

Abstract
Neurons rely on action potentials, or spikes, to relay information. Pathological changes in spike generation likely contribute to certain enigmatic features of neurological disease, like paroxysmal attacks of pain and muscle spasm. Paroxysmal symptoms are characterized by abrupt onset and short duration, and are associated with abnormal spiking although the exact pathophysiology remains unclear. To help decipher the biophysical basis for ‘paroxysmal’ spiking, we replicated afterdischarge (i.e. continued spiking after a brief stimulus) in a minimal conductance-based axon model. We then applied nonlinear dynamical analysis to explain the dynamical basis for initiation and termination of afterdischarge. A perturbation could abruptly switch the system between two (quasi-)stable attractor states: rest and repetitive spiking. This bistability was a consequence of slow positive feedback mediated by persistent inward current. Initiation of afterdischarge was explained by activation of the persistent inward current forcing the system to cross a saddle point that separates the basins of attraction associated with each attractor. Termination of afterdischarge was explained by the attractor associated with repetitive spiking being destroyed. This occurred when ultra-slow negative feedback, such as intracellular sodium accumulation, caused the saddle point and stable limit cycle to collide; in that regard, the active attractor is not truly stable when the slowest dynamics are taken into account. The model also explains other features of paroxysmal symptoms, including temporal summation and refractoriness.
Introduction
Clinical medicine has a long history of describing disease symptoms in exquisite detail. This is especially true in neurology, where careful evaluation of negative (i.e. loss-of-function) and positive (i.e. gain-of-function) symptoms plays a crucial role in clinical diagnosis (Donaghy et al 2001). A subset of symptoms can be distinguished as paroxysmal based on their abrupt onset and relatively brief duration (seconds to minutes). A common example is the explosive pain of trigeminal neuralgia (Henderson 1967), which can occur spontaneously or be triggered, for example by mechanical stimulation. The pain invariably subsides after several seconds and is followed by a refractory period during which pain attacks are absent or relatively more difficult to provoke, although they can nonetheless recur up to several hundred times per day. Similar paroxysmal episodes occur in multiple sclerosis (MS) in a remarkable diversity of forms, including pain, paresthesia (numbness), pruritus (itchyness), diploplia (double vision), dysarthria (difficulty speaking), weakness and muscle spasm (Ostermann and Westerberg 1975, Twomey and Espir 1980). In MS and other diseases, including trigeminal neuralgia and neuropathic pain following nerve injury, paroxysmal symptoms are indirectly linked to demyelination. Negative symptoms tend to parallel disease progression (i.e. spikes do not propagate through newly demyelinated regions of axon), whereas positive paroxysmal symptoms reflect functional recovery of the demyelinated axon, but to the point of becoming hyperexcitable (Waxman 1982, 2006, Waxman et al 1995). Consistent with a ‘rebalancing’ mechanism, antiepileptic drugs like carbamazepine can relieve paroxysmal symptoms, but often do so at the expense of worsening negative symptoms (Sakurai and Kanazawa 1999).
Electrophysiological data in humans and animal models have linked positive neurological symptoms with abnormal neuronal discharge (Ochoa and Torebjork 1980, Calvin et al 1982, Baldissera et al 1994, Felts et al 1995); by extension, paroxysmal symptoms likely reflect ectopic discharge with a paroxysmal pattern, i.e. abrupt onset and limited duration (Devor et al 2002). That discharge pattern does not require recurrent synaptic connectivity, like in network-based seizures, as evident by the occurrence of such activity in the periphery where there is no recurrent connectivity. Thus, paroxysmal discharge is likely to originate at the level of single axons, although this does not preclude it from spreading ephaptically to neighboring axons (Seltzer and Devor 1979, Devor and Wall 1990). It is nonetheless notable that activity in single primary afferent fibers can be perceived (Ochoa and Torebjork 1983), and more recent evidence suggests that activity in single or small sets of central neurons may be similarly consequential (Wolfe et al 2010).
