively, and an overall AUC of 0.954 for CONSeQuence rank. For this reason, CONSeQuence rank is the default method supported on our accompanying web site (http://king.smith.man.ac.uk/CONSeQuence).
We also evaluated prediction performance when missed cleavage peptides were entirely removed from both positive and negative training/testing sets, and the same cross-validation protocol was applied. A similar result was observed, with CONSeQuence outperforming both PeptideSieve and ESPPredictor with respect to PPV at any given sensitivity. At CONS4 a PPV of 0.7 is attained at a high sensitivity of 0.74, whereas PeptideSieve with a threshold of 0.9 achieves a PPV of 0.69 with sensitivity of 0.15 and ESPPredictor with a threshold of 0.9 achieves a PPV of 0.86 at a sensitivity of 0.03 (data not shown).
Broadly equivalent prediction performance is obtained from both data sets, although notably the general performance of PeptideSieve and ESPPredictor is improved on data set 1 and closer to the CONSeQuence performance (Fig. 3A). This data set was selected as an alternative to the PeptideAtlas derived data set because it involves a rigorous manual selection of both the positive and negative entries. It is more problematic to select the latter from a repository of peptide identifications because there is an assumption that an absence of a given peptide in the database means it does not readily ionize and is not usually detected. However, this could arise from a variety of reasons, including sampling effects, the low abundance of a protein or a post-translational modification that has not been accounted for. A given peptide may therefore be intrinsically “detectable,” yet remain unobserved. We, and other authors (2, 20), have attempted to mitigate for these factors by selecting frequently observed proteins of high coverage but this is not expected to be perfect. Therefore, it is encouraging to see comparable and good quality predictions from both data sets.
Consensus Prediction on the Complete Yeast Proteome
We evaluated the actual coverage of putative Q-peptides we would obtain when applying the CONSeQuence binary prediction on the complete yeast proteome, obtained from the Saccharomyces Genome Database (35) (downloaded on 8th March 2010), digested in silico and filtered using the same length and missed cleavage constraints of data set 2. The resulting 710,720 tryptic peptides were then submitted to the CONSeQuence binary predictor trained on data set 2. As expected, increasing voting levels (CONS1–4) of the binary predictor produces fewer “flyers” per protein (Figs. 4A–D). Interestingly, even at the low CONS1 level there are still 44 proteins that do not contain any peptides that are predicted to “fly” (<0.7% of the proteome).
Fig. 4.

Histogram showing the proportion of proteins with a minimum number of suitable Q-peptides in the complete yeast proteome. The required voting level increases from 1 to 4 prediction algorithms across panels A to D. The black blocks indicate the proportion of the proteome with peptides that are predicted to “fly” and can be used for quantification (based on sequence composition and mass). The white blocks show the proportion of the proteome that has peptides predicted to “fly”, but are otherwise unsuitable as Q-peptides for quantification owing to sequence features or nonunique mass.
The predicted peptide “flyers” for each CONS level were filtered for use as Q-peptides as described in previous publications (6, 9), removing those with incompatible amino acids such at N-terminal glutamine to prevent pyroglutamic acid formation, AspPro dipetides (which can undergo hydrolysis and subsequent internal fragmentation) and AsnGly dipetides (which are liable to undergo deamidation). This was carried out to assess the content of the proteome that could be used for absolute quantification using QconCAT or AQUA. A similar trend was demonstrated with the prediction of the Q-peptides, though the decrease in number of peptides with increased voting level is more apparent than when considering all “flyers.” Nevertheless, encouragingly, at the high confidence level of CONS4, ∼64% of proteins have at least two Q-peptides per protein that are predicted to “fly.” These results reinforce the necessity to develop a predictor with good sensitivity in order to retain sufficient candidate peptides for selection of reference standards in quantification experiments. It is possible to improve performance in terms of PPV beyond the 0.80 mark (see Fig. 3B and 3C), although the sensitivity becomes impractically low much beyond this point.
