

Abstract
Background
Epithelia act as physical barriers protecting living organisms and their organs from the surrounding environment. Simple epithelial tissues have the capacity to efficiently repair wounds through a resealing mechanism. The known molecular mechanisms underlying this process appear to be conserved in both vertebrates and invertebrates, namely the involvement of the transcription factors Grainy head (Grh) and Fos. In Drosophila, Grh and Fos lead to the activation of wound response genes required for epithelial repair. ERK is upstream of this pathway and known to be one of the first kinases to be activated upon wounding. However, it is still unclear how ERK activation contributes to a proper wound response and which molecular mechanisms regulate its activation.
Methodology/Principal Findings
In a previous screen, we isolated mutants with defects in wound healing. Here, we describe the role of one of these genes, hole-in-one (holn1), in the wound healing process. Holn1 is a GYF domain containing protein that we found to be required for the activation of several Grh and Fos regulated wound response genes at the wound site. We also provide evidence suggesting that Holn1 may be involved in the Ras/ERK signaling pathway, by acting downstream of ERK. Finally, we show that wound healing requires the function of EGFR and ERK signaling.
Conclusions/Significance
Based on these data, we conclude that holn1 is a novel gene required for a proper wound healing response. We further propose and discuss a model whereby Holn1 acts downstream of EGFR and ERK signaling in the Grh/Fos mediated wound closure pathway.
Introduction
Epithelial tissues form a critical barrier between the external environment and an organism's internal organs. All epithelia have developed robust methods for maintaining tissue integrity during natural processes such as cell turnover, as well as restoring barrier function when tissues are damaged. The wound healing process can differ between developmental stages and can also vary among tissues and involve the cooperation of several cell types such as neutrophils and macrophages. One important mechanism, called “purse-string” wound closure, is conserved in epithelial tissues of several animal species including Drosophila, chick, mouse, and human [1]. This process involves a rapid assembly of an actomyosin contractile cable in the epithelial cells bordering the wound [2]. Concurrently, these cells extend actin-based protrusions, such as filopodia and lamellipodia, into the wound site. As the contractile cable cinches the wound closed, the wound bordering cells simultaneously elongate in the direction of the wound and contract the edge of the wound. In the final stages of wound closure, lamellipodia and filopodia are required for knitting the wound bordering cells together to form a seamless epithelium. The signaling cascades that regulate the concerted epithelial resealing process as a whole are just beginning to be unraveled. The known molecular mechanisms appear to be conserved in both vertebrates and invertebrates, namely the involvement of the transcription factor Grainy head (Grh) and of the JNK signaling cascade, transduced by AP-1 (Jun/Fos) transcriptional complexes [3]–[7]. In the fly, the expression of some genes at the wound site is dependent on functional Grh and AP-1 binding sites in their promoter region [5], [8]. These observations are consistent with abnormal wound healing in grh, basket/JNK or jra/Jun mutants and activation of JNK signaling pathway at wound sites [3], [5], [9]. The upstream signals activating the cells surrounding the wound are still unknown, but it is established that extracellular signal-regulated kinase (ERK) is phosphorylated upon wounding, an event required at wound sites for a robust closure response [5]. It is also known that ERK can phosphorylate the afore mentioned wound response transcription factors Grh and Fos both in vivo and in vitro [10]–[12]. Furthermore, recent data demonstrated that Stitcher (Stit), a target of Grh transcriptional regulation, encodes a receptor tyrosine kinase (RTK) also capable of inducing ERK phosphorylation in wounded epithelia [13]. All together these data have led to the proposal that a Grh-dependent positive feedback loop likely functions as an amplification mechanism to ensure efficient epidermal wound repair [5], [13].
In a previous screen, we isolated mutants displaying defects in wound healing [9]. One of these identified loci, CG5198, is predicted to be involved in processes that are likely associated with wound healing. Specifically, the human homologue of CG5198, CD2 Binding Protein 2 (CD2BP2), binds to the adhesion molecule CD2 and induces cytokine production in T cells, a key component of the mammalian immune response [14], [15]. In a Drosophila cell culture system, CG5198 was found to be involved in the phagocytosis of fungi and bacteria, suggesting a possible role in innate immunity [16]. In other work CD2BP2 has been referred to as U5-52K and is proposed to mediate the assembly of the core spliceosome, a protein complex required for the proper processing of all intron containing RNA transcripts [17]–[19]. As the Drosophila homologue of CD2BP2 was not previously described, we named the CG5198 locus hole-in-one (holn1), in honor of the wound healing defect attributed to the mutant holn1c07150.
In this work we further describe the role of Holn1 in the wound healing process. We reveal the requirement of Holn1 for transcriptional regulation of known ERK/Grh/Fos dependent wound response loci surrounding the wound site. We provide phenotypic evidence suggesting that Holn1 may be involved in other developmental processes requiring Ras signaling. Finally, we analyze the behavior of Epidermal Growth Factor Receptor (EGFR)/ERK signaling mutants and show that reduced EGFR and ERK signaling leads to wound closure defects. We propose a model whereby Holn1 acts downstream of EGFR and ERK signaling in the Grh/Fos mediated wound closure pathway.
Results
holn1 mRNA is expressed ubiquitously in the Drosophila embryo and Holn1 protein localizes to the nucleus
In situ hybridization of embryos revealed that holn1 mRNA is maternally deposited (Fig 1A,B) and remains weak and ubiquitous throughout embryonic development (Fig 1C,D). Importantly, holn1 is expressed in the epidermis at stage 14/15 (Fig 1D), placing it in the right place at the right time to be involved in healing the laser induced wounds implemented in our wounding assay [9]. Expression of GFP-tagged Holn1 (UAS>GFP-holn1) using the epidermal driver e22c>gal4 revealed the nuclear localization of GFP-Holn1 (Fig 1E–J), consistent with the observed distribution of its human homologue CD2BP2 [17], [19], [20]. We noted that GFP-Holn1 signal is reduced in heterochromatin regions, as detected by overlay with areas of intense DAPI staining (see arrowheads in Fig 1G–I).
Figure 1. Expression of Holn1 in wild-type embryos.

