

Abstract
Background
Mitochondrial dysfunction due to respiratory chain impairment is a key feature in pathogenesis of Friedreich ataxia. Friedreich ataxia affects the nervous system, heart and pancreas.
Methods
We assessed hepatic mitochondrial function by 13C-methionine-breath-test in 16 Friedreich ataxia patients and matched healthy controls.
Results
Patients exhaled significantly smaller amounts of 13CO2 over 90 minutes. Maximal exhaled percentage dose of 13CO2 recovery was reduced compared to controls.
Conclusions
13C-methionine-breath-test indicates subclinical hepatic mitochondrial dysfunction in Friedreich ataxia but did not correlate with GAA repeat lengths, disease duration or disease severity.
Keywords: 13C-methionine, breath test, Friedreich, Ataxia, neurodegeneration
1. Background
Friedreich ataxia (FRDA) is an autosomal recessive neurodegenerative disorder caused by expanded GAA triplet repeats located in the first intron of the FXN gene coding for frataxin on chromosome 9 [1]. Frataxin is a mitochondrial protein involved in biogenesis of iron-sulfur clusters (ISCs). ISCs serve as prosthetic group in several enzymes of the mitochondrial energy metabolism including aconitase and complexes I, II and III of the respiratory chain that are impaired in FRDA [2]. Frataxin mRNA was found to show a broad expression pattern, including tissues with a high metabolic rate, like liver, kidney, brown fat and heart [3]. In addition to mitochondrial dysfunction, increase of reactive oxygen species (ROS)s is regarded as key feature in FRDA pathogenesis [4]. The length of the shorter of the two GAA repeats is thought to determine the residual amount of frataxin and thereby influencing age at onset of symptoms and disease severity [5,6]. Additionally, repeat length influences the development of diabetes and cardiomyopathy [5].
Several clinical rating scales have been proposed to assess disease severity in FRDA. The "scale for the assessment and rating of ataxia" (SARA) was evalutated in FRDA with high interrater reliability and practicability [7]. For future trials, especially for a possible neuroprotective treatment, there is a need for biomarkers linked to pathogenesis of FRDA. Beside others, quantification of frataxin, proton magnetic resonance spectroscopy (MRS) of the brain and phosphorus MRS in calf muscle [8] as well as atrophy of spinal cord and the medulla [9] were described as potential biomarkers in FRDA.
Methionine, an essential amino acid, is mainly metabolized in the liver which is remarkable rich in mitochondria [10]. Excess of methionine methyl groups is metabolised via sarcosine (N-methylglycine) and mitochondrial oxidation to CO2 [10]. Since the highest specific activity of methionine adenosyltransferase in mammals occurs in hepatic tissue, methyl-13C-methionine breath test (MeBT) has been proposed for the assessment of hepatic mitochondrial function in vivo [11]. Meanwhile MeBT is established for non-invasive, easy to perform and cheap quantification of oxidative capacity of liver mitochondria and was used for drug monitoring in HIV and Hepatitis C [12,13]. Further applications include detecting of drug and alcohol related liver toxicity, liver steatosis and cirrhosis [10,14]. The method has not been used in neurological disorders so far.
The aim of this study was to investigate mitochondrial liver function of FRDA patients and to explore if the MeBT is feasible to reflect disease severity in FRDA.
2. Methods
16 patients all with genetically confirmed Friedreich ataxia and homozygous for the GAA expansion were recruited from the Department of Neurology, St. Josef-Hospital, Ruhr-University Bochum and the Ataxia Clinic, Department of Neurodegeneration, University of Tübingen, Germany. Healthy volunteers were matched to patients by age, gender and body surface area (BSA; see Fischer Analysen Instrumente GmbH, Leipzig, Germany). All participants gave written informed consent to the study. The study was approved by the ethic committee of the Ruhr-University Bochum, (registration-number 2718). Exact GAA repeat lengths in the FXN gene were available in 14 of 16 FRDA patients. Two patients had a late onset of the disease (28 and 30 years), only one was still able to walk independently. Disease severity was assessed using SARA (table 1) [7]. Six FRDA participants were free of medication, four were on proton pump inhibitors (PPIs) and beta-blockers, two respectively on baclofen 5 or 10 mg, ACE-inhibitors, digitoxin and one respectively on buprenorphine 0.2 mg, ropinirole 4 mg, olmesartan 20 mg, trospium chloride 60 mg, etoricoxib 60 mg, clonazepam 1 mg, citalopram 10 mg, amitriptyline 50 mg, gabapentin 800 mg, prednisone 7.5 mg, hydrochlorothiazide 25 mg, acetylsalicylic acid 300 mg daily. Three FRDA patients got antioxidants; one took L-carnitine 1500 mg, coenzyme Q(10) 400 mg and vitamine E 1000 mg, one idebenone 2250 mg and one L-carnitine 1500 mg. All participants paused with their medication for at least 12 hours prior to the MeBT. Exclusion criteria were concurrent liver diseases, excessive alcohol consumption (50 g/d of ethanol), severe other diseases, underage and pregnancy.