Prediction Comparison on Peptide Atlas From Other Organisms
We also evaluated the general performance of the predictor trained on yeast data applied to other proteomes. Two data sets were constructed for Caenorhabditis elegans and Drosophila melanogaster, using the same method used to create data set 2. This produced a total of 795 “flyers” and 6707 “nonflyers” covering 210 proteins for C. elegans and 1060 “flyers” and 10702 “nonflyers” covering 443 proteins for D. melanogaster. The two data sets were submitted to the CONSeQuence predictor (binary and rank) trained on data set 2, as well as PeptideSieve and ESPPredictor, and precision-recall curves of the predictions from all three are shown in Fig. 5. In both cases CONSeQuence (binary and rank) outperforms both PeptideSieve and ESPPredictor. Although ESPPredictor achieves a high PPV (78%) at the 0.9 threshold for the C. elegans proteome, this is at a markedly lower SN compared with both CONSeQuence predictors, and the CONSeQuence rank predictor outperforms this anyway (90% PPV at higher sensitivity).
Fig. 5.

Comparison of prediction performance of CONSeQuence, PeptideSieve, and ESPPredictor using precision-recall curves of the predictions for Caenorhabditis elegans (A) and Drosophila melanogaster (B). Depicted are the Sensitivity and PPV for increasing PeptideSieve and ESPPredictor score cut-offs (0.1 to 0.9) and the increasing “number of votes required” (1 to 4) of CONSeQuence binary and increasing PPV (0.1 to 0.9) for CONSeQuence rank.
As a further test, CONSeQuence binary was applied to more complete PeptideAtlas builds, the Drosophila melanogaster July 2009 build and the Caenorhabditis elegans May 2008 build, considering 45,119 and 73,159 fully tryptic peptides, respectively. The proteome sequences to which the peptides were mapped were also downloaded from PeptideAtlas, then subjected to in silico digestion filtered by the same length and missed cleavage constraints as for data set 2, providing a total of 4,849,151 and 3,197,578 peptides for D. melanogaster and C. elegans respectively. CONseQuence binary prediction performance, trained on data set 2, was measured using PPV and an enrichment factor. Here, we adopt a more simplistic measure of detectability (“flyers”) as any observed peptide in the PeptideAtlas build. The PPV was calculated for CONS1–4 as the number of predicted “flyers” that are present in the PeptideAtlas build divided by the total number of predicted “flyers” (Fig. 6, bars on primary y axis). Although the PPV increases with CONS level, the values appear to be low, suggesting over-prediction. However, there is a caveat because the PeptideAtlas builds are presumed to comprehensively cover all proteotypic peptides from all proteins that would be regularly observed. Clearly, this is unlikely to be the case, particularly as the number of tryptic peptides in the build constitutes only ∼1 and ∼2% of the total tryptic peptides available in D. melanogaster and C. elegans respectively. This is lower than the corresponding number in the S. cerevisiae April 2009 PeptideAtlas build, which contains over 6% of the potential tryptic peptides of the proteome (data not shown), indicating a deeper coverage of the yeast proteome. Assuming that future revisions of the builds within PeptideAtlas will contain more peptide identifications using higher quality tandem MS data, one can expect that the PPV observed here will increase.
Fig. 6.

Prediction performance for complete proteomes, plotting PPV and enrichment factor over random for Drosophila melanogaster (A) and Caenorhabditis elegans (B). The bars on the primary y axis (left) show the PPV for CONS1–4 and the lines on the secondary y axis (right) show the enrichment factor for CONS1–4.
An associated assessment of the performance is the calculation of an enrichment factor. At each CONS predictor level this was calculated as the ratio of the number of build peptides predicted to “fly” to the expected (random) number (Fig. 6, lines on secondary y axis). The latter random number of “flyers” was calculated as the total number of peptides in the PeptideAtlas build multiplied by NCONS/Ntotal, where NCONS is the number of “flyers” predicted from a given consensus prediction, and Ntotal is the total number of peptides in the entire proteome. The observed enrichment is typically an order of magnitude (×4–×17) above random and follows the same trend as the PPV for both builds across the four consensus levels. This demonstrates that, moving from CONS1 to CONS4, CONSeQuence binary predicts many more build peptides to “fly” than expected from random selection and that the proportion of predicted “flyers” that are in the build increases.
These two sets of results show that the CONSeQuence predictors, despite being trained exclusively on yeast proteome data, generalize well to peptide detectability prediction in other organisms while maintaining the same improved prediction performance compared with other algorithms.
DISCUSSION
CONSeQuence has been developed as a reliable approach for predicting peptides suitable for use as internal standards for absolute protein quantification by ESI-MS. Significantly, it has been demonstrated to outperform other predictor tools currently available, both on the species on which it has been trained and importantly on other unrelated species, maintaining similarly high levels of performance. We suspect this improvement stems from a more rigorous approach to the definition of the negative (“nonflyers”) peptide data set, and a modest improvement from consensus prediction. We restricted our negative data set to proteins with high sequence coverage and eliminated peptides from consideration if they have been observed even once with a modest significance score.