(A–D) Expression pattern of holn1 RNA in wild-type embryos. (A,C) Sense control in situ hybridization showing lack of staining in stage 5 (A) and stage 14 (C) embryos. (B) holn1 anti-sense RNA probe shows strong maternal contribution of holn1 RNA in stage 5 embryo. (D) holn1 RNA expression is weak and ubiquitous in stage 14 embryo, enriched slightly in the nerve cord and present in the epidermis. Dorsal is to the top and anterior to the left. stg., stage. (E–J) Expression of GFP-Holn1 in the embryonic ventral epidermis under the control of the epidermal driver e22c>gal4. (E,F) GFP-Holn1 is expressed in the nuclei of ventral epidermis cells. (G–J) Magnified view of embryo shown in E,F. (E–G) Merged channels. (H) DAPI shows nuclear staining. (I) GFP-Holn1 localization in the cell nuclei. (J) Phalloidin marks filamentous actin at the cell cortex. Arrowheads in (G–I) indicate regions where heterochromatin is more condensed. All images are single Z slices. Scale bar in (E,F) = 20 µm, and in (G–J) = 10 µm.
holn1 mutants have wound healing defects
In our screen [9], we uncovered the wound healing defects of the lethal mutant holn1c07150, caused by the insertion of a piggyBac transposable element after nucleotide 878 of the holn1 ORF (Fig 2A). This inserted element results in a missense mutation leading to a K to N switch in amino acid position 293, immediately followed by a stop codon likely truncating the C-terminal GYF domain [21]. The GYF domain is the only recognizable functional domain of Holn1 and is characterized as being a protein-protein interacting domain with affinity towards proline-rich regions [22]. In the human Holn1 homologue, the GYF domain is responsible for interactions both with CD2 and with spliceosome components [15], [17], [22]. To confirm that the wound healing defects seen in the holn1c07150 mutant are indeed due to a disruption in Holn1 function caused by the piggyBac transposable element, we remobilized this element by precise excision [21]. We observed a complete restoration of wound healing capacity (Fig 2B) and viability (data not shown) upon precise excision of the piggyBac element. Also, upon expression of wild-type holn1 (pUASp>holn1) under control of the epidermal driver e22c>gal4 in holn1c07150 mutant background, we observed a rescue of the wound healing phenotype of holn1c07150 (Fig 2B).
Figure 2. holn1c07150 mutant phenotype is caused by holn1 loss-function.

(A) Gene (upper panel) and protein (lower panel) schematic views of Holn1. UTR's are shown in white and coding regions in grey and black. holn1LL07287 is inserted in the 5′UTR region whereas holn1c07150 is inserted in the third coding exon, in the GYF domain region (black). (B,C) Wound healing phenotypes (% open of open wounds) 16 hours post wounding. (B) Precise excision of piggyBac element (holn1 jumpout, light grey) and expression of one copy of holn1 transgene in holn1c07150 mutants (holn1c07150,e22c>holn1, white bar) restore wound healing defects observed in holn1c07150 mutants (dark grey). (C) holn1c07150/holn1LL07287 transheterozygote embryos (spotty bar) show similar percentage of open wounds to holn1c07150 homozygous mutants (light grey bar), in contrast to wild-type (black bar), holn1LL07287 homozygous mutants (striped bar), holn1c07150 heterozygotes (dark grey), and holn1LL07287 heterozygotes (white bar). Fisher's exact test showed significant different between groups (**, p<0.01; ***, p<0.0001).
We obtained a second lethal allele of holn1, holn1LL07287, which results from a piggyBac insertion in the 5 UTR of the gene [23]. holn1LL07287 fails to complement the lethality of holn1c07150. We performed the wounding assay on transheterozygous holn1c07150/ holn1LL07287 embryos and found a phenotype similar to that of holn1c07150 homozygotes (29% open wounds in transheterozygotes, 23% in holn1c07150 homozygotes, Fig 2C). Interestingly, homozygous holn1LL07287 mutants displayed a weak wound healing defect (7% open wounds, Fig 2C). It is important to note that, in this wounding assay, we were only able to score fully developed hatching larvae for wound healing defects. We observed that 41% of homozygous holn1LL07287 embryos died before hatching, during late embryogenesis, whereas in holn1LL07287/ holn1c07150 transheterozygotes and holn1c07150 homozygotes we observed this embryonic lethality phenotype in only 25% and 15% of embryos, respectively (Fig. S1). This suggests that holn1LL07287 might be a stronger allele than holn1c07150 and the early lethality phenotype is dependent on the number of copies of the holn1LL07287 allele.
Taken together, we conclude that holn1 gene is indeed required for proper wound closure.
Holn1 is required for efficient wound closure but not for wound edge actomyosin cable formation
To analyze the wound healing phenotype of holn1 mutants in more detail, we performed time-lapse live recordings of the wound closure process in holn1c07150 mutants, upon laser wounding of the ventral epidermis. We analyzed holn1c07150 mutants, as the holn1LL07287 mutants die earlier making interpretation of the results more difficult. We observed that both control and holn1c07150 mutant embryos assembled a contractile cable containing actin and myosin within minutes upon wounding (Fig 3A–D, Movies S1, S2). Actin-containing cell protrusions also form during wound closure in both cases (see arrows in Movies S1, S2). On the other hand, soon after the actomyosin cable has formed, the wound closure process slows down in holn1 mutants when compared to controls (Fig 3E). Whereas holn1 mutant embryos take on average 194 minutes to close 7000 µm-diameter wounds, control embryos take 128 minutes (Fig 3E, n = 3). Together these data indicate that holn1 is required for efficient wound closure, but not for the immediate assembly of the actomyosin cable.
Figure 3. Time course of wound closure in control and holn1c07150 mutant embryos.

(A–B) Stills of wound closure process in e22c>cherry-moesin control (A) and e22c>cherry-moesin,holn1c07150 mutant embryos (B) at 1, 20, 50, 110, and 200 minutes after wounding. At 20 minutes after wounding, an actin cable is already formed in both control (A) and mutant embryo (B). Control embryo has its wound closed at 110 minutes after wounding, whereas mutant embryo only close around 200 minutes after wounding. (C,D) Stills of wounds in myosin-GFP control (C) and myosin-GFP,holn1c07150 mutant embryos (D) at 20 minutes after wounding. A myosin cable is observed at the wound edge in both control and mutant embryos. (E) Plot showing average wound diameter of closing wounds in control (n = 3) and holn1c07150 mutant embryos (n = 3). Both in control and mutant embryos, wounds start as 7000 µm2-diameter holes and expand in size during the first 10 minutes after wounding. After that, wound size decreases exponentially, but slower in mutants than in controls. Error bars represent the standard error of the mean for each time point. Average time of wound closure is significantly higher in mutants (194.3 minutes) compared to controls (127.7 minutes; unpaired t test, p<0.05). Images in (A–D) are projections of spinning disc confocal time-lapse recordings. Scale bar = 20 µm.
holn1c07150 genetically interacts with a constitutively active Ras allele, and RNAi knock-down as well as holn1c07150 phenocopy Ras overactivation phenotypes
To gain insight on the possible pathways where Holn1 might be playing a role, we analyzed the phenotype of holn1c07150 as well as of holn1 RNAi knockdown in the adult fly. We were prompted to do this because we observed that rare homozygous holn1c07150 escaper flies (obtained when growing a recombinant stock at 18°C; see Materials and methods) showed clear developmental phenotypes characteristic of EGFR/Ras/ERK pathway mutants. In contrast to heterozygous flies (Fig 4A, left), the holn1c07150 homozygous escapers displayed a subtle rough eye phenotype (Fig 4A, right), reminiscent of the oomatidia fusion phenotype observed in flies expressing a constitutively active form of Ras under the direct control of the sevenless (sev) eye specific promoter (sev>ras1V12) (Fig 4B, left) [24]. It is known that EGFR and subsequent Ras activation induce a signaling cascade that is involved in various aspects of organism development, including morphogenesis of the eye, wing and thorax [25], [26]. We performed a classic genetic interaction test and observed that one copy of holn1c07150 dominantly enhanced the sev>ras1V12-induced rough eye phenotype (Fig 4B, right), when compared to sev>ras1V12 alone (Fig 4B, left). This result indicates that the holn1c07150 mutation causes an increased activation of the Ras signaling pathway, suggesting that wild type Holn1 might function as an inhibitor of this pathway.
