

Abstract
Nitric oxide (NO) has been widely recognized as a positive regulator of tumorigenesis and cancer progression through its ability to regulate important proteins in various signal transduction pathways. S-nitrosylation, or covalent attachment of NO to protein sulphydryl groups, has gained prominence as an important mechanism by which NO modulates physiologic and pathologic cellular responses. In this article, we discuss S-nitrosylation of two key apoptosis-regulatory proteins of the intrinsic and extrinsic death pathways, namely B-cell lymphoma-2 (Bcl-2) and FLICE-inhibitory protein (FLIP). These proteins have been shown to be upregulated in a variety of tumors and have been implicated with cancer chemoresistance through dysregulation of apoptosis. S-nitrosylation of these proteins precludes their ubiquitination and subsequent degradation by the proteasome, thus accentuating their anti-apoptotic effect which is critical in the context of tumorigenic potential and cancer progression. We propose that such post-translational modifications of proteins by NO may be a general mechanism that tumor cells exploit to tilt the scales towards survival and proliferation by evading cell death.
Keywords: nitric oxide, S-nitrosylation, apoptosis, FLIP, Bcl-2, ubiquitination
Introduction
Nitric oxide (NO) is a short-lived gaseous free radical which predominantly functions as a messenger and effector molecule. NO is produced by many mammalian cells and is critical for numerous biological processes, including apoptosis [1-3]. Consequently, NO becomes an important factor in cancer development as apoptosis is impaired during carcinogenesis [2, 3]. In mammals, NO is synthesized by a family of NO synthases (NOS) which exist in several isoforms, including neuronal (type I or nNOS), inducible (type II or iNOS), and endothelial (type III or eNOS) [4, 5]. Of these, types I and III are found to constitutively express NO at low levels in a calcium-dependent manner [6], whereas as type II, which is induced primarily by inflammatory cytokines and gram-negative endotoxins, produces relatively high amounts of NO and is independent of intracellular calcium levels [4]. Interestingly, various studies have shown that all three isoforms of NOS are involved in promoting or inhibiting the etiology of cancer.
NOS activity has been associated with tumor grade, proliferation rate and expression of important signaling components associated with cancer development [7]. The presence of NOS has been assessed in a various human malignant tumors including brain, breast, lung, prostate, bladder and pancreatic carcinomas, mesotheliomas, Kaposi's sarcoma, salivary tumors and oral squamous cell carcinomas [7-10]. Various studies have shown NO to be an important component of the tumor microenvironment, where it is produced either by tumor cells, endothelial cells in the tumor microvasculature, or stromal cells within the tumors [11]. Due to its lipophilic nature, NO can rapidly cross cell membranes and enter intracellular compartments, where it can exert its action even when produced by a neighboring cell, thus mediating interactions between tumor and host cells. However, the functional role of NO in tumor biology is complex and remains to be fully defined.
Research over the past couple of decades suggests that NO can have opposite effects, depending upon the level of NO induction, the temporal-spatial mode of NO action, intracellular targets of NO, and other environmental and pathophysiological conditions. While a number of reports indicate that the presence of NO in tumor cells or in their microenvironment is detrimental to tumor cell survival and metastatic ability, numerous clinical and experimental studies suggest a promoting role of NO in tumor progression and metastasis [1]. Thus, a dichotomy in effects of NO is observed, leading researchers to suggest that NO may exert a biphasic response such that when NO levels go beyond a critical concentration that would be suitable for tumor growth and survival, growth arrest and/or apoptotic/necrotic pathways are initiated [12].
Although NO has been shown to have a direct effect on secondary metastasis, it can also promote carcinogenesis, leading to formation of tumors due to transformation of normal cells [13]. This is because NO has been shown to exert pleitropic effects including cell proliferation, smooth-muscle relaxation, neurotransmission, toxicity, differentiation and apoptosis, all critical factors which when dysregulated may lead to tumorigenesis [14]. The redox state and chemistry of NO facilitate its interaction with various proteins thus regulating various intracellular and intercellular signaling events [15]. Recent evidence indicate that NO activates a complex network of responses leading to apoptosis via mitochondrial, death receptor, p38/mitogen-activated protein kinase (p38 MAPK), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-Siah1 cascades [16-19]. The anti-apoptotic effect of NO can be mediated through a number of mechanisms, such as caspase inactivation, induction of cellular tumor antigen p53 (p53) gene expression, upregulation of cellular FLIP, and overexpression of Bcl-2 and Bcl-XL with subsequent inhibition of cytochrome c release from the mitochondria [20-25]. One of the key mechanisms by which NO regulates the function of various target proteins is through S-nitrosylation [15].
Protein S-nitrosylation
The reversible coupling of a nitroso moiety to a reactive cysteine thiol leads to the formation of S-nitrosothiol (SNO), the process commonly known as S-nitrosylation [26-27]. Although protein modifications induced by NO were initially detected only on protein-bound transition metals (such as that observed for the haem Fe+2 of guanylate cyclase), research over the past decade has shown that the principal target of cellular NO is the thiol group of cysteine residues contained in signature motifs of substrate proteins and peptides, and that NO substantially regulates numerous signal transduction pathways via S-nitrosylation of proteins [28-29]. For instance, NO inhibits the function of nuclear factor-kappa B (NF-κB), c-Jun N-terminal kinase (JNK) and protein kinase C and activates Ras and ryanodine receptor via S-nitrosylation [30-34]. However, substrate specificity that dictates the propensity of certain cysteine residues to get nitrosylated (or de-nitrosylated) over others depends upon a number of factors including the electrostatic environment, orientation of aromatic residues, hydrophobicity, proximity of target thiols to redox centers and protein-protein interactions [29, 35]. Also, the source of the coupled NO moiety could be provided by NO itself, other NO oxides, metal-NO complexes or various SNOs. Although specific enzymes that exclusively mediate S-nitrosylation have not been characterized, several agents have been identified that play an important role in catalysis of this process [36]. Such post-translational modification of proteins can positively or negatively regulate various signaling pathways, proteins, and metabolic processes [29, 35]. One of the most important processes regulated by S-nitrosylation is apoptosis [37].