Although performance to date has centered on ESI-MS data, it is straightforward to apply the same general principles to other approaches including MALDI and gel-based data. Application of this predictor will undoubtedly ease the quantitative analytical workflow.
Importantly, development of this tool has further unraveled some of the physicochemical features that significantly influence peptide ion detectability in an ESI-MS experiment. The roles of size, hydrophobicity and charge have already been demonstrated to be influential, (2, 18, 20, 36, 37) and our data lends further support. Positive charge is the top ranked feature with a KL distance of 0.92 (supplemental Table 1) and negatively influences peptide detectability, concurring with Aebersold and colleagues who demonstrated that Asp/Glu are represented in significantly higher levels in proteotypic peptides from Multidimensional Protein Identification Technology-ESI experiments (2). Indeed, pI value was one of the most influential parameters identified during manual interrogation of data set 1 (data not shown) and resulted in peptides with low pI values (≤4.5) being optimally selected as Q-peptides prior to development of this computational method. Hydrophobicity is also represented by the “buriability” feature, which is ranked as the third most important (supplemental Table 1). We examined this in more detail in relation to protein structure, by matching our positive and negative peptides in data set 2 to known tertiary structures. The 536 proteins used in data set 2 were matched to the Protein Data Bank (PDB) database using BLAST (38), retaining the top hit when the percentage identity was >80% and alignment length >75% of the query sequence. The coordinates for each peptide (“flyers”/“nonflyers”) were then overlaid onto the matching PDB sequence via the alignment, where possible. In total, 359 “flyers” from 138 proteins and 1447 “nonflyers” from 143 proteins were unambiguously mapped to structures, using the biological assembly matches downloaded from the PDB (39). Mean residue relative accessibility scores were calculated for each mapped peptide using NACCESS (available from http://www.bioinf.manchester.ac.uk/naccess/). The distribution of mean accessibility scores for the two peptide data sets is shown in Fig. 7. The positive “flyer” peptides show a significantly lower mean accessibility score than the “non-flyers” (p < 0.0001), suggesting that “flyers” are preferentially buried within the three-dimensional tertiary structure. Indeed, these data suggest that protein structure per se plays no overt role in defining proteotypic peptides, and that neither tertiary structure, residual structure in denatured proteins nor hydrophobic aggregates are generally limiting for peptide detection via MS. Therefore, there is no apparent intrinsic reason that proteolytic digestion is likely to be rate-limiting.
Fig. 7.

Mean residue accessibility for peptides assigned as “flyers” and “nonflyers” in data set 2 which have been mapped to tertiary structures in the Protein Data Bank. Distributions are significantly different with t = 5.0414 at p < 0.0001.
Although tertiary structure does not appear to have a direct effect, we have additionally identified a major role of local structure in peptide “detectability,” with 23 of the top 50 features being directly related to secondary structure. Interestingly, of the eight features relating specifically to helical structure in the top 30 parameters, seven have a negative influence on peptide detectability. Conversely, parameters for coil exert a positive influence, suggesting that less regulated peptide structure may be optimal for LC-ESI. It has been demonstrated previously that peptides with α-helical structure have reduced ionization efficiency in MALDI (40), however to our knowledge this feature has not previously been demonstrated to influence efficiency of ionization by electrospray. Interestingly, Russell and colleagues (41) have evidence for a singly protonated model peptide suggesting that whereas computer simulations predict helical conformation to be the lowest energy structure, random coil and turn conformers are more prevalent. The data presented here lead us to speculate that differences in either the extent and/or rate of desolvation of helical peptide ions during electrospray may be the reason for this phenomenon. However, the efficiency of gas-phase ion production from small highly charged droplets cannot be distinguished from potential differences in decomposition of the desolvated ions in these experiments. For the peptides to be included in data set 2 as “flyers,” they would have to have been both ionized and fragmented efficiently (for subsequent identification using search algorithms). These structural features could therefore be regulating either the number of gas-phase ions generated, or resulting in differences in the internal energies of the desolvated ions which may subsequently be influencing the extent of low energy decomposition and thus identification.