Figure 4. holn1c07150 genetic interaction with ras1V12, holn1c07150 homozygous escaper phenotypes and RNAi phenotypes in adult tissues.

(A) Flies expressing one copy of the holn1c07150 mutant chromosome have eyes with a wild type appearance, i.e. organized lattice of ommatidia (A, left). In contrast, holn1c07150 homozygous escaper flies (A, right) display a weak rough eye phenotype. (A, inserts) Digital zooms of the upper right region of the eye. (B) One copy of the sev>ras1V12 chromosome results in a rough eye phenotype (B, left). This rough eye phenotype is enhanced when combined with one copy of holn1c07150 (B, right). (C) holn1c07150 homozygous escaper (C, middle) and RNAi pupae ubiquitously expressing dsRNA#27369 directed against holn1 RNA (C, left) have extra and/or misplaced (C, middle and right, #5) and sometimes missing macrochaetae (C, right, 3*) on the scutellum, when compared to wild type flies (C, left). Wild type macrochaetae are arbitrarily labeled 1–4 (C, left) for comparison to the corresponding macrochaetae in mutant flies (C, middle and right panels). (D) RNAi knockdown of holn1 RNA using the wing specific driver ms1096>gal4 in combination with 2 independent dsRNAs directed against holn1: UAS>holn1-dsRNA#27369 (D, middle; arrows indicate ectopic veins) and UAS>holn1-dsRNA#110281 (D, right). Scale bar = 50 µm.
Moreover, reducing holn1 levels by expressing dsRNA [27] directed against holn1 in the developing wing resulted in a range of phenotypes from wild type looking (less common, not shown) to a smaller, cylindrically curved wing with increased number of veins (Fig 4D, middle), or a blistered wing with much of the surface converted to vein material (Fig 4D, right). In contrast, control wings expressing dsRNA directed against GFP always showed a wild type appearance (Fig 4D, left). EGFR/Ras/ERK signaling is also known to specify the vein regions of the adult wing [26]. The extra vein phenotype observed when knocking down holn1 using RNAi is also typical of increased EGFR/Ras/ERK signaling during wing development [11], [28], supporting the hypothesis that Holn1 plays a role in this pathway.
Adult thorax macrochaete development is also dependent on the EGFR/Ras/MAPK pathway [25]. We observed that the rare holn1c07150 homozygous escaper flies and late pupae often had extra and/or misplaced or missing macrochaetae on the scutellum (Fig 4C, middle; macrochaete #5 is misplaced or extra), when compared to wild type pupae (Fig 4C, left). We confirmed that this phenotype was due to defects in Holn1 expression by performing RNAi to reduce holn1 levels in a wild type background. Ubiquitous knockdown of holn1 using actin5c>gal4 caused late pupal lethality. We noted that the pupae appeared fragile and fell apart just before hatching, or died during eclosion. When removing the pupal case, we observed a scutellar macrochaete phenotype identical to that seen in the homozygous escapers (Fig 4C, right; macrochaete #3* is missing, and #5 is misplaced/extra). A similar scutellar phenotype was reported when overexpressing EGFR using apterous>gal4, although the increase in macrochaete number was ubiquitous throughout the thorax in those experiments [25].
Taken together, these data are consistent with Holn1 involvement in the EGFR/Ras/ERK signaling pathway.
holn1c07150 mutants show reduced activation of wound reporter genes downstream of wound healing transcriptional pathways
ERK phosphorylation occurs downstream of RTK and Ras activation [29]. It has been shown that phosphorylation of ERK occurs at wound sites in the Drosophila embryo as well as in cell culture systems [5], [30]. As we observed a genetic interaction between Holn1 and Ras signaling, we asked whether Holn1 would have an influence on ERK phosphorylation around the wound site in our system. We observed that, as previously shown [5], [13], ERK appears to be strongly activated in the cells immediately surrounding the wound, as shown by the detection of the diphosphorylated form of ERK (dpERK) by immunostaining (Fig 5A–F). This activation was observed as early as 15 minutes after wounding (data not shown), reaching maximum levels 30 minutes after wounding (Fig 5A). One hour after wounding, ERK phosphorylation decreased around the wound edge and was undetectable two hours after wounding (Fig 5B,C). Surprisingly, we observed a similar pattern of ERK activation in holn1c07150 mutants when compared to wild type (Fig 5D–F). This shows that Holn1 is not required for ERK phosphorylation at the wound edge epithelium, suggesting that Holn1 functions in parallel to or downstream of ERK activation.
Figure 5. ERK and wound reporter activation in wounded control and holn1c07150 mutant embryos.

(A–F) dpERK immunostaining in ventral epidermis of wounded wild-type (A–C) and holn1c07150 mutant embryos (D–F) 30 minutes (A,D), 1 hour (B,E), and 2 hours after wounding (C,F). In both controls and mutants, dpERK is activated in cells surrounding the wound (dashed line) 30 minutes and 1 hour after wounding, and it disappears 2 hours after wounding. (G,H) Ddc-GFP activation in cells around wound region (red arrows) in control (G) and mutant (H) embryos. (I) The number of embryos that activate the wound reporter Ddc-GFP 5 hours after wounding is significantly lower in mutants (34% of embryos, n = 44) compared to controls (78% of embryos, n = 50; p<0.0001). (J,K) Msn-DsRed activation in cells around wound region (red arrows) in control (J) and mutant (K) embryos. (L) The number of embryos that activate the wound reporter Msn-DsRed 5 hours after wounding is significantly lower in mutants (25% of embryos, n = 125) compared to controls (74% of embryos, n = 27; p<0.0001). Dashed lines in (A–F), wound edge; mpw, minutes post wounding; hpw, hours post wounding. Red cross in (G,H,J,K), wound site. Scale bar = 20 µm, except in (J) = 18 µm.
It has been recently reported that phosphorylation of ERK upon wounding occurs upstream of a transcriptional activation pathway involved in epidermal wound repair [5], [10]. Particularly, the transcription factors Grh and Fos have been shown to act downstream of ERK and to induce the transcription of genes required for cuticle repair [5], [8]. One of the Grh/Fos target genes, dopa decarboxylase (Ddc), encodes an enzyme involved in the production of highly reactive quinones involved in crosslinking chitin and cuticle proteins during the construction and repair of the cuticular barrier [31]. Another factor induced by ERK, Grh and Fos upon wounding is the kinase misshapen (msn) [8]. To determine whether Holn1 is involved in the induction of wound reporter genes downstream of ERK, we compared activation of Ddc and msn in control and holn1c07150 mutant embryos by using previously described tagged reporters for these genes [5], [8]. In control embryos we observed activation of Ddc-GFP and msn-DsRed 5 hours after wounding as previously reported, whereas in holn1c07150 mutants the activation of Ddc-GFP and msn-DsRed was significantly decreased (Fig 5G–L). These observations suggest that Holn1 is required for activation of Ddc and Msn upon wounding. Together, these data suggest that Holn1 might act downstream of ERK in the regulation of Ddc and Msn transcription to promote wound healing.