S-nitrosylation and apoptosis
Apoptosis is a highly regulated process leading to cell death characterized by cell shrinkage, membrane blebbing and DNA fragmentation [38]. Caspases, a family of cysteine proteases, are central regulators of apoptosis. Caspase-8 and -9 are key pro-apoptotic proteins of the two major pathways regulating apoptosis, viz., the extrinsic (death receptor) pathway and the intrinsic (mitochondria) pathway. The extrinsic pathway of apoptosis is induced via signaling through a family of death receptors including Fas (CD95/APO-1) and tumor necrosis factor receptor-1 (TNFR-1). Death receptor ligands characteristically initiate signaling by recruiting specialized adaptor proteins such as Fas-associated death domain (FADD) to their cytosolic death domains, resulting in the recruitment of procaspase-8 to the death-inducing signaling complex (DISC) causing activation of caspase-8 [39-41]. Activated caspase-8 stimulates apoptosis through the activation of effector caspase-3 either directly or indirectly through the intrinsic death pathway. The intrinsic pathway is induced by pro-apoptotic Bcl-2 family of proteins such as Bid, Bax and Bad that reside in the cytosol but translocate to the mitochondria following death signaling. In the mitochondria, these proteins promote the release of cytochrome c, which then binds to apoptotic protease activating factor 1 (Apaf 1) and forms an activation complex (apoptosome) with procaspase-9 leading to its cleavage and activation in the form of caspase-9 [42-43]. Caspase-9 activates effector caspases such as caspase-3 leading to apoptosis.
The apoptotic pathways are regulated at different levels by various anti-apoptotic proteins including FLIP and Bcl-2. FLIP is the key regulator of the extrinsic death receptor pathway. This anti-apoptotic protein is recruited to the DISC upon stimulation where it prevents procaspase-8 recruitment/processing and subsequent induction of apoptosis through the extrinsic pathway [44-46]. Bcl-2 is a key apoptosis-regulatory protein of the mitochondrial death pathway [42]. Formation of heterodimers with pro-apoptotic proteins and inhibition of mitochondrial membrane transition and cytochrome c release are some of the mechanisms by which Bcl-2 exerts its anti-apoptotic effect [47-48]. Several studies have demonstrated that the overexpression of Bcl-2 and FLIP increases resistance to apoptotic cell death induced by various DNA-damaging agents, which is an important feature of malignant cells [49-51].
The anti-apoptotic function of these proteins is closely associated with their expression levels which are regulated by various mechanisms, including dimerization, transcription, post-translational modification, and degradation. Degradation of these anti-apoptotic proteins is mainly mediated via the ubiquitin-proteasomal pathway, which is a major system for selective protein removal in eukaryotic cells [52, 53]. Accumulating evidence also suggests that this degradation pathway is a key pathway of apoptosis regulation by the anti-apoptotic proteins under various apoptosis and stress conditions (20-22, 52). The degradation process involves selective modification of ε-NH2 groups of lysine residues in the proteins by ubiquitination which targets them for degradation by the proteasome complex [52-53]. Various factors such as specific structural features, phosphorylation or a partially conserved sequence motif are implicated in making proteins susceptible to degradation [54]. However, the physiological signals that lead to protein recognition by ubiquitin and degradation by proteasome are unclear.
Over the past few years, our group has focused on the role of NO in apoptosis regulation. As mentioned earlier, NO is known to regulate apoptosis through S-nitrosylation of various pro- and anti-apoptotic proteins. For instance, nitrosylation of caspases including caspase-8, caspase-9 and caspase-3 at their active site has been shown to inhibit their enzymatic activity [24, 55]. Here, we present two case studies dealing with S-nitrosylation of key anti-apoptotic proteins of the death receptor and mitochondrial pathways, namely FLIP and Bcl-2 respectively, and discuss their role and mechanism of apoptosis regulation as well as their linkage to carcinogenesis.
Case Study I: NO and FLIP (death receptor pathway)
As mentioned earlier, FLIP is a key apoptosis-regulatory protein of the death receptor pathway. FLIP is involved in rendering cells resistant to death receptor-mediated apoptosis in various cell types [44, 56-58] and elevated expression of this protein has been associated with tumor cells that can escape from immune surveillance in vivo [59]. Furthermore, downregulation of FLIP by cytotoxic agents has been shown to sensitize cells to death receptor-mediated apoptosis [60]. Thus, FLIP represents a potential crucial step in tumorigenesis and a promising target for drug development against cancer.