Here, we sought to determine how paroxysmal discharge can arise in single axons. Specifically, our aim was to replicate paroxysmal afterdischarge (i.e. continued spiking after stimulation) in as simple a model as possible, and then to analyze that model to gain insight into the basic underlying mechanisms. This minimal modeling approach is motivated by the fact that our understanding of the phenomena (i.e. paroxysmal symptoms) is comparatively good relative to our patchy understanding of potential mechanisms. This contrasts a biophysically detailed modeling approach in which experimentally identified changes in channel expression, for example, are incorporated into a model (which becomes progressively more complicated as a result) to determine how those changes impact neuronal function. Moreover, we placed specific emphasis on how various ion currents and other processes interact. Indeed, paroxysmal discharge has the hallmarks of a highly nonlinear phenomenon, which points to nonlinear interaction between underlying mechanisms as a key factor in its genesis. According to dynamical analysis in our minimal models, (1) paroxysmal discharge can only occur when a system is bistable (at least transiently), (2) initiation occurs when a perturbation activates a positive feedback process that abruptly switches the system between attractor states, and (3) termination typically occurs when an even slower negative feedback process destroys the attractor associated with repetitive spiking. Temporal summation and refractoriness can also be explained within this framework. Although not biologically detailed, our modeling nonetheless provides significant insight into how biologically realistic mechanisms can, in theory at least, interact to produce paroxysmal neuronal discharge.
Methods
Our starting model was based on the Morris–Lecar model (Morris and Lecar 1981, Rinzel et al 1998) and has been described in detail (Prescott et al 2008, Coggan et al 2010). It is a single compartment, conductance-based model. The 3D model is described as
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
where V is the voltage and w and z are the variables controlling time- and voltage-dependent activation of gK and gNaP, respectively; gNa activates instantaneously and m was therefore always at steady state. In equations (4) and (5), x corresponds to m, w, or z. The following parameters were used in all simulations: C = 2 μF cm−2, EL = −70 mV, ENa = +50 mV, EK = −100 mV, βm = −1.2 mV, γm = 18 mV, βw = −10 mV, γw = 10 mV, ϕw = 0.15, βz = −45 mV, γz = 10 mV, ϕz = 0.05 (unless otherwise stated), delayed rectifier potassium conductance gK = 20 mS cm−2, and leak conductance gL = 2 mS cm−2. Fast sodium conductance gNa and persistent sodium conductance gNaP were varied as indicated in the text.
For bifurcation analysis in the V–w system, the 3D model described above was reduced to a 2D model by treating z as a parameter rather than as a variable. Conversely, in later simulations, we expanded our model to 4D by tracking dynamic changes in intracellular sodium [Na+]i with the equation
| (6) |
The above equation describes intracellular sodium accumulation on the basis of influx through fast and persistent Na+ channels and assumes exponential decay toward a steady-state value of 17.5 mM with a time constant τNa = 100 ms. The surface area-to-volume ratio is defined by SAV = 2/r based on a cylindrical compartment with radius r and no ends, i.e. compartment ends are assumed to connect to other compartments rather than contributing membrane exposed to extracellular fluid. Under these conditions, compartment length is irrelevant. r = 0.5 μm unless otherwise indicated. F is the Faraday constant 96 485 C mol−1. In the 4D model, ENa was continuously updated according to ENa = 25 ln ([Na+]o/[Na+]i), where extracellular sodium concentration [Na+]o was assumed constant at 138 mM.
Unless otherwise stated, ‘evoked’ spikes were triggered by instantaneously resetting V to 0 mV and then letting the system evolve freely. This method, rather than injecting a current pulse, was chosen to mimic abrupt spike initiation that occurs when a spike is propagated from a remote initiation site (which we cannot formally simulate in our single compartment model) and represents a consistent, unbiased form of perturbation. Equations were numerically integrated in XPP (Ermentrout 2002) using the Euler method with a 0.01–0.05 ms time step. Nullclines were also calculated in XPP. To calculate a nullcline at time t, all variables not associated with the nullcline (e.g. w and z for the V-nullcline) were held constant at their value at time t. Bifurcation analysis was conducted in AUTO using the XPP interface. For good introductions to dynamical systems theory and the analysis techniques used in this study, see Izhikevich (2007) or Strogatz (1998).
The multicompartment Hodgkin–Huxley model shown in figure 1 was based on the model in Coggan et al (2010). Parameters are reported in the figure legend. In part F of that figure, the Na+ and K+ reversal potentials were updated according to the Nernst equation rather than being fixed parameters, as in the original model, but no other changes were made.
Figure 1.