EGFR/ERK signaling regulates wound healing
Considering that Holn1 appears to be involved in Ras signaling in adult epithelia and that it influences the transcription of genes known to be transcribed downstream of ERK activation, we asked whether the ERK pathway itself was also required during epithelial wound healing. To this end, we analyzed the wound healing phenotypes of EGFRt1 and ERK/rolled(rl)10a homozygous mutant embryos. EGFRt1 is a homozygous viable hypomorphic mutant for EGFR [32] whereas ERK/rl10a is described as a strong loss-of-function allele of ERK/rl [33]. We found that both mutants have wound closure defects, where EGFRt1 show 37% open wounds and ERK/rl10a 28% (Fig 6). These data indicate that activation of EGFR/ERK signaling is necessary for proper wound healing to occur.
Figure 6. EGFR and ERK are required for wound healing.
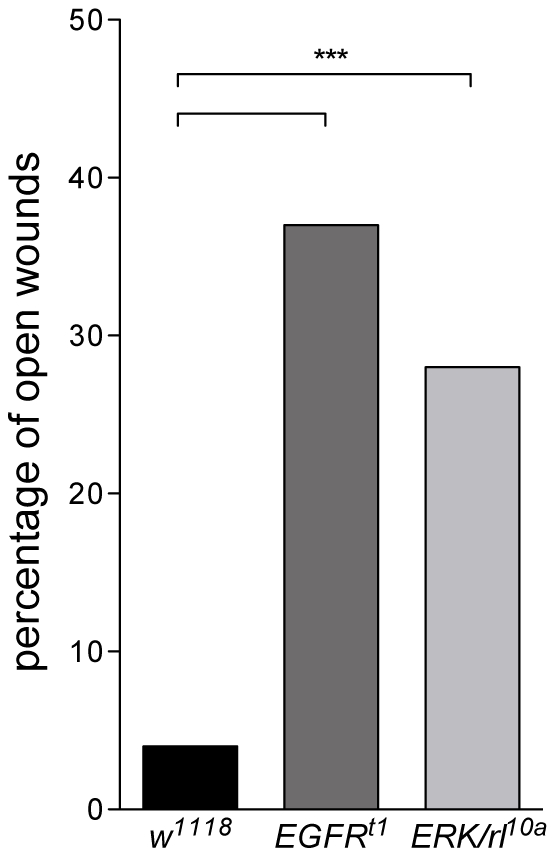
EGFRt1 and ERK/rl10a mutants show a significantly higher percentage of open wounds 16 hours post wounding (EGFRt1, 37%; ERK/rl10a, 28%) compared to wild-type (w1118, 5%). n[w1118] = 138; n[EGFRt1] = 70; n[ERK/rl10a] = 71. Fisher's exact test showed significant different between groups (*** = p<0.0001).
Discussion
This work describes the novel gene holn1 and reveals its involvement during embryonic wound healing. We have found that Holn1 is acting downstream of ERK activation in the ERK/Grainy head pathway when activated during the wound healing process.
holn1c07150 is a loss-of-function allele of Holn1
The holn1c07150 allele is an insertion of a piggyBac element in the coding region of the holn1 gene [21]. This inserted sequence putatively leads to the production of a premature stop codon within the GYF domain region, shown to be key to the known function of the human homologue of this protein, CD2BP2 [15], [17], [22]. We showed that the lethality and wound healing defects in holn1c07150 could be rescued by ubiquitous expression of pUASp-holn1 wild-type construct, as well as by precise excision of the piggyBac element. Furthermore, we observed that holn1 c07150 heterozygotes do not show wound healing defects or other mutant phenotypes, indicating that this is not a dominant mutation. Together, these results suggest that this allele is a loss-of-function mutation.
The second holn1 allele, holn1LL07287, results from a piggyBac element insertion in the 5′ UTR region of the gene [23], fails to complement holn1c07150, and therefore possibly leads to a loss-of-function mutation as well. We observed that holn1LL07287 homozygous mutants have two fold higher percentage of dead embryos compared to holn1c07150, making holn1LL07287 a stronger allele. The percentage of dead embryos in holn1LL07287 homozygotes and holn1LL07287/holn1c07150 transheterozygotes embryos is the same, suggesting that the increase in lethality is independent of genetic background effects. Interestingly, surviving holn1LL07287 homozygote embryos do not display wound healing defects. We believe that these surviving embryos are likely to have developed a mechanism to compensate for Holn1 loss. Although beyond the scope of this work, experiments analyzing Holn1 protein levels in the different mutants would help to further characterize each allele.
Holn1 is involved in the Ras signaling pathway during eye development and is required for proper thorax and wing development in adult flies
This study reveals that the holn1c07150 allele genetically interacts with a key component of the EGFR/Ras/ERK eye development pathway in the adult fly. In particular, we found that the holn1c07150 mutation enhances the rough eye phenotype induced by constitutively active Ras (Ras1V12). This suggests that wild type Holn1 might normally be a suppressor of the EGFR/Ras/ERK pathway. In support of a role of Holn1 in eye development, an analogous, albeit much weaker rough eye phenotype was observed in holn1c07150 homozygous escaper flies.
We provide further phenotypic evidence that Holn1 is involved in additional developmental processes known to be regulated by the EGFR/Ras/ERK pathway. Namely, the phenotypes observed in the adult wing and late pupal/adult thorax tissues resulting from either holn1c07150 mutant or holn1 RNAi knockdown phenocopy those previously observed upon overactivation of the EGFR/Ras/ERK pathway (Fig 4C,D) [11], [25], [28]. Together, these data suggest that Holn1 might be a suppressor of the EGFR/Ras/ERK pathway during adult development.
These results provided a connection between a gene required for Drosophila wound healing and the EGFR mediated signaling pathway, which prompted us to test whether this pathway is also required during the wound closure.
EGFR/ERK signaling is required for proper wound healing
Loss of Holn1 function in the holn1c07150 mutants, as well as reduced ERK signaling in EGFRt1 and ERK/rl10a mutants, impaired the wound healing process. Moreover, ERK activation was detected shortly after wounding and maintained at least until one hour after wounding (Mace, et al. 2005, this study). Although previous studies have proposed that ERK activation is required for proper wound healing [5], [13], direct evidence for this has never been provided. Mace and co-workers have shown that inhibiting ERK phosphorylation by injecting a drug against MAP kinase kinase (MEK) into the perivitelline space of embryos leads to reduced activation of the wound reporter gene Ddc, whereas the wound closure at the cellular level has not been addressed [5]. In another study, it has been shown that ERK phosphorylation during wound healing is partially dependent on the RTK Stit, although other RTK(s) must be also involved, as ERK phosphorylation still occurs in the stit mutant [13].