Table 1.
Baseline group statistics of FRDA patients and healthy controls
| Parameter | FRDA (n = 16) | controls (n = 16) |
|---|---|---|
| Age [yr] | 43.8 ± 9.3 (28-59) | 43.1 ± 8.7 (28-60) |
| Gender (male/female) | 10/6 | 10/6 |
| Height [m] | 1.72 ± 0.1 (1.59-1.83) | 1.73 ± 0,1 (1.57-1.89) |
| Weight [kg] | 73.9 ± 9.9 (58-95) | 74.8 ± 9 (56-94) |
| BSA [m2] | 1.89 ± 0,2 (1.61-2.20) | 1.9 ± 0.2 (1.61-2.16) |
| GAA shorter allel * | 402 ± 127 (220-560) | -- |
| GAA larger allel * | 798.5 ± 250.6 (250-1110) | -- |
| GAA product both alleles * |
394492.9 ± 245596.8 (62500-954600) |
-- |
| Onset of the disease [yr] | 17.2 ± 5.9 (7-30) | -- |
| Disease duration [yr] | 26.7 ± 8.7 (8-42) | -- |
| SARA | 26.5 ± 7 (9.5-36) | -- |
Values are given as mean ± SD; range (min-max) in brackets; Abbreviations: BSA - body surface area, yr - years, SARA - scale for the assessment and rating of ataxia. *Exact GAA allel length was available for n = 14
The 13C-methionine breath test was prepared as established by Banasch et al 2008 [15]. 13C-methionine breath test was run after an overnight fasting. All subjects received [methyl-13C]-labelled methionine (L-methionine-13C, 99% atom isotopic enrichment; Cambridge Isotope, Andover, MA) in a dose of 2 mg/kg of body weight dissolved in 100 ml of water. Breath samples were collected before substrate administration at baseline and then every 10 minutes for 90 minutes in 50 ml closed aluminized plastic breath bags. During the test patients were requested to sit relaxed. The collected breath samples were analysed by measuring the 13C/12C isotope ratio via nondispersive isotope selective infrared spectroscopy (IRIS; 13C Wagner Analysen Technik, Bremen, Germany). Results were expressed as delta (δ) 13C/12C and as delta over baseline (DOB). PDB-standard from South Carolina (RatioPDB = 0.0112372) was used for the analysis of the data. The results were expressed as percentage dose of 13C recovery (PDR) over time for each time interval, maximum PDR (PDRmax) and cumulative PDR (cPDR) after different minutes of testing time up to 90 min (cPDR90).
Statistical analysis was performed using the software program SPSS statistics 18. All measured parameters and clinical data are presented as mean ± SD. Normality of distribution of the data was tested by one-sample Kolmogorov-Smirnov test. For testing the significance of differences between the two groups, independent t test procedure was used. Homogeneity of variance was shown by Levene's test. For specification of relationship between 13C-methionine breath test results and FRDA rating scales as well as clinical data of patients a stepwise linear regression model was performed. The significance level of the F value for accepting an independent variable to the linear regression model was chosen as p = 0.05 and for exclusion as p = 0.1.
3. Results
Demographic, clinical and genetic characteristics of FRDA participants and matched healthy controls are provided in table 1. All variables showed normal distribution. Independent t-test for scale parameter and Fisher exact homogeneity Chi-square test for categorical parameter revealed no significant differences (t-test: p > 0.1, Chi-square test: p = 1) between the FRDA group and the control group.