Patterns of excitability in a multicompartment Hodgkin–Huxley axon model. (A) Schematic of axon morphology. A single action potential (spike) was evoked upstream, in the soma, while monitoring the response in a distant region of simulated demyelination, shown in gray. For simulation results in panels C–F, spikes recorded in the demyelinated region are shown in gray and spikes recorded in upstream or downstream nodes (shown only in C) are in black. (B) Cartoon representation of different outcomes of demyelination. Normal direction of propagation is from left to right. (C) In a normal axon, the spike is propagated at uniform speed (top). When a demyelinated zone was introduced into the axon model, spike propagation failed, i.e. there is no (gray) spike recorded in the mid-point of that region (middle). Compensatory changes (i.e. upregulation of Na+ conductance and/or downregulation of leak conductance) resulted in recovery of spike propagation although at a reduced rate because conduction through the demyelinated region is not saltatory (bottom). Moreover, overcompensation can produce different degrees of hyperexcitability, as shown in parts D–F. (D) In this example, a single evoked spike triggered prolonged afterdischarge in the demyelinated region. (E) For slightly less overcompensation, multiple evoked spikes were required to trigger afterdischarge. In both D and E, afterdischarge continued indefinitely. (F) The model was made more realistic by calculating transmembrane ion flux and using that information to update Na+ and K+ concentrations and their associated reversal potentials, ENa and EK. In this modified model, afterdischarge stopped after a variable number of spikes, which depended on the precise model parameters. Conductance densities (in S cm−2) were as follows: (C, top) gNa = 1.5, gNaP = 0.002, gK = 1.6, gL = 0.07; (C, middle) gNa = 0.15, gNaP = 0.0002, gK = 0.16, gL = 0.07; (C, bottom) gNa = 0.15, gNaP = 0.0002, gK = 0.16, gL = 0.004; (D) gNa = 0.15, gNaP = 0.0002, gK = 0.16, gL = 0.0029; (E) gNa = 0.15, gNaP = 0.0002, gK = 0.16, gL = 0.0031; (F) gNa = 0.8, gNaP = 0.0013, gK = 0.75, gL = 0.029.
Results
Patterns of abnormal excitability in a realistic axon model
We started by considering the effects of changing ion channel density in a multicompartmental Hodgkin–Huxley model of an axon (figure 1(A)) in order to understand the compensatory changes in axonal excitability that occur following focal demyelination. Figure 1(B) summarizes the different possible outcomes of demyelination and compensation. When axonal membrane that is normally ensheathed in myelin becomes exposed, that membrane must actively contribute to spike propagation since saltatory conduction between nodes is interrupted. In the immediate aftermath of demyelination, Na+ channel density in this newly exposed membrane is low (Waxman 1982), which results in failed spike propagation (figure 1(C) middle). As the ratio of Na+ and leak channels is re-adjusted, propagation recovers but is nonetheless slowed (figure 1(C) bottom). However, imperfect re-balancing of ion channel density can produce axonal hyperexcitability (figures 1(D)–(F)). In figure 1(D), the evoked spike propagates through the area of demyelination but, in the process, triggers an afterdischarge that continues indefinitely. In figure 1(E), afterdischarge is not elicited unless a burst of evoked spikes propagate through the demyelinated region. This latter scenario is interesting insofar as the axon can function normally under most conditions despite harboring an occult abnormality; specifically, sparsely occurring spikes are normally propagated and afterdischarge is triggered only if a (rare) burst of spikes occurs. In both figures 1(D) and (E), however, afterdischarge continued indefinitely, inconsistent with the self-limiting nature of paroxysmal symptoms. Continuous afterdischarge reflects an oversimplification in the starting model, because if ion concentrations are updated on the basis of transmembrane ion flux, then afterdischarge does indeed terminate due to ion accumulation (figure 1(F)).
This multicompartment axon model thus replicates several features of demyelination-induced pathology. Using this model, known or presumed changes in ion channel expression can be simulated to help ascertain their effects on axonal excitability. One caveat with this biophysical approach is that unless one can rule out or strictly control for other molecular changes, those other changes may compete, cooperate, or interfere with the ‘change of interest’. Indeed, changes in multiple ion channels occur following demyelination and other forms of nerve injury (Waxman 2006, Costigan et al 2009, Dib-Hajj et al 2010). Nonlinear interactions can easily cause one to over- or underestimate the contribution made by any one change. Furthermore, mechanistic understanding of afterdischarge requires that one understands those interactions. Dynamical systems theory provides the tools to characterize them.