Our work shows for the first time that both activation of the RTK EGFR, as well as the activation of ERK, canonically found downstream of EGFR signaling [34], are required during wound healing in Drosophila (Fig 7). Further experiments will be necessary to determine whether additional RTKs are involved in wound closure. For instance, it would be interesting to see the effect of a simultaneous knockdown of Stit and EGFR function on ERK phosphorylation and wound closure.
Figure 7. Schematic model of proposed Holn1 function during wound healing.

Several signals are generated upon wounding, which activate different RTKs at the cell membrane, such as EGFR and Stitcher. These RTKs activate the ERK pathway leading to activation of Grh and Fos transcription factors, which subsequently induce the expression of cuticle repair genes (Ddc and Msn). One hypothesis for Holn1 function is that Holn1 is involved in the transcription and/or splicing of components of the ERK/Grh/Fos pathway in the nucleus and thereby contribute to proper wound healing.
Holn1 acts downstream or in a parallel pathway to ERK during wound healing
Interestingly, we observed that holn1 mutants showed similar levels of ERK activation to wild-type embryos upon wounding. This result leads us to propose that Holn1 functions as an activator of the pathway downstream of, or in parallel to, EGFR/ERK signaling during wound healing. In contrast, Holn1 appears to have repressor activity during adult fly development. Indeed, the activity of the Ras pathway is known to have distinct outcomes depending both on levels of Ras signaling as well as on the tissue context [26], [35].
In addition, we observed that loss of Holn1 function results in reduced activation of known wound reporter genes, Ddc and msn, both previously described to be transcribed downstream of ERK activation and of Grh and Fos transcription factors upon wounding. Grh phosphorylation by ERK was recently shown to be required for Ddc and Msn activation and for the re-establishment of an epithelial barrier after injury [10]. Furthermore, Fos is known to be phosphorylated and activated by ERK during wing vein patterning and neuronal differentiation [11]. Therefore, Holn1 is probably acting in the ERK/Grh/Fos pathway upstream of Ddc and Msn and downstream of ERK during wound healing (Fig 7). This is in agreement with the nuclear localization of GFP-Holn1 protein and its predicted role in mRNA splicing. In the canonical EGFR/Ras/ERK pathway, the activated form of ERK translocates to the nucleus and phosphorylates its targets, thereby inducing gene expression [34]. Human and yeast homologues of Holn1 are known to associate with spliceosome components during early stages of spliceosome assembly [19], [17]. The initial response to wounding appears to involve the transcription of several genes [5], [6]. It is therefore conceivable that Holn1 contributes to efficient wound healing by acting in the nucleus to promote the splicing of components involved in the ERK/Grh/Fos pathway. Consistent with an important role of mRNA splicing in regulating the ERK pathway, a recent report has shown that the exon junction complex (EJC) is required for the splicing of specific introns in the ERK/rl gene, and that knockdown of components of the EJC lead to an overall reduction in ERK expression [36]. We show that Holn1 knockdown does not seem to affect ERK phosphorylation levels indicating that Holn1 probably does not play an essential role in ERK splicing. However, it is possible that our immunostaining assay is not sensitive enough to detect a subtle change in ERK protein levels. To clarify this, it would be necessary to use a quantitative method to measure ERK expression levels, such as real time PCR. It has also been shown that different splicing variants of Grh are expressed during embryonic development [37]. Holn1 could also be involved in the splicing of this transcription factor during wound healing and during development in general, yielding holn1 mutant embryos without certain Grh isoforms that may be required for a rapid response. Another hypothesis is that Holn1 works in a distinct pathway that acts in parallel to the ERK/Grh/Fos pathway. One could begin to address this challenging question by testing if holn1 mutations affect the expression levels and/or splicing patterns of known components of the ERK/Grh/Fos pathway. Another possible approach would be to determine Holn1 protein-protein interaction partners in the Drosophila embryo using tagged Holn1 to immunoprecipitate associated proteins and determine their identity with mass spectrometry analysis.
We have shown that Holn1 is not required for the initial rapid response to wound infliction, i.e. the formation of the actomyosin cable within minutes of wounding (this work and [2]) and the phosphorylation of ERK, which is also detectable soon after wounding (this work and [5]). This observation is consistent with Holn1 playing an indirect role in the mechanics of wound closure by regulating the mRNA levels of genes required for this process, such as those involved in rapid and productive cable contraction. Interestingly, the actin cable was present in all the wound closure mutants isolated in our previous screen that we have analyzed in more detail, suggesting that regulatory events downstream of cable formation dominate the wound closure process [9]. In any case, it is clear that Holn1 is required to perform some additional function needed to sustain the closure process, as holn1 mutants take on average 1.5 times longer to close a wound compared to wild type embryos. A similar delay in wound closure was previously reported for rho1 GTPase mutants, which do not form an actin cable, but can still close small wounds, albeit 2 times slower than wild type embryos [2]. Aside from its possible role in the epithelial hole closure process, Holn1 could also be involved in cuticle repair. As mentioned above, Grh and ERK activity are required for the re-establishment of the epithelial permeability barrier after injury [10]. Thus, Holn1 might be involved in this process by regulating the ERK/Grh pathway.
In the future, just as the holn1c07150 mutation uncovered a connection with the EGF/Ras/ERK signaling pathway and wound healing, microarray analysis of wounded holn1c07150 embryos would identify genes that are likely activated downstream of this wound closure pathway. Performing the same experiment using an alternative splicing array as in [38] would further reveal if Holn1 plays a role in wound dependent splicing events.
Materials and Methods
Fly strains and genetics
Crosses were performed at 25°C on standard medium. The w1118 strain was used as control. All strains used were purchased from Bloomington Stock Center (Indiana, USA) unless stated otherwise. piggyBac(PB)CG5198c07150/CyO and piggyBac(SAstopDsRed)LL07287/CyO (Drosophila Genetic Resource Center, Kyoto; [23], renamed respectively holn1c0715 0 and holn1LL07287 in this work, were rebalanced with CyO-CTG.
Remobilization of the transposable element present in holn1c07150 was performed as previously described [21]. The revertant line was confirmed by PCR and sequencing, and homozygous flies were viable.
The following recombinants and stocks were generated using standard methods: 1) For the rescue experiments, holn1c07150,e22c>gal4/CTG; holn1c07150,pUASp>holn1(6M)/CTG; holn1c07150/CyO; da>gal4/TM6b. 2) For genetic interaction tests: sev>Ras1.V12. 3) For Gal4 expression: actin5C>gal4/TM6b (strong ubiquitous expression), e22c>gal4 (ubiquitous expression starting at stage 12, stronger in the ectoderm), ms1096>gal4 (wing disc/adult wing), da>gal4 (moderate ubiquitous expression). 4) For live reporter lines: UAS>cherry-moesin/CTG to label actin [39], sqh-GFP42 to label myosin [40], and Ddc-GFP and Msn-DsRed for wound reporters [5], [8]. UAS>cherry-moesin/CTG, sqh-GFP42 and Msn-DsRed transgenes were recombined with holn1c07150 mutation and for Ddc-GFP the holn1c07150/CTG; Ddc-GFP stock was generated.