Activation of Fas (CD95) death receptor by its ligand (FasL) induces apoptotic cell death of susceptible cells. To study the role of NO in the apoptotic process and its regulatory mechanism, we analyzed the expression levels of key apoptosis proteins known to be involved in Fas death signaling, including Fas death receptor, FADD adaptor protein, and the antiapoptotic protein FLIP, in human lung epithelial cells following FasL stimulation [22]. Among these, only the level of FLIP was affected by the FasL treatment. FasL induced rapid downregulation of FLIP and its overexpression by ectopic gene transfection inhibited the apoptotic effect of FasL, indicating the important role of FLIP in the apoptotic process. FasL treatment also induced rapid generation of cellular NO and its inhibition by the NO scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethyl-imidazoline-1-oxy-3-oxide (PTIO) or iNOS inhibitor aminoguanidine (AG) strongly inhibited the downregulation of FLIP and apoptosis induced by FasL. In contrast, treatment of the cells with NO donor, sodium nitroprusside (SNP) or dipropylenetriamine (DPTA) NONOate, showed opposite effects. NO was further found to exert its effect on FLIP through inhibition of protein ubiquitination and proteasomal degradation which was induced by the FasL treatment. Therefore, our data suggested that NO exerted its anti-apoptotic effect by interfering with the FLIP degradation mechanism.
The mechanism by which NO inhibits FLIP degradation was shown to involve protein S-nitrosylation. To demonstrate this, cells expressing ectopic FLIP were treated with FasL in the presence or absence of NO modulators, and cell lysates were immunoprecipitated and analyzed by Western blot using anti-S-nitrosocysteine antibody. The result showed that the NO donor DPTA NONOate and SNP were able to induce S-nitrosylation of FLIP, whereas the NO inhibitors AG and PTIO inhibited the nitrosylation (Fig. 1). These results suggested that S-nitrosylation may be a key mechanism utilized by NO to regulate FLIP ubiquitination and proteasomal degradation. In order to delineate the mechanism of FLIP S-nitrosylation, we determined the domain(s) of FLIP which is responsible for its nitrosylation by constructing a series of FLIP deletion mutants (Δ1- Δ4). These mutants were individually transfected into cells and their S-nitrosylation by NO was determined in FasL-treated cells (Fig. 2). Although partial deletion of the caspase-like domain of FLIP (Δ1) showed no effect on S-nitrosylation induced by the NO donor SNP, complete deletion of this domain (Δ2) as well as the death effector domain 2 (Δ3) strongly inhibited the S-nitrosylation. Immunoprecipitation and ubiquitination studies further showed that the NO donor SNP was able to inhibit FasL-induced ubiquitination of FLIP and its Δ1 mutant, but not the Δ2 and Δ3 mutants (Fig. 3A), indicating the protective role of S-nitrosylation on FLIP ubiquitination.
Figure 1. S-nitrosylation of FLIP by NO.
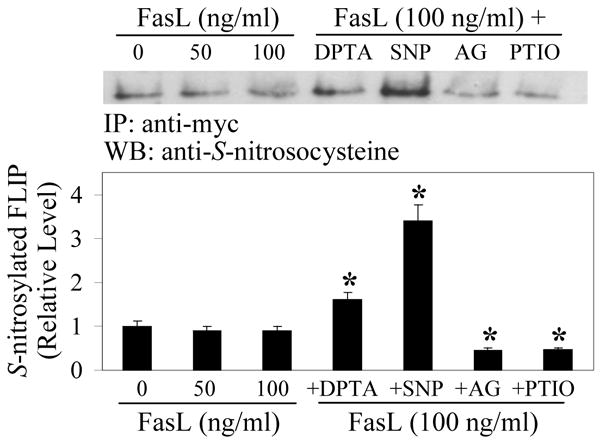
Human lung epithelial BEAS-2B cells were transiently transfected with myc-FLIP plasmid and treated 1 day later with FasL (0-100 ng/ml) in the presence or absence of the NO donor SNP (300 μg/ml) or DPTA NONOate (200 μM), or with the NO inhibitor AG (100 μg/ml) or PTIO (100 μM) for 2 h. Cell lysates (60 μg protein) were immunoprecipitated (IP) using myc antibody and analyzed by Western blot (WB) using S-nitrosocysteine antibody. The immunoblot signals were quantified by densitometry and mean data from independent experiments (one of which is shown here) were normalized to the result obtained in non-treated control. Plots are mean ± S.D. (n = 3). *p < 0.05 versus FasL-treated control. (Reproduced from ref. 22 with permission from the Journal of Biological Chemistry).
Figure 2. The caspase-like domain of FLIP is required for S-nitrosylation.

A, Schematic structures of FLIP and various constructs (Δ1-Δ4) used in this study. DED stands for death effector domain. Amino acids present in each construct are labeled. Asterisks indicate Cys254 and Cys259 to alanine mutations in the caspase-like domain. B, S-nitrosylation of FLIP and its mutants was analyzed by transient transfection and immunoprecipitation with myc antibody as described in Fig. 1. The density of S-nitrosylated bands was determined by densitometry and normalized against FasL-treated control bands. Plots are mean ± S.D. (n = 3). *p < 0.05 versus myc-FLIP-transfected control. (Reproduced from ref. 22 with permission from the Journal of Biological Chemistry).
Figure 3. S-nitrosylation of Cys254 and Cys259 inhibits FLIP ubiquitination and degradation induced by FasL.