Care should be taken when attempting to use these top ranked features in isolation to interpret peptide ionization properties, as many are interlinked. For example, the second feature, the helix termination parameter at position j+1 (42) (also described as the end-cap value), is governed by the prevalence of Lys/Arg/His/Gly and Glu/Asp/Pro as positive and negative contributing factors respectively. This feature could thus be indicating either that the end-cap value or that the basicity of the peptide (or a combination of both) is negatively regulating peptide detectability and indeed this parameter appears to be more representative of peptide charge (42).
The observation and detection of a peptide is also governed by other properties in an LC-ESI experiment, including the binding and elution of these peptides to a reversed-phase column, as demonstrated by the influence of hydrophobicity related parameters, and the likelihood to be generated by proteolysis. Indeed, a notable feature not apparently selected for the machine learning model is missed cleavages. Missed cleavages affect the detectability of a peptide both through the presence of internal basic side-chains and through the cleavage context at the N- and C termini of the peptide in the native protein. Internal missed cleavages are indicative of partial proteolysis which confounds the ability of the peptide to be used for absolute quantitative purposes. In the machine learning protocol they are indirectly accounted for by peptide Arg/Lys proportions (mean:R and mean:K), and both are present in the top 20 of the 50 features used, and are seen to have a negative effect on detectability. However, the missed cleavage context in the endogenous protein is dependent on the sequence outside the peptide itself and this leads to a problem for machine learning where identical peptides can occur in multiple different cleavage contexts. Such degenerate peptides would, however, clearly be unsuitable for quantification purposes. For QconCAT design purposes we take an alternative approach in which we apply a missed cleavage predictor to the set of predicted “flyers” independently of CONSeQuence. This allows contextual sequences at the N- and C termini of the Q-peptides in their native proteins which are predicted to be missed to be down-weighted or eliminated as candidate QconCAT peptides.
Acknowledgments
We thank Dr. J. Siepen for computational support and Professor A. Doig for helpful discussion.
Footnotes
* This work was supported by the Biotechnology and Biological Sciences Research Council, via several grants to SJH and SJG (BB/F004605/1, BB/G009058/1, BB/C007735/1) and the Engineering and Physical Sciences Research Council (EP/D013615/1). CE is supported by a Royal Society Dorothy Hodgkin Research Fellowship.
 This article contains supplemental Table S1 and Figs. S1 and S2.
This article contains supplemental Table S1 and Figs. S1 and S2.
1 The abbreviations used are:
- LC
- Reversed-phase liquid chromatography
- ANN
- Artificial Neural Networks
- ESI
- Electrospray ionization
- GP
- Genetic Programming
- KL
- Kullback-Leibler
- MALDI
- Matrix assisted laser desorption/ionization
- PDB
- Protein Data Bank
- PPV
- Positive predictive value
- QqQ
- Triple quadrupole
- Q-ToF
- Hybrid quadrupole time-of-flight
- SN
- Sensitivity
- SVM
- Support Vector Machines.
REFERENCES
- 1. Blüthgen N., Bruggeman F. J., Legewie S., Herzel H., Westerhoff H. V., Kholodenko B. N. (2006) Effects of sequestration on signal transduction cascades. Febs. J. 273, 895–906 [DOI] [PubMed] [Google Scholar]
- 2. Mallick P., Schirle M., Chen S. S., Flory M. R., Lee H., Martin D., Ranish J., Raught B., Schmitt R., Werner T., Kuster B., Aebersold R. (2007) Computational prediction of proteotypic peptides for quantitative proteomics. Nat. Biotechnol. 25, 125–131 [DOI] [PubMed] [Google Scholar]
- 3. Marko-Varga G., Lindberg H., Löfdahl C. G., Jönsson P., Hansson L., Dahlbäck M., Lindquist E., Johansson L., Foster M., Fehniger T. E. (2005) Discovery of biomarker candidates within disease by protein profiling: principles and concepts. J. Proteome Res. 4, 1200–1212 [DOI] [PubMed] [Google Scholar]