The homozygous escaper fly eyes depicted in figure 4A arise from recombinant stock holn1c07150, P(neoFRT)40A when grown at 18°C, generated using standard methods. The ERK/rl10a allele was kindly provided by E. Hafen.
Generation of Holn1 transgenics
To generate pUASp>holn1 rescue construct/transgenic flies, cDNA of holn1 coding region was amplified using Pfu Taq polymerase (Promega) from plasmid GH13760 (DGRC) using primers: forward = TCctcgagCTTGATAAAATGGCGAGCAAAAG (adding a Xho1 site at the 5′end) and reverse = TCtctagaCTACAAGTACAAATCGAAATCTATG (adding an Xba1 site at the 3′end). This fragment was cloned into Topo vector pCR2.1 (Invitrogen) and then subcloned into pUASp vector utilizing XbaI and NotI restriction sites. Colonies were tested by PCR using the above primers. One colony was selected and presence of CG5198 insert was confirmed with restriction digest. This construct was used to obtain transgenic flies (BestGene Inc, USA). The obtained homozygous viable fly strain pUASp>holn1 (6M) was used for recombination with the mutant chromosome holn1c07150.
The GFP-Holn1 construct was created using the Gateway® Technology with Clonase™ II (Invitrogen). holn1 attB-flanked DNA was amplified from plasmid GH13760 (DGRC) using primers For = ggggacaagtttgtacaaaaaagcaggcttcatggcgagcaaaagaaagc and rev = ggggacccatttgtacaagaaagctggctcctacaagtacaaatcg and recombined into an attP-containing donor vector to generate an entry holn1 clone, using BP Clonase™ II (Invitrogen). This entry clone was next recombined with pPGW destination vector from the Drosophila GatewayTM Vector Collection developed by the Murphy lab, using LR Clonase™ II (Invitrogen). The resulting construct with the pUASp promoter sequence and GFP tag sequence directly upstream of the holn1 start codon was sequence verified and sent to BestGene Inc (USA) for injection. Transformants were selected by eye color and GFP expression.
Transgenic RNAi
Transgenic fly stocks containing Gal4 inducible inverted repeat constructs specifically targeting holn1 were obtained from the Vienna Drosophila Research Center (VDRC), stocks 27369, 27370 (not shown, results identical to 27369, although slightly less penetrant), and 110281. All three lines used gave similar results, have no predicted off target hits and are pupal lethal when expressed using actin5c>gal4 [27] (VDRC and this work). As a control we used a transgenic fly expressing an inverted repeat construct directed against GFP (pUAS>Avic-GFP.dsRNA.R). Expression of this construct ablates expression of NLS-GFP at both 25°C and 29°C (S. Prag, unpublished observations). Flies homozygous for the RNAi constructs were crossed to flies expressing ms1096>gal4 or actin5c>gal4/TM3-TTG and selected against GFP and Sb for analysis. All RNAi crosses were performed at 29°C or 18°C.
Wing preparations and dissection of late pupa
For wing preparations, 1 to 3 day old flies were anesthetized and placed in 95% ethanol. Wings were dissected in ethanol with forceps, transferred to 100% isopropanol on a glass slide, dried briefly, and covered with a drop of Euparal mountant (Fischer Scientific). Pupae of the proper genotype were identified using GFP marked balancer selection and stuck to a slide with double-sided tape. The pupal case was carefully cut away using forceps.
Wounding assay
The wounding assay was performed on stage 15 embryos as described [9], except that the nitrogen laser-pumped dye laser was connected to an Andor Revolution spinning disc confocal microscope (Andor Technology).
In situ hybridization and immunochemistry
Standard protocol for in vitro transcription of DIG labeled mRNA (using DIG RNA labeling mix, Roche) was followed using linearized holn1 cDNA (clone GH13760, DGRC) as a template. Whole-mount in situ hybridization was carried out using standard methods [41].
For whole-mount immunochemistry, stage 15 embryos were fixed and stained essentially as described [9], except that fixation was performed in 4% formaldehyde in phosphate buffer (PBS) and blocking was performed over night. We used the following primary antibodies: mouse anti-armadillo 1:50 (Developmental Studies Hybridoma Bank), and rabbit anti-dpERK 1:100 (Santa Cruz Biotechnology). Secondary antibodies and probes used were: Alexa anti-rabbit 488 1:250, Alexa anti-mouse 568, Alexa 594 Phalloidin 1:200 (Invitrogen), and DAPI 1 µg/ml (Sigma-Aldrich).
Confocal imaging
For live imaging, embryos were collected as described [9], and mounted on their ventral side on glass bottom culture dishes (MatTek Corporation; USA) coated with double-sided tape, on Halocarbon carbon oil 700 (Sigma-Aldrich). Stage 15 live embryos were wounded as described above. The laser power used to wound control versus holn1 mutant embryos for live imaging was lower than the one used for 16 hour after wounding observation, in order to inflict smaller wounds that would close during imaging procedure. Imaging was performed at 25°C using an Andor Revolution spinning disc confocal microscope (Andor Technology). Individual Z-slices with a step size of 1 µm were taken every 1 minute for 4 hours. For imaging of Ddc-GFP and Msn-DsRed reporters [5], [8], embryos were allowed to develop 5 to 5.5 hours at 25°C after wounding. Percentage of embryos showing wound reporter activation was quantified as previously described [5]. Stained and wound reporter embryos were imaged using a Zeiss LSM 510 Meta or Zeiss LSM 710 confocal microscope, and scanned with 1 µm between slices. All images shown are Z-projections, except stated otherwise, and processed using ImageJ (NIH) and Photoshop (Adobe).
Scanning Electron Microscopy
Flies were anesthetized and frozen at −20°C for 30 minutes or −80°C for days. Fly heads were removed with a razor blade and mounted on a metal thumb-tac coated with nailpolish. Samples were then sputter coated with gold using a JEOL JFC-1200 machine and imaged within a few days on the Scanning Electron Microscope JEOL JSM-5200LV at 150 or 200X. Images were processed using Photoshop (Adobe).
Supporting Information
Percentage of dead embryos observed after wounding. The graph shows the percentage of dead embryos (unhatched larvae) observed 16 hours post wounding. holn1LL07287 homozygous show significantly higher percentage of dead embryos when compared to other genotypes, including holn1LL07287 heterozygotes and holn1c07150/holn1LL07287 transheterozygotes. Fisher's exact test showed significant different between groups (*<0.05**, p<0.01; ***, p<0.0001).
(TIF)
Time-lapse showing wound closure process in control e22c>cherry-moesin embryo depicted in Figure 3A . The first time point (t = 0min) was taken just before wounding. Actin cable formation starts around 10 minutes after wounding and appears to be completely formed and continuous before 20 minutes after wounding. The wound is closed at 110 minutes after wounding. Arrows point towards membrane protrusions. Images were taken in a spinning disc confocal microscope. Images are Z projections of 23 slices (total = 23 µm) scanned at 1-minute intervals. Scale bar = 20 µm.