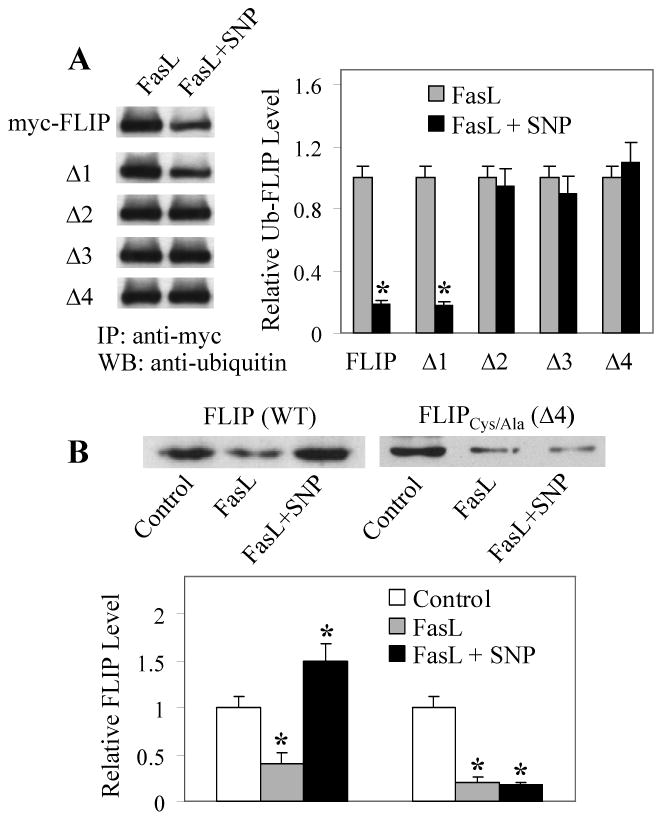
A, BEAS-2B cells were transiently transfected with ubiquitin and myc-FLIP or its mutant plasmids. One day after the transfection, cells were treated with FasL (100 ng/ml) in the presence or absence of SNP (300 μg/ml) for 2 h and cell lysates were prepared for immunoprecipitation using myc antibody. The immunoprecipitated proteins were analyzed by Western blot with antibody against ubiquitin. Band densities were normalized against FasL-treated controls. Plots are mean ± S.D. (n = 3). *p < 0.05 versus FasL-treated controls. B, Cells were transiently transfected with myc-FLIP or Δ4 mutant plasmid and then treated 1 day later with FasL (100 ng/ml) in the presence or absence of SNP (300 μg/ml) for 12 h. Cell lysates were immunoprecipitated with myc antibody and analyzed by Western blot using antibody against FLIP. Band densities were normalized against non-treated controls. Plots are mean ± S.D. (n = 3). *p < 0.05 versus non-treated controls. (Reproduced from ref. 22 with permission from the Journal of Biological Chemistry).
The results of this study also suggested that the amino acid sequence difference between Δ1 and Δ2 (or the amino acid sequence 233-328 of the caspase-like domain) was essential for the S-nitrosylation of FLIP. Since S-nitrosylation involves the transfer of NO group to cysteine residues, we mutated the two cysteines present in this region (Δ4) to determine whether these mutations interfere with the S-nitrosylation of FLIP. We found that mutations of these cysteine residues (Cys-254 and Cys-259) resulted in a complete inhibition of FLIP nitrosylation (Fig. 2B). Such mutations also inhibited the protective effect of NO on FLIP ubiquitination (Fig. 3A), supporting the role of S-nitrosylation in the ubiquitination process. As expected, our result showed that NO was able to prevent FLIP degradation by FasL, as indicated by its increased expression in the presence of the NO donor SNP (Fig. 3B). In contrast, the NO donor had no protective effect on the non-nitrosylable FLIP mutant (Δ4), suggesting that S-nitrosylation of FLIP, in addition to preventing ubiquitination, also protected this molecule from FasL-induced degradation.
Together, our data provide evidence that FasL can induce downregulation of FLIP through ubiquitin-proteasomal degradation. NO negatively regulates this process through its ability to nitrosylate the protein at Cys-254 and Cys-259, which prevents its degradation through the ubiquitin pathway. Since increased FLIP expression has been associated with refractory tumors that escape immune surveillance, and inhibition of FLIP is effective in rendering tumor cells responsive to chemotherapy, the knowledge gained from this study could have important implications in the pathogenesis of cancers and their treatment.
Case Study II: NO and Bcl-2 (mitochondrial pathway)
Bcl-2 is a key apoptosis-regulatory protein of the mitochondrial death pathway [42]. The oncogenic potential of Bcl-2 protein is well characterized with its overexpression reported in 70% of breast cancer, 30-60% of prostate cancer, and 90% of colorectal cancer [61-62]. Recent evidence have shown elevated expression levels of Bcl-2 and NO in various cancer cells [48-50, 62-64]; however, the potential role of NO in the regulation of Bcl-2 and the underlying mechanism are largely unknown. In our study, we investigated the role of NO in apoptotic cell death induced by the carcinogenic metal chromium [Cr(VI)] which has been shown to induce apoptosis through Bcl-2-dependent mitochondrial death pathway [20, 65]. Like FLIP, Bcl-2 was found to be downregulated through ubiquitin-proteasomal degradation and NO inhibited this degradation. To determine the role of S-nitrosylation in this process, cells expressing ectopic Bcl-2 were treated with Cr(VI) in the presence or absence of NO modulators, and cell lysates were immunoprecipitated and analyzed for S-nitrosylation by Western blotting. We found that Cr(VI) induced S-nitrosylation of Bcl-2 and this effect was inhibited by the NO inhibitor PTIO and AG, and enhanced by the NO donor SNP and DPTA NONOate (Fig. 4A). These results suggest that NO, through its ability to S-nitrosylate Bcl-2, may interfere with the ubiquitination process and inhibit proteasomal degradation of the protein. To test this possibility, cells were treated with a known inhibitor of S-nitrosylation, dithiothreitol (DTT), and its effects on S-nitrosylation and ubiquitination of Bcl-2 were examined. The results showed that DTT was able to prevent S-nitrosylation of Bcl-2 by Cr(VI) (Fig. 4A) and inhibited the effect of NO donors on Bcl-2 ubiquitination in Cr(VI)-treated cells (Fig. 4B). These results indicate that S-nitrosylation might be a key mechanism utilized by NO to regulate ubiquitination and degradation of Bcl-2. To confirm this finding and to determine the cysteine residue(s) involved in the S-nitrosylation process, we constructed Bcl-2 mutant plasmids replacing the two cysteines in Bcl-2 with alanines (C158A and C229A) and introduced them into cells by gene transfection. The results showed that mutations of one or both cysteine residues completely inhibited the S-nitrosylation of Bcl-2 by Cr(VI) (Fig. 5A). Such mutations also led to increased Bcl-2 ubiquitination (Fig. 5B), thus confirming that S-nitrosylation of Bcl-2 prevents its ubiquitination and subsequent degradation. S-nitrosylation of Bcl-2 was also found to be induced under various apoptotic and stress conditions, including exposure to death ligand and glutathione depletion [20], suggesting that it may be a general mechanism of apoptosis regulation under diverse conditions.