- 4. Pan S., Rush J., Peskind E. R., Galasko D., Chung K., Quinn J., Jankovic J., Leverenz J. B., Zabetian C., Pan C., Wang Y., Oh J. H., Gao J., Zhang J., Montine T., Zhang J. (2008) Application of Targeted Quantitative Proteomics Analysis in Human Cerebrospinal Fluid Using a Liquid Chromatography Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Tandem Mass Spectrometer (LC MALDI TOF/TOF) Platform. J. Proteome Res. 7, 720–730 [DOI] [PubMed] [Google Scholar]
- 5. Barr J. R., Maggio V. L., Patterson D. G., Jr., Cooper G. R., Henderson L. O., Turner W. E., Smith S. J., Hannon W. H., Needham L. L., Sampson E. J. (1996) Isotope dilution–mass spectrometric quantification of specific proteins: model application with apolipoprotein A-I. Clin. Chem. 42, 1676–1682 [PubMed] [Google Scholar]
- 6. Beynon R. J., Doherty M. K., Pratt J. M., Gaskell S. J. (2005) Multiplexed absolute quantification in proteomics using artificial QCAT proteins of concatenated signature peptides. Nat. Methods 2, 587–589 [DOI] [PubMed] [Google Scholar]
- 7. Gerber S. A., Rush J., Stemman O., Kirschner M. W., Gygi S. P. (2003) Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. U.S.A. 100, 6940–6945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Malmström J., Beck M., Schmidt A., Lange V., Deutsch E. W., Aebersold R. (2009) Proteome-wide cellular protein concentrations of the human pathogen Leptospira interrogans. Nature 460, 762–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pratt J. M., Simpson D. M., Doherty M. K., Rivers J., Gaskell S. J., Beynon R. J. (2006) Multiplexed absolute quantification for proteomics using concatenated signature peptides encoded by QconCAT genes. Nat. Protoc. 1, 1029–1043 [DOI] [PubMed] [Google Scholar]
- 10. Rivers J., Simpson D. M., Robertson D. H., Gaskell S. J., Beynon R. J. (2007) Absolute multiplexed quantitative analysis of protein expression during muscle development using QconCAT. Mol. Cell. Proteomics 6, 1416–1427 [DOI] [PubMed] [Google Scholar]
- 11. Mirzaei H., McBee J. K., Watts J., Aebersold R. (2008) Comparative evaluation of current peptide production platforms used in absolute quantification in proteomics. Mol. Cell. Proteomics 7, 813–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Picotti P., Rinner O., Stallmach R., Dautel F., Farrah T., Domon B., Wenschuh H., Aebersold R. (2010) High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nat. Methods 7, 43–46 [DOI] [PubMed] [Google Scholar]
- 13. Brönstrup M. (2004) Absolute quantification strategies in proteomics based on mass spectrometry. Expert Rev. Proteomics 1, 503–512 [DOI] [PubMed] [Google Scholar]
- 14. Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 15. Li J., Taraszka J. A., Counterman A. E., Clemmer D. E. (1999) Influence of solvent composition and capillary temperature on the conformations of electrosprayed ions: unfolding of compact ubiquitin conformers from pseudonative and denatured solutions. Int. J. Mass Spectrom. 185, 37–47 [Google Scholar]
- 16. Krause E., Wenschuh H., Jungblut P. R. (1999) The dominance of arginine-containing peptides in MALDI-derived tryptic mass fingerprints of proteins. Anal. Chem. 71, 4160–4165 [DOI] [PubMed] [Google Scholar]
- 17. Gay S., Binz P. A., Hochstrasser D. F., Appel R. D. (2002) Peptide mass fingerprinting peak intensity prediction: extracting knowledge from spectra. Proteomics 2, 1374–1391 [DOI] [PubMed] [Google Scholar]
- 18. Sanders W. S., Bridges S. M., McCarthy F. M., Nanduri B., Burgess S. C. (2007) Prediction of peptides observable by mass spectrometry applied at the experimental set level. BMC Bioinformatics 8, S23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kuster B., Schirle M., Mallick P., Aebersold R. (2005) Scoring proteomes with proteotypic peptide probes. Nat. Rev. Mol. Cell Biol. 6, 577–583 [DOI] [PubMed] [Google Scholar]