(AVI)
Time-lapse showing wound closure process in the e22c>cherry-moesin,holn1c07150 mutant embryo depicted in Figure 3B . The first time point (t = 0 min) was taken just before wounding. Actin cable formation starts around 15 minutes after wounding and appears to be completely formed and continuous before 30 minutes after wounding. The wound is closed at 200 minutes after wounding. Arrows point towards membrane protrusions. Images were taken in a spinning disc confocal microscope. Images are Z projections of 23 slices (total = 23 µm) scanned at 1-minute intervals. Scale bar = 20 µm.
(AVI)
Acknowledgments
We would like to thank W. McGinnis for the Ddc-GFP and Msn-DsRed transgenic lines, and to E. Hafen for the rl10a mutant. We are grateful to Ângela Dias and Ana Cláudia Nunes for precious technical help, to the IMM bioimaging facility for microscopy support and to Telmo José Gonçalves Nunes for scanning electron microscopy assistance. We also thank S. Prag and J. Cordeiro for critically reading this manuscript.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by Fundação para a Ciência e a Tecnologia (FCT) PTDC/SAU/OBD/65872/2006, European Research Fund, ERC-208631-RESEAL, and Marie Curie/FP7. JAG was supported by a FCT fellowship, SFRH/BD/21895/2005, LC by a FCT fellowship, SFRH/BPD/62896/2009, and by Marie Curie Intra-European Fellowship, IC by a FCT fellowship, SFRH/BPD/17729/2004, and ACS by a FCT fellowship, SFRH/BPD/24976/2005. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Garcia-Fernandez B, Campos I, Geiger J, Santos AC, Jacinto A. Epithelial resealing. Int J Dev Biol. 2009;53:1549–56. doi: 10.1387/ijdb.072308bg. [DOI] [PubMed] [Google Scholar]
- 2.Wood W, Jacinto A, Grose R, Woolner S, Gale J, et al. Wound healing recapitulates morphogenesis in Drosophila embryos. Nature Cell Biol. 2002;4:907–12. doi: 10.1038/ncb875. [DOI] [PubMed] [Google Scholar]
- 3.Galko MJ, Krasnow MA. Cellular and genetic analysis of wound healing in Drosophila larvae. PLoS Biol. 2004;2:E239. doi: 10.1371/journal.pbio.0020239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li G, Gustafson-Brown C, Hanks SK, Nason K, Arbeit JM, et al. c-Jun is essential for organization of the epidermal leading edge. Dev Cell. 2003;4:865–77. doi: 10.1016/s1534-5807(03)00159-x. [DOI] [PubMed] [Google Scholar]
- 5.Mace KA, Pearson JC, McGinnis W. An epidermal barrier wound repair pathway in Drosophila is mediated by grainy head. Science. 2005;308:381–5. doi: 10.1126/science.1107573. [DOI] [PubMed] [Google Scholar]
- 6.Rämet M, Lanot R, Zachary D, Manfruelli P. JNK signaling pathway is required for efficient wound healing in Drosophila. Dev Biol. 2002;241:145–56. doi: 10.1006/dbio.2001.0502. [DOI] [PubMed] [Google Scholar]
- 7.Ting SB, Caddy J, Hislop N, Wilanowski T, Auden A, et al. A homolog of Drosophila grainy head is essential for epidermal integrity in mice. Science. 2005;308:411–413. doi: 10.1126/science.1107511. [DOI] [PubMed] [Google Scholar]
- 8.Pearson JC, Juarez MT, Kim M, Drivenes Ø, McGinnis W. Multiple transcription factor codes activate epidermal wound-response genes in Drosophila. Proc Natl Acad Sci USA. 2009;106:2224–9. doi: 10.1073/pnas.0810219106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campos I, Geiger JA, Santos AC, Carlos V, Jacinto A. Genetic screen in Drosophila melanogaster uncovers a novel set of genes required for embryonic epithelial repair. Genetics. 2010;184:129–40. doi: 10.1534/genetics.109.110288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim M, McGinnis W. Proc Natl Acad Sci USA; 2010. Phosphorylation of Grainy head by ERK is essential for wound-dependent regeneration but not for development of an epidermal barrier. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ciapponi L, Jackson DB, Mlodzik M, Bohmann D. Drosophila Fos mediates ERK and JNK signals via distinct phosphorylation sites. Genes Dev. 2001;15:1540–53. doi: 10.1101/gad.886301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liaw GJ, Rudolph KM, Huang JD, Dubnicoff T, Courey a J, et al. The torso response element binds GAGA and NTF-1/Elf-1, and regulates tailless by relief of repression. Genes Dev. 1995;9:3163–3176. doi: 10.1101/gad.9.24.3163. [DOI] [PubMed] [Google Scholar]
- 13.Wang S, Tsarouhas V, Xylourgidis N, Sabri N, Tiklová K, et al. The tyrosine kinase Stitcher activates Grainy head and epidermal wound healing in Drosophila. Nature Cell Biol. 2009;11:890–5. doi: 10.1038/ncb1898. [DOI] [PubMed] [Google Scholar]
- 14.Freund C, Kühne R, Yang H, Park S, Reinherz EL, et al. Dynamic interaction of CD2 with the GYF and the SH3 domain of compartmentalized effector molecules. EMBO J. 2002;21:5985–95. doi: 10.1093/emboj/cdf602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishizawa K, Freund C, Li J, Wagner G, Reinherz EL. Identification of a proline-binding motif regulating CD2-triggered T lymphocyte activation. Proc Natl Acad Sci USA. 1998;95:14897–902. doi: 10.1073/pnas.95.25.14897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stroschein-Stevenson SL, Foley E, O'Farrell PH, Johnson AD. Identification of Drosophila gene products required for phagocytosis of Candida albicans. PLoS Biol. 2006;4:e4. doi: 10.1371/journal.pbio.0040004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laggerbauer B, Liu S, Makarov E, Vornlocher H-P, Makarova O, et al. The human U5 snRNP 52K protein (CD2BP2) interacts protein with U5-102K (hPrp6), a U4/U6.U5 tri-snRNP bridging protein, but dissociates upon tri-snRNP formation. RNA. 2005;11:598–608. doi: 10.1261/rna.2300805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kofler M, Motzny K, Beyermann M, Freund C. Novel interaction partners of the CD2BP2-GYF domain. J Biol Chem. 2005;280:33397–402. doi: 10.1074/jbc.M503989200. [DOI] [PubMed] [Google Scholar]
- 19.Kofler M, Heuer K, Zech T, Freund C. Recognition sequences for the GYF domain reveal a possible spliceosomal function of CD2BP2. J Biol Chem. 2004;279:28292–28297. doi: 10.1074/jbc.M402008200. [DOI] [PubMed] [Google Scholar]
- 20.Heinze M, Kofler M, Freund C. Investigating the functional role of CD2BP2 in T cells. Int Immunol. 2007;19:1313–8. doi: 10.1093/intimm/dxm100. [DOI] [PubMed] [Google Scholar]