3.1. 13C-Methionine Breath Test
Significant decay of mitochondrial function was found in FRDA patients in comparison to controls for the cumulative percentage dose rate over 90 minutes of exhaled 13CO2 (cPDR90 [%] ± SEM: 5.61% ± 0.61% vs. 7.45% ± 0.4%; p = 0.018; Figure 1B). Additionally, the maximal exhaled percentage dose of 13CO2 recovered was reduced in the FRDA patient group (PDRmax ± SEM: 6.82%/h ± 0.81%/h vs. 9.71%/h ± 0.6%/h; p = 0.007; at 33 ± 10.5 min, see Figure 1A). In an additional explorative analysis, independent t-test was calculated for the cPDR of each time point. Here differences of cPDR between FRDA and matched controls were found already after test duration of 20 minutes (Figure 1B). No differences were observed between drug-free FRDA patients (n = 6; cPDR90 [%] ± SEM: 6.99% ± 0.67%, PDRmax ± SEM: 8.65%/h ± 0.93%/h) and patients on medication (n = 10; cPDR90 [%] ± SEM: 4.79% ± 0.8%, PDRmax ± SEM: 5.72%/h ± 1.05%/h). No differences were observed between patients with antioxidants (n = 3; cPDR90 [%] ± SEM: 5.86% ± 1.24%, PDRmax ± SEM: 6.86%/h ± 1.42%/h) and patients without antioxidants (n = 13; cPDR90 [%] ± SEM: 5.56% ± 2.69%, PDRmax ± SEM: 6.81%/h ± 3.56%/h; p = 0.855, 0.979 respectively).
Figure 1.
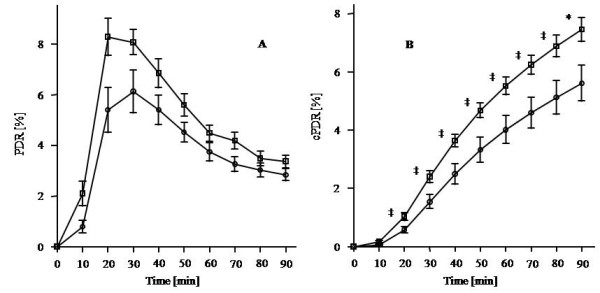
Mean recovered 13CO2 as function of time (PDR [%/h]) (A) and mean cumulative exhaled 13CO2 (cPDR [%]) (B) of 16 FRDA patients (circle) and 16 healthy controls (square); Error bares = ± SEM; * - significant difference between the groups (p < 0.05); ‡ - explorative difference between the groups (p < 0.05).
Liver metabolism as assessed by cPDR and PDRmax did not correlate with genetic markers (expanded GAA repeats and their interaction) nor with age, age at onset of symptoms or duration of disease and did not reflect disease severity as assessed by SARA using multiple linear regression analysis.
4. Discussion
The present cross-sectional study is the first to show subclinical liver affection in FRDA. Since participants with concurrent liver diseases or excessive alcohol consumption were excluded, none of the participants was suffering from a clinical manifest liver affection. Using the 13C-methionine breath test, however, we could clearly demonstrate mitochondrial dysfunction in the liver in vivo. This is in accordance with histophalogical findings, describing an increased iron deposition not only in FRDA heart, but also in liver and spleen in a pattern consistent with a mitochondrial location [16]. Down regulation or defect of citric acid cycle enzymes like aconitase, defects in complex I/II/III of the endoxidation and thus a failure of redox-equivalent oxidation necessary for the citric acid cycle are potential mechanisms. The reduced mitochondrial function in MeBT confirms the importance of frataxin for mitochondrial energy metabolism in FRDA [2].
Subclinical, impaired mitochondrial liver function should be considered when applying drugs with potential mitochondrial toxicity like valproate or propofol that are known to exert critical effects in other mitochondrial disorders like polymerase gamma (POLG) mutations [17]. One case with fatal liver failure associated with valproate therapy in a patient with FRDA has been described so far [18].