Figure 4. Effects of NO on S-nitrosylation and ubiquitination of Bcl-2.

A, Effect of NO modulators on Bcl-2 S-nitrosylation in Cr(VI)-treated cells. Human lung epithelial H460 cells overexpressing myc-Bcl-2 were either left untreated or pretreated with SNP (500 μg/ml), DPTA NONOate (400 μM), AG (300 μM), PTIO (300 μM), or DTT (10 mM) for 1 h. The cells were then treated with Cr(VI) (20 μM) for 3 h and cell lysates were prepared for immunoprecipitation using anti-myc antibody. The resulting immune complexes were analyzed by Western blot using S-nitrosocysteine antibody. Densitometry was performed to determine the relative S-nitrosocysteine levels after reprobing the membranes with anti-Bcl-2 antibody. B, Effect of NO modulators on Cr(VI)-induced Bcl-2 ubiquitination. Cells overexpressing myc-Bcl-2 were treated with the indicated test agents in the presence of lactacystin (10 μM) to prevent proteasome-mediated Bcl-2 degradation. Cell lysates were then immunoprecipitated with anti-myc antibody and the immune complexes were analyzed for ubiquitin. Data are mean ± S.D. (n = 4). *p < 0.05 versus Cr(VI)-treated controls. #p < 0.05 versus NO-modulated controls. (Reproduced from ref. 20 with permission from the Journal of Biological Chemistry).
Figure 5. Effect of cysteine mutations on S-nitrosylation and ubiquitination of Bcl-2.

A, H460 cells were transiently transfected with myc-tagged wild-type Bcl-2 plasmid or with myc-tagged C158A, C229A, or C158A/C229A mutant plasmid. Thirty six hours later, the cells were treated with or without Cr(VI) (20 μM) for 3 h and cell lysates were prepared for immunoprecipitation using anti-myc antibody. The immune complexes were analyzed for S-nitrosocysteine by Western blotting. Densitometry was performed to determine the relative S-nitrosocysteine levels after reprobing of the membranes with anti-Bcl-2 antibody. B, Immunoprecipitates were prepared as described above and analyzed for ubiquitin by Western blotting. Data are mean ± S.D. (n = 4). *p < 0.05 versus non-treated controls. (Reproduced from ref. 20 with permission from the Journal of Biological Chemistry).
S-nitrosylation and carcinogenesis
Resistance to apoptosis is a key feature of cancer cells and is involved in the pathogenesis of cancer. It is also a major cause of failure for many drug therapies against cancer. Molecular alterations that lead to apoptosis resistance can be initiated or promoted by S-nitrosylation of anti-apoptotic proteins such as Bcl-2 and FLIP. This process is mediated by NO and results in upregulation of the proteins which is observed in many types of tumors. Thus, S-nitrosylation conveys a key influence of NO on apoptosis signaling and may provide a key mechanism for apoptosis resistance and carcinogenesis. However, direct evidence linking S-nitrosylation and carcinogenesis are lacking. Our ongoing studies support the role of S-nitrosylation of anti-apoptotic proteins in the pathologic development of cancer (unpublished data). We observed that long-term exposure of non-tumorigenic lung epithelial cells to non-toxic concentrations of carcinogenic metal Cr(VI) led to apoptosis-resistant and malignant transformed phenotype. The transformed cells exhibited anchorage-independent growth, loss of contact inhibition, increased cell invasion and migration activities, key characteristics of cancer cells. These cells also exhibited increased NO production and elevated expression of S-nitrosylated Bcl-2, supporting the role of S-nitrosylation in the carcinogenic process. The increase in S-nitrosylated Bcl-2 level was also accompanied by a parallel increase in total Bcl-2 level, suggesting positive regulation of Bcl-2 by S-nitrosylation through protein stabilization. Since resistance to apoptosis is a hallmark of neoplastic evolution, selection of cells that are resistant to apoptotic cell death by S-nitrosylation may be a key determining factor in cancer progression.
Summary
NO has multitude effects in tumor biology and our studies support the role of NO in carcinogenesis through its ability to nitrosylate important anti-apoptotic proteins such as Bcl-2 and FLIP. These proteins are key regulators of apoptosis and their importance in the development of apoptosis resistance and tumorigenesis has been established. Since increased NO production and anti-apoptotic protein expression have been associated with several human tumors, NO may be one of the key regulators of cell death resistance and cancer development through its ability to S-nitrosylate the anti-apoptotic proteins. Our work provides insights into the molecular mechanisms of apoptosis resistance and carcinogenesis. Our finding on the novel function of NO through S-nitrosylation of anti-apoptotic proteins may have important implications in the treatment of cancer and its development.