- 20. Fusaro V. A., Mani D. R., Mesirov J. P., Carr S. A. (2009) Prediction of high-responding peptides for targeted protein assays by mass spectrometry. Nat. Biotechnol. 27, 190–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deutsch E. W., Lam H., Aebersold R. (2008) PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO Rep. 9, 429–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Siepen J. A., Keevil E. J., Knight D., Hubbard S. J. (2007) Prediction of missed cleavage sites in tryptic peptides aids protein identification in proteomics. J. Proteome Res. 6, 399–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Keller A., Nesvizhskii A. I., Kolker E., Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 24. Kawashima S., Pokarowski P., Pokarowska M., Kolinski A., Katayama T., Kanehisa M. (2008) AAindex: amino acid index database, progress report 2008. Nucleic Acids Res. 36, D202–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Doig A. J., Chakrabartty A., Klingler T. M., Baldwin R. L. (1994) Determination of free energies of N-capping in alpha-helices by modification of the Lifson-Roig helix-coil therapy to include N- and C-capping. Biochemistry 33, 3396–3403 [DOI] [PubMed] [Google Scholar]
- 26. Cortes C., Vapnik V. (1995) SUPPORT-VECTOR NETWORKS. Machine Learning 20, 273–297 [Google Scholar]
- 27. Ben-Hur A., Ong C. S., Sonnenburg S. R., Schölkopf B., Rätsch G. (2008) Support Vector Machines and Kernels for Computational Biology. PLoS Comput. Biol. 4, e1000173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Joachims T. (1999) Transductive inference for text classification using Support Vector Machines. In: Bratko I., Dzeroski S. eds. Machine Learning, Proceedings, pp. 200–209, Morgan Kaufmann Pub Inc, San Francisco [Google Scholar]
- 29. Breiman L. (2001) Random forests. Machine Learning 45, 5–32 [Google Scholar]
- 30. Koza J. R. ed. (1992) Genetic Programming: On the Programming of Computers by Means of Natural Selection., MIT Press, Cambridge, MA [Google Scholar]
- 31. Wedge D., Lau K. W., Gaskell S. J., Hubbard S. J., Kell D. B., Eyers C. E. (2007) Peptide detectability prediction following ESI mass spectrometry using genetic programming. GECCO 2007: Proceedings of the 9th annual conference on Genetic and Evolutionary Computation [Google Scholar]
- 32. Banzhaf W. (1998) Genetic programming : an introduction : on the automatic evolution of computer programs and its applications, Morgan Kaufmann Publishers, San Francisco, CA [Google Scholar]
- 33. Haykin S. ed. (1998) Neural Networks: A Comprehensive Foundation., 2nd ed. Ed., Pearson Education [Google Scholar]
- 34. Marquardt D. W. (1963) An algorithm for least-squares estimation of nonlinear parameters. J. Soc. Industr. Appl. Math. 11, 431–441 [Google Scholar]
- 35. Botstein D., Cherry J. M. (1997) Molecular linguistics: Extracting information from gene and protein sequences. Proc. Natl. Acad. Sci. U.S.A. 94, 5506–5507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tang H., Arnold R. J., Alves P., Xun Z., Clemmer D. E., Novotny M. V., Reilly J. P., Radivojac P. (2006) A computational approach toward label-free protein quantification using predicted peptide detectability. Bioinformatics 22, e481–488 [DOI] [PubMed] [Google Scholar]
- 37. Webb-Robertson B. J., Cannon W. R., Oehmen C. S., Shah A. R., Gurumoorthi V., Lipton M. S., Waters K. M. (2010) A support vector machine model for the prediction of proteotypic peptides for accurate mass and time proteomics. Bioinformatics 26, 1677–1683 [DOI] [PubMed] [Google Scholar]
- 38. Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T. L. (2009) BLAST+: architecture and applications. BMC Bioinformatics 10, 421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wenschuh H., Halada P., Lamer S., Jungblut P., Krause E. (1998) The ease of peptide detection by matrix-assisted laser desorption/ionization mass spectrometry: the effect of secondary structure on signal intensity. Rapid Commun. Mass Spectrom. 12, 115–119 [DOI] [PubMed] [Google Scholar]
- 41. Tao L., Dahl D. B., Pérez L. M., Russell D. H. (2009) The contributions of molecular framework to IMS collision cross-sections of gas-phase peptide ions. J. Am. Soc. Mass Spectrom. 20, 1593–1602 [DOI] [PubMed] [Google Scholar]
- 42. Finkelstein A. V., Badretdinov A. Y., Ptitsyn O. B. (1991) Physical reasons for secondary structure stability: alpha-helices in short peptides. Proteins 10, 287–299 [DOI] [PubMed] [Google Scholar]
- 43. Hellberg S., Sjöstrom M., Skagerberg B., Wold S. (1987) Peptide quantitative structure-activity relationships, a multivariate approach. J. Med. Chem. 30, 1126–1135 [DOI] [PubMed] [Google Scholar]