Contrary to our expectation, no correlations between MeBT and disease severity, age of onset or disease duration could be established. Although inverse correlation of clinical severity and repeat lengths has repeatedly been reported in FRDA,[5,6,19] we found no correlation of MeBT results with the number of GAA repeats or their interaction. Thus, measuring of hepatic mitochondrial activity by MeBT does not seem to be a good biomarker on the follow-up of natural history of the disease and the response to drugs in clinical trials.
One limitation of our study is that an influence of co-commitant medication on the MeBT outcome cannot be completely excluded. However, no differences were observed between drug-free FRDA patients (n = 6) and patients on medication (n = 10) and an impairment of mitochondrial metabolism is not described for the listed drugs [11,12,14].
5. Conclusions
Taken together, the data of our pilot study using the MeBT in FRDA indicate that FRDA patients exhale significantly smaller amounts of 13CO2 compared with healthy controls indicating a subclinical liver affection in FRDA.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
Stüwe 1C, 2B, 3A; Goetze 2A, 2B, 2C, 3B; Banasch 1C, 3B; Arning 1C, 2C, 3B; Schmidt 1A, 2C, 3B; Schöls 1B, 2A, 2C, 3B; Saft 1A, 1B, 1C, 2A, 2C, 3A, 3B
1. Research project: A. Conception, B. Organization, C. Execution;
2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique;
3. Manuscript: A. Writing of the first draft, B. Review and Critique;
All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Contributor Information
Sven H Stüwe, Email: sven-stuewe@t-online.de.
Oliver Goetze, Email: Oliver.Goetze@usz.ch.
Larissa Arning, Email: Larissa.Arning@ruhr-uni-bochum.de.
Matthias Banasch, Email: MBanasch@gmx.de.
Wolfgang E Schmidt, Email: wolfgang.e.schmidt@rub.de.
Ludger Schöls, Email: Ludger.Schoels@uni-tuebingen.de.
Carsten Saft, Email: carsten.saft@ruhr-uni-bochum.de.
Acknowledgements
We are grateful to all patients for participation.
Funding
Carsten Saft was supported by a FoRUM grand, University of Bochum (AZ: K040-2009). Ludger Schöls was supported by a BMBF grant to mitoNET (01GM0864). Oliver Götze was supported by DFG (Gö 13582/1).
Ethics approval
The study was approved by the ethics committee of the University of Bochum.
References
- Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A. et al. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science New York, NY. 1996;271(5254):1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- Rotig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet. 1997;17(2):215–217. doi: 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- Koutnikova H, Campuzano V, Foury F, Dolle P, Cazzalini O, Koenig M. Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin. Nature genetics. 1997;16(4):345–351. doi: 10.1038/ng0897-345. [DOI] [PubMed] [Google Scholar]
- Schulz JB, Dehmer T, Schols L, Mende H, Hardt C, Vorgerd M, Burk K, Matson W, Dichgans J, Beal MF. et al. Oxidative stress in patients with Friedreich ataxia. Neurology. 2000;55(11):1719–1721. doi: 10.1212/wnl.55.11.1719. [DOI] [PubMed] [Google Scholar]
- Filla A, De Michele G, Cavalcanti F, Pianese L, Monticelli A, Campanella G, Cocozza S. The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. American journal of human genetics. 1996;59(3):554–560. [PMC free article] [PubMed] [Google Scholar]
- Schols L, Amoiridis G, Przuntek H, Frank G, Epplen JT, Epplen C. Friedreich's ataxia. Revision of the phenotype according to molecular genetics. Brain. 1997;120(Pt 12):2131–2140. doi: 10.1093/brain/120.12.2131. [DOI] [PubMed] [Google Scholar]