Acknowledgments
This work was supported by the National Institutes of Health Grant R01- HL076340.
Abbreviations
- NO
nitric oxide
- Bcl-2
B-cell lymphoma-2
- FLIP
FLICE-inhibitory protein
- NOS
NO synthases
- SNO
S-nitrosothiol
- DNA
deoxyribonucleic acid
- NF-κB
nuclear factor-kappa B
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- p53
cellular tumor antigen p53
- DISC
death-inducing signaling complex
- FADD
Fas-associated death domain
- TNFR-1
tumor necrosis factor receptor-1
- Apaf 1
apoptotic protease activating factor 1
- PTIO
2-(4-carboxyphenyl)-4,4,5,5-tetramethyl-imidazoline-1-oxy-3-oxide
- AG
aminoguanidine
- SNP
sodium nitroprusside
- DPTA
dipropylenetriamine
- FasL
fas ligand
- Cr(VI)
hexavalent chromium
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mocellin S, Bronte V, Nitti D. Nitric oxide, a double edged sword in cancer biology: searching for therapeutic opportunities. Medicinal Research Reviews. 2007;27:317–352. doi: 10.1002/med.20092. [DOI] [PubMed] [Google Scholar]
- 2.Chang WC, Chapkin RS, Lupton JR. Predictive value of proliferation, differentiation and apoptosis as intermediate markers for colon tumorigenesis. Carcinogenesis. 1997;18:721–730. doi: 10.1093/carcin/18.4.721. [DOI] [PubMed] [Google Scholar]
- 3.Morin PJ, Vogelstein B, Kinzler KW. Apoptosis and APC in colorectal tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:7950–7954. doi: 10.1073/pnas.93.15.7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu W, Liu LZ, Loizidou M, Ahmed M, Charles IG. The role of nitric oxide in cancer. Cell Research. 2002;12:311–320. doi: 10.1038/sj.cr.7290133. [DOI] [PubMed] [Google Scholar]
- 5.Wink DA, Vodovotz Y, Laval J, Laval F, Dewhirst MW, Mitchell JB. The multifaceted roles of nitric oxide in cancer. Carcinogenesis. 1998;19:711–721. doi: 10.1093/carcin/19.5.711. [DOI] [PubMed] [Google Scholar]
- 6.Knowles RG, Moncada S. Nitric oxide synthases in mammals. The Biochemical Journal. 1994;298(Pt 2):249–258. doi: 10.1042/bj2980249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumour progression. Nature Reviews Cancer. 2006;6:521–534. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- 8.Thomsen LL, Miles DW. Role of nitric oxide in tumour progression: lessons from human tumours. Cancer Metastasis Reviews. 1998;17:107–118. doi: 10.1023/a:1005912906436. [DOI] [PubMed] [Google Scholar]
- 9.Lala PK, Orucevic A. Role of nitric oxide in tumor progression: lessons from experimental tumors. Cancer Metastasis Reviews. 1998;17:91–106. doi: 10.1023/a:1005960822365. [DOI] [PubMed] [Google Scholar]
- 10.Lancaster JR, Jr, Xie K. Tumors face NO problems? Cancer Research. 2006;66:6459–6462. doi: 10.1158/0008-5472.CAN-05-2900. [DOI] [PubMed] [Google Scholar]
- 11.Lala PK, Chakraborty C. Role of nitric oxide in carcinogenesis and tumour progression. Lancet Oncology. 2001;2:149–156. doi: 10.1016/S1470-2045(00)00256-4. [DOI] [PubMed] [Google Scholar]
- 12.Ridnour LA, Thomas DD, Donzelli S, Espey MG, Roberts DD, Wink DA, Isenberg JS. The biphasic nature of nitric oxide responses in tumor biology. Antioxidants & Redox Signaling. 2006;8:1329–1337. doi: 10.1089/ars.2006.8.1329. [DOI] [PubMed] [Google Scholar]
- 13.Sawa T, Ohshima H. Nitrative DNA damage in inflammation and its possible role in carcinogenesis. Nitric Oxide. 2006;14:91–100. doi: 10.1016/j.niox.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 14.Hirst D, Robson T. Targeting nitric oxide for cancer therapy. The Journal of Pharmacy & Pharmacology. 2007;59:3–13. doi: 10.1211/jpp.59.1.0002. [DOI] [PubMed] [Google Scholar]
- 15.Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T, Singel DJ, Loscalzo J. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:444–448. doi: 10.1073/pnas.89.1.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukuo K, Hata S, Suhara T, Nakahashi T, Shinto Y, Tsujimoto Y, Morimoto S, Ogihara T. Nitric oxide induces upregulation of Fas and apoptosis in vascular smooth muscle. Hypertension. 1996;27:823–826. doi: 10.1161/01.hyp.27.3.823. [DOI] [PubMed] [Google Scholar]
- 17.Hara MR, Snyder SH. Nitric oxide-GAPDH-Siah: a novel cell death cascade. Cellular & Molecular Neurobiology. 2006;26:527–538. doi: 10.1007/s10571-006-9011-6. [DOI] [PubMed] [Google Scholar]
- 18.Kuzushima M, Mogi M, Togari A. Cytokine-induced nitric-oxide-dependent apoptosis in mouse osteoblastic cells: involvement of p38MAP kinase. Archives of Oral Biology. 2006;51:1048–1053. doi: 10.1016/j.archoralbio.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 19.Li CQ, Robles AI, Hanigan CL, Hofseth LJ, Trudel LJ, Harris CC, Wogan GN. Apoptotic signaling pathways induced by nitric oxide in human lymphoblastoid cells expressing wild-type or mutant p53. Cancer Research. 2004;64:3022–3029. doi: 10.1158/0008-5472.can-03-1880. [DOI] [PubMed] [Google Scholar]