- 21.Thibault ST, Singer MA, Miyazaki WY, Milash B, Dompe NA, et al. A complementary transposon tool kit for Drosophila melanogaster using P and piggyBac. Nat Genet. 2004;36:283–287. doi: 10.1038/ng1314. [DOI] [PubMed] [Google Scholar]
- 22.Kofler MM, Freund C. The GYF domain. FEBS J. 2006;273:245–56. doi: 10.1111/j.1742-4658.2005.05078.x. [DOI] [PubMed] [Google Scholar]
- 23.Schuldiner O, Berdnik D, Levy JM, Wu JS, Luginbuhl D, et al. piggyBac-based mosaic screen identifies a postmitotic function for cohesin in regulating developmental axon pruning. Dev Cell. 2008;14:227–38. doi: 10.1016/j.devcel.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karim F, Chang HC, Themen M, Wassarman DA, Laverty T, et al. A Screen for Genes That Function Downstream of Rasl During Drosophila Eye Development. Genetics. 1996;143:315–329. doi: 10.1093/genetics/143.1.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Culí J, Martín-Blanco E, Modolell J. The EGF receptor and N signalling pathways act antagonistically in Drosophila mesothorax bristle patterning. Development. 2001;128:299–308. doi: 10.1242/dev.128.2.299. [DOI] [PubMed] [Google Scholar]
- 26.Shilo B-Z. Regulating the dynamics of EGF receptor signaling in space and time. Development. 2005;132:4017–27. doi: 10.1242/dev.02006. [DOI] [PubMed] [Google Scholar]
- 27.Dietzl G, Chen D, Schnorrer F, Su K-C, Barinova Y, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- 28.Sturtevant MA, Roark M, Bier E. The Drosophila rhomboid gene mediates the localized formation of wing veins and interacts genetically with components of the EGF-R signaling pathway. Genes Dev. 1993;7:961–73. doi: 10.1101/gad.7.6.961. [DOI] [PubMed] [Google Scholar]
- 29.Gabay L, Seger R, Shilo BZ. MAP kinase in situ activation atlas during Drosophila embryogenesis. Development. 1997;124:3535–41. doi: 10.1242/dev.124.18.3535. [DOI] [PubMed] [Google Scholar]
- 30.Dieckgraefe BK, Weems DM, Santoro SA, Alpers DH. ERK and p38 MAP kinase pathways are mediators of intestinal epithelial wound-induced signal transduction. Biochem Biophys Res Commun. 1997;233:389–94. doi: 10.1006/bbrc.1997.6469. [DOI] [PubMed] [Google Scholar]
- 31.Wright TRF. Phenotypic Analysis of the Dopa decarboxylase Gene Cluster Mutants in Drosophila melanogaster. J Hered. 1996;87:175–190. doi: 10.1093/oxfordjournals.jhered.a022983. [DOI] [PubMed] [Google Scholar]
- 32.Clifford R, Schüpbach T. Molecular analysis of the Drosophila EGF receptor homolog reveals that several genetically defined classes of alleles cluster in subdomains of the receptor protein. Genetics. 1994;137:531–50. doi: 10.1093/genetics/137.2.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Biggs WH, Zavitz KH, Dickson B, Straten A van der, Brunner D, et al. The Drosophila rolled locus encodes a MAP kinase required in the sevenless signal transduction pathway. EMBO J. 1994;13:1628–35. doi: 10.1002/j.1460-2075.1994.tb06426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shilo B. Signaling by the Drosophila epidermal growth factor receptor pathway during development. Exp Cell Res. 2003;284:140–149. doi: 10.1016/s0014-4827(02)00094-0. [DOI] [PubMed] [Google Scholar]
- 35.Lesokhin M, Yu SY, Katz J, Baker NE. Several levels of EGF receptor signaling during photoreceptor specification in wild-type, Ellipse, and null mutant Drosophila. Dev Biol. 1999;205:129–44. doi: 10.1006/dbio.1998.9121. [DOI] [PubMed] [Google Scholar]
- 36.Roignant J-Y, Treisman JE. Exon Junction Complex Subunits Are Required to Splice Drosophila MAP Kinase, a Large Heterochromatic Gene. Cell. 2010;143:238–250. doi: 10.1016/j.cell.2010.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uv AE, Harrison EJ, Bray SJ. Tissue-specific splicing and functions of the Drosophila transcription factor Grainyhead. Mol Cell Biol. 1997;17:6727–35. doi: 10.1128/mcb.17.11.6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blanchette M, Green RE, Brenner SE, Rio DC. Global analysis of positive and negative pre-mRNA splicing regulators in Drosophila. Genes Dev. 2005;19:1306–1314. doi: 10.1101/gad.1314205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Millard TH, Martin P. Dynamic analysis of filopodial interactions during the zippering phase of Drosophila dorsal closure. Development. 2008;135:621–6. doi: 10.1242/dev.014001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Royou A, Field C, Sisson JC, Sullivan W, Karess R. Reassessing the Role and Dynamics of Nonmuscle Myosin II during Furrow Formation in Early Drosophila Embryos. Mol Biol Cell 15: 2004;838 - 850 doi: 10.1091/mbc.E03-06-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lehmann R, Tautz D. In Situ Hybridization to RNA. Methods Cell Biol. 1994;44:597–598. doi: 10.1016/s0091-679x(08)60933-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Percentage of dead embryos observed after wounding. The graph shows the percentage of dead embryos (unhatched larvae) observed 16 hours post wounding. holn1LL07287 homozygous show significantly higher percentage of dead embryos when compared to other genotypes, including holn1LL07287 heterozygotes and holn1c07150/holn1LL07287 transheterozygotes. Fisher's exact test showed significant different between groups (*<0.05**, p<0.01; ***, p<0.0001).
(TIF)
Time-lapse showing wound closure process in control e22c>cherry-moesin embryo depicted in Figure 3A . The first time point (t = 0min) was taken just before wounding. Actin cable formation starts around 10 minutes after wounding and appears to be completely formed and continuous before 20 minutes after wounding. The wound is closed at 110 minutes after wounding. Arrows point towards membrane protrusions. Images were taken in a spinning disc confocal microscope. Images are Z projections of 23 slices (total = 23 µm) scanned at 1-minute intervals. Scale bar = 20 µm.
(AVI)
Time-lapse showing wound closure process in the e22c>cherry-moesin,holn1c07150 mutant embryo depicted in Figure 3B . The first time point (t = 0 min) was taken just before wounding. Actin cable formation starts around 15 minutes after wounding and appears to be completely formed and continuous before 30 minutes after wounding. The wound is closed at 200 minutes after wounding. Arrows point towards membrane protrusions. Images were taken in a spinning disc confocal microscope. Images are Z projections of 23 slices (total = 23 µm) scanned at 1-minute intervals. Scale bar = 20 µm.
(AVI)