- Schmitz-Hubsch T, du Montcel ST, Baliko L, Berciano J, Boesch S, Depondt C, Giunti P, Globas C, Infante J, Kang JS. et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology. 2006;66(11):1717–1720. doi: 10.1212/01.wnl.0000219042.60538.92. [DOI] [PubMed] [Google Scholar]
- Vorgerd M, Schols L, Hardt C, Ristow M, Epplen JT, Zange J. Mitochondrial impairment of human muscle in Friedreich ataxia in vivo. Neuromuscul Disord. 2000;10(6):430–435. doi: 10.1016/S0960-8966(00)00108-5. [DOI] [PubMed] [Google Scholar]
- Della Nave R, Ginestroni A, Giannelli M, Tessa C, Salvatore E, Salvi F, Dotti MT, De Michele G, Piacentini S, Mascalchi M. Brain structural damage in Friedreich's ataxia. Journal of neurology, neurosurgery, and psychiatry. 2008;79(1):82–85. doi: 10.1136/jnnp.2007.124297. [DOI] [PubMed] [Google Scholar]
- Milazzo L, Piazza M, Sangaletti O, Gatti N, Cappelletti A, Adorni F, Antinori S, Galli M, Moroni M, Riva A. [13C]Methionine breath test: a novel method to detect antiretroviral drug-related mitochondrial toxicity. J Antimicrob Chemother. 2005;55(1):84–89. doi: 10.1093/jac/dkh497. [DOI] [PubMed] [Google Scholar]
- Armuzzi A, Candelli M, Zocco MA, Andreoli A, De Lorenzo A, Nista EC, Miele L, Cremonini F, Cazzato IA, Grieco A. et al. Review article: breath testing for human liver function assessment. Aliment Pharmacol Ther. 2002;16(12):1977–1996. doi: 10.1046/j.1365-2036.2002.01374.x. [DOI] [PubMed] [Google Scholar]
- Banasch M, Emminghaus R, Ellrichmann M, Schmidt WE, Goetze O. Longitudinal effects of hepatitis C virus treatment on hepatic mitochondrial dysfunction assessed by C-methionine breath test. Aliment Pharmacol Ther. 2008;28(4):443–449. doi: 10.1111/j.1365-2036.2008.03745.x. [DOI] [PubMed] [Google Scholar]
- Banasch M, Frank J, Serova K, Knyhala K, Kollar S, Potthoff A, Brockmeyer NH, Goetze O. Impact of antiretroviral treatment on (13)C-methionine metabolism as a marker of hepatic mitochondrial function: a longitudinal study. HIV Med. 2010. [DOI] [PubMed]
- Wutzke KD, Forberger A, Wigger M. Effect of alcohol consumption on the liver detoxication capacity as measured by [13C]methacetin- and [methyl-13C]methionine-breath tests. Isotopes Environ Health Stud. 2008;44(2):219–226. doi: 10.1080/10256010802066372. [DOI] [PubMed] [Google Scholar]
- Banasch M, Knyhala K, Kollar S, Serova K, Potthoff A, Schlottmann R, Schmidt WE, Brockmeyer NH, Goetze O. Disease- and treatment-related predictors of hepatic mitochondrial dysfunction in chronic HIV infection assessed by non-invasive (13)C-methionine breath test diagnostic. Eur J Med Res. 2008;13(9):401–408. [PubMed] [Google Scholar]
- Bradley JL, Blake JC, Chamberlain S, Thomas PK, Cooper JM, Schapira AH. Clinical, biochemical and molecular genetic correlations in Friedreich's ataxia. Human molecular genetics. 2000;9(2):275–282. doi: 10.1093/hmg/9.2.275. [DOI] [PubMed] [Google Scholar]
- Kam PC, Cardone D. Propofol infusion syndrome. Anaesthesia. 2007;62(7):690–701. doi: 10.1111/j.1365-2044.2007.05055.x. [DOI] [PubMed] [Google Scholar]
- Konig SA, Schenk M, Sick C, Holm E, Heubner C, Weiss A, Konig I, Hehlmann R. Fatal liver failure associated with valproate therapy in a patient with Friedreich's disease: review of valproate hepatotoxicity in adults. Epilepsia. 1999;40(7):1036–1040. doi: 10.1111/j.1528-1157.1999.tb00814.x. [DOI] [PubMed] [Google Scholar]
- Delatycki MB, Paris DB, Gardner RJ, Nicholson GA, Nassif N, Storey E, MacMillan JC, Collins V, Williamson R, Forrest SM. Clinical and genetic study of Friedreich ataxia in an Australian population. American journal of medical genetics. 1999;87(2):168–174. doi: 10.1002/(SICI)1096-8628(19991119)87:2<168::AID-AJMG8>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]