- 20.Azad N, Vallyathan V, Wang L, Tantishaiyakul V, Stehlik C, Leonard SS, Rojanasakul Y. S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal degradation. A novel antiapoptotic mechanism that suppresses apoptosis. The Journal of Biological Chemistry. 2006;281:34124–34134. doi: 10.1074/jbc.M602551200. [DOI] [PubMed] [Google Scholar]
- 21.Chanvorachote P, Nimmannit U, Stehlik C, Wang L, Jiang BH, Ongpipatanakul B, Rojanasakul Y. Nitric oxide regulates cell sensitivity to cisplatin-induced apoptosis through S-nitrosylation and inhibition of Bcl-2 ubiquitination. Cancer Research. 2006;66:6353–6360. doi: 10.1158/0008-5472.CAN-05-4533. [DOI] [PubMed] [Google Scholar]
- 22.Chanvorachote P, Nimmannit U, Wang L, Stehlik C, Lu B, Azad N, Rojanasakul Y. Nitric oxide negatively regulates Fas CD95-induced apoptosis through inhibition of ubiquitin-proteasome-mediated degradation of FLICE inhibitory protein. The Journal of Biological Chemistry. 2005;280:42044–42050. doi: 10.1074/jbc.M510080200. [DOI] [PubMed] [Google Scholar]
- 23.Delikouras A, Hayes M, Malde P, Lechler RI, Dorling A. Nitric oxide-mediated expression of Bcl-2 and Bcl-xl and protection from tumor necrosis factor-alpha-mediated apoptosis in porcine endothelial cells after exposure to low concentrations of xenoreactive natural antibody. Transplantation. 2001;71:599–605. doi: 10.1097/00007890-200103150-00004. [DOI] [PubMed] [Google Scholar]
- 24.Kim YM, Talanian RV, Billiar TR. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. The Journal of Biological Chemistry. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- 25.Mannick JB, Hausladen A, Liu L, Hess DT, Zeng M, Miao QX, Kane LS, Gow AJ, Stamler JS. Fas-induced caspase denitrosylation. Science. 1999;284:651–654. doi: 10.1126/science.284.5414.651. [DOI] [PubMed] [Google Scholar]
- 26.Stamler JS. Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 27.Stamler JS, Toone EJ, Lipton SA, Sucher NJ. (S)NO signals: translocation, regulation, and a consensus motif. Neuron. 1997;18:691–696. doi: 10.1016/s0896-6273(00)80310-4. [DOI] [PubMed] [Google Scholar]
- 28.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nature Reviews Molecular Cellular Biology. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 29.Stamler JS, Toone EJ, Lamas S, Fang FC. Nitrosylation. the prototypic redox-based signaling mechanism. Cell. 2001;106:675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 30.DelaTorre A, Schroeder RA, Kuo PC. Alteration of NF-kappa B p50 DNA binding kinetics by S-nitrosylation. Biochemical & Biophysical Research Communications. 1997;238:703–706. doi: 10.1006/bbrc.1997.7279. [DOI] [PubMed] [Google Scholar]
- 31.Gopalakrishna R, Chen ZH, Gundimeda U. Nitric oxide and nitric oxide-generating agents induce a reversible inactivation of protein kinase C activity and phorbol ester binding. The Journal of Biological Chemistry. 1993;268:27180–27185. [PubMed] [Google Scholar]
- 32.Park HS, Huh SH, Kim MS, Lee SH, Choi EJ. Nitric oxide negatively regulates c-Jun N-terminal kinase/stress-activated protein kinase by means of S-nitrosylation. Proceedings of the National Acadamy of Sciences of the United States of America. 2000;97:14382–14387. doi: 10.1073/pnas.97.26.14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teng KK, Esposito DK, Schwartz GD, Lander HM, Hempstead BL. Activation of c-Ha-Ras by nitric oxide modulates survival responsiveness in neuronal PC12 cells. The Journal of Biological Chemistry. 1999;274:37315–37320. doi: 10.1074/jbc.274.52.37315. [DOI] [PubMed] [Google Scholar]
- 34.Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 35.Lane P, Hao G, Gross SS. S-nitrosylation is emerging as a specific and fundamental posttranslational protein modification: head-to-head comparison with O-phosphorylation. Science's STKE. 2001;2001:RE1. doi: 10.1126/stke.2001.86.re1. [DOI] [PubMed] [Google Scholar]
- 36.Gaston BM, Carver J, Doctor A, Palmer LA. S-nitrosylation signaling in cell biology. Molecular Interventions. 2003;3:253–263. doi: 10.1124/mi.3.5.253. [DOI] [PubMed] [Google Scholar]
- 37.Benhar M, Stamler JS. A central role for S-nitrosylation in apoptosis. Nature Cell Biology. 2005;7:645–646. doi: 10.1038/ncb0705-645. [DOI] [PubMed] [Google Scholar]
- 38.Wyllie AH, Kerr JF, Currie AR. Cell death: the significance of apoptosis. International Review of Cytology. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 39.Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- 40.Wallach D, Varfolomeev EE, Malinin NL, Goltsev YV, Kovalenko AV, Boldin MP. Tumor necrosis factor receptor and Fas signaling mechanisms. Annual Reviews of Immunology. 1999;17:331–367. doi: 10.1146/annurev.immunol.17.1.331. [DOI] [PubMed] [Google Scholar]
- 41.Yuan J. Transducing signals of life and death. Current Opinions in Cell Biology. 1997;9:247–251. doi: 10.1016/s0955-0674(97)80069-5. [DOI] [PubMed] [Google Scholar]
- 42.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 43.Reed JC. Cytochrome c: can't live with it--can't live without it. Cell. 1997;91:559–562. doi: 10.1016/s0092-8674(00)80442-0. [DOI] [PubMed] [Google Scholar]
- 44.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 45.Klas C, Debatin KM, Jonker RR, Krammer PH. Activation interferes with the APO-1 pathway in mature human T cells. International Immunology. 1993;5:625–630. doi: 10.1093/intimm/5.6.625. [DOI] [PubMed] [Google Scholar]
- 46.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 47.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 48.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 49.Ben-Ezra JM, Kornstein MJ, Grimes MM, Krystal G. Small cell carcinomas of the lung express the Bcl-2 protein. American Journal of Pathology. 1994;145:1036–1040. [PMC free article] [PubMed] [Google Scholar]
- 50.Ikegaki N, Katsumata M, Minna J, Tsujimoto Y. Expression of bcl-2 in small cell lung carcinoma cells. Cancer Research. 1994;54:6–8. [PubMed] [Google Scholar]
- 51.Jiang SX, Sato Y, Kuwao S, Kameya T. Expression of bcl-2 oncogene protein is prevalent in small cell lung carcinomas. Journal of Pathology. 1995;177:135–138. doi: 10.1002/path.1711770206. [DOI] [PubMed] [Google Scholar]
- 52.Breitschopf K, Haendeler J, Malchow P, Zeiher AM, Dimmeler S. Posttranslational modification of Bcl-2 facilitates its proteasome-dependent degradation: molecular characterization of the involved signaling pathway. Molecular Cell Biology. 2000;20:1886–1896. doi: 10.1128/mcb.20.5.1886-1896.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hochstrasser M. Ubiquitin-dependent protein degradation. Annual Reviews of Genetics. 1996;30:405–439. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- 54.Hershko A, Ciechanover A. The ubiquitin system. Annual Reviews of Biochemistry. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 55.Mannick JB, Schonhoff C, Papeta N, Ghafourifar P, Szibor M, Fang K, Gaston B. S-Nitrosylation of mitochondrial caspases. Journal of Cell Biology. 2001;154:1111–1116. doi: 10.1083/jcb.200104008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abedini MR, Qiu Q, Yan X, Tsang BK. Possible role of FLICE-like inhibitory protein (FLIP) in chemoresistant ovarian cancer cells in vitro. Oncogene. 2004;23:6997–7004. doi: 10.1038/sj.onc.1207925. [DOI] [PubMed] [Google Scholar]
- 57.Lee SH, Kim HS, Kim SY, Lee YS, Park WS, Kim SH, Lee JY, Yoo NJ. Increased expression of FLIP, an inhibitor of Fas-mediated apoptosis, in stomach cancer. APMIS. 2003;111:309–314. doi: 10.1034/j.1600-0463.2003.1110203.x. [DOI] [PubMed] [Google Scholar]
- 58.Tanaka T, Yoshimi M, Maeyama T, Hagimoto N, Kuwano K, Hara N. Resistance to Fas-mediated apoptosis in human lung fibroblast. The European Respiratory Journal. 2002;20:359–368. doi: 10.1183/09031936.02.00252602. [DOI] [PubMed] [Google Scholar]
- 59.Medema JP, de Jong J, van Hall T, Melief CJ, Offringa R. Immune escape of tumors in vivo by expression of cellular FLICE-inhibitory protein. Journal of Experimental Medicine. 1999;190:1033–1038. doi: 10.1084/jem.190.7.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kinoshita H, Yoshikawa H, Shiiki K, Hamada Y, Nakajima Y, Tasaka K. Cisplatin (CDDP) sensitizes human osteosarcoma cell to Fas/CD95-mediated apoptosis by down-regulating FLIP-L expression. International Journal of Cancer. 2000;88:986–991. doi: 10.1002/1097-0215(20001215)88:6<986::aid-ijc23>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 61.Buolamwini JK. Novel anticancer drug discovery. Current Opinion in Chemical Biology. 1999;3:500–509. doi: 10.1016/S1367-5931(99)80073-8. [DOI] [PubMed] [Google Scholar]
- 62.Osford SM, Dallman CL, Johnson PW, Ganesan A, Packham G. Current strategies to target the anti-apoptotic Bcl-2 protein in cancer cells. Current Medicinal Chemistry. 2004;11:1031–1039. doi: 10.2174/0929867043455486. [DOI] [PubMed] [Google Scholar]
- 63.Carlisle DL, Pritchard DE, Singh J, Patierno SR. Chromium(VI) induces p53-dependent apoptosis in diploid human lung and mouse dermal fibroblasts. Molecular Carcinogenesis. 2000;28:111–118. doi: 10.1002/1098-2744(200006)28:2<111::aid-mc7>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 64.Haendeler J, Hoffmann J, Tischler V, Berk BC, Zeiher AM, Dimmeler S. Redox regulatory and anti-apoptotic functions of thioredoxin depend on S-nitrosylation at cysteine 69. Nature Cell Biology. 2002;4:743–749. doi: 10.1038/ncb851. [DOI] [PubMed] [Google Scholar]
- 65.Shi X, Chiu A, Chen CT, Halliwell B, Castranova V, Vallyathan V. Reduction of chromium(VI) and its relationship to carcinogenesis. The Journal of Toxicological & Environmental Health Part B Critical Reviews. 1999;2:87–104. doi: 10.1080/109374099281241. [DOI] [PubMed] [Google Scholar]