Initiation of afterdischarge
To apply dynamical systems theory, we explored a simplified model capable of replicating the hyperexcitability patterns identified in the biologically detailed model described above. Figure 2 demonstrates that afterdischarge initiation can be explained on the basis of interaction between three variables: V (which controls activation of gNa), w (which controls activation of gK), and z (which controls activation of gNaP). Co-evolution of those variables is plotted against time in figure 2(A), but to investigate how those variables interact with each other, we plotted variables against one another to create phase planes (figure 2(B)). Nullclines represent locations in phase space where a given variable remains constant. The points at which nullclines intersect provide valuable information about how the system will evolve within the context of that phase space, as we will show. However, because our model is 3D, there are three potential interactions: V–w, V–z, and w–z. Because w and z do not interact directly, we are left with two component subsystems. Each can be investigated separately because V and w interact on a faster timescale than V and z.
Figure 2.

Visualizing the interaction between multiple variables. We constructed a one compartment Morris–Lecar-type model to investigate the initiation and termination of afterdischarge. For initial tests shown here, gNaP = 1.0 mS cm−2 and gNa = 20 mS cm−2. (A) Time series show the co-evolution of V (which controls instantaneous activation of gNa), w (which controls activation of gK) and z (which controls activation of gNaP). Both w and z are voltage dependent but do not change instantaneously with V. A single spike was evoked at time 0 by re-setting V to 0 mV and then letting the system evolve freely. To visualize how those variables interact, we can plot variables against one another (as in B and C) rather than against time (as in A). Furthermore, rather than trying to visualize interactions in 3D space, dynamical interactions can be analyzed in terms of the component 2D systems; in other words, we ask how V and z interact (on a relatively slow timescale) and how V and w interact (on a faster timescale during which z stays relatively constant). (B) The first interaction is visualized on the V–z phase-plane. The system normally rests at the stable fixed point in the bottom left corner. The V–z trajectory is seen to pass on the far side (relative to the stable fixed point) of the saddle point (sp) following the evoked spike. Having traversed the saddle point, the system moves toward its other attractor but enters a stable limit cycle and spikes repetitively rather than converging on a stable fixed point. (C) The onset of repetitive spiking can be understood from snapshots of the V–w plane at different time points indicated in A. At t = 0 ms, z = 0, which means gNaP is not activated; under these conditions, the V- and w-nullclines intersect at the stable fixed point. At t = 30 ms, the evoked spike has caused an increase in z, which means gNaP contributes an inward current. That current shifts the V-nullcline (green) from the solid position toward the dashed position. Upon sufficient shift, the intersection between the V- and w-nullclines is destabilized by a Hopf bifurcation and repetitive spiking ensues. Thus, generation of individual spikes depends on fast interaction between V and w, but the slow process responsible for a bifurcation in the V–w subsystem reflects changes in z. (D) The saddle point represents the border between two attractor states to which z can evolve following a perturbation. If z approaches the saddle point from the far side, the system will move toward its ‘active’ attractor, causing it to spike repetitively. If z approaches the saddle point from the near side, the system will return to its ‘quiet’ attractor or resting state. ‘Near’ and ‘far’ are defined relative to the quiet attractor.
We start by considering the interaction between V and z by analyzing the V–z phase plane (figure 2(B); see figure 2(D) for summary). The V- and z-nullclines intersect at three points, which are referred to as fixed points. The system usually rests at the stable fixed point in the bottom left corner. However, following an evoked spike, the V–z trajectory can return to the resting state, or it can move to a different attractor that happens to be a stable limit cycle. To be clear, the trajectory moves toward the fixed point in the top-right corner of the V–z phase plane, but is diverted by interactions occurring in the V–w phase plane (see below). The decision to switch attractors is based on whether the trajectory stays above or dips below the middle fixed point, which is known as a saddle point. This system is therefore bistable, with one attractor corresponding to a rest state and the other to a state of repetitive spiking; we refer to them as the ‘quiet’ and ‘active’ attractor, respectively. The saddle point represents the threshold separating the attractors.
An important question is why the active attractor in the 3D system is associated with repetitive spiking. This can be answered by analysis of the V–w plane (figure 2(C)). The V- and w-phase planes intersect at a stable fixed point, which, in the absence of any perturbation, is where the neuron normally rests. Following a perturbation, the V–w system would return to this fixed point were it not for slow activation of gNaP (controlled by z), which introduces an inward (depolarizing) current. That current shifts the V-nullcline vertically on the V–w phase plane. If the V-nullcline gets shifted far enough, its intersection with the w-nullcline will be destabilized through a Hopf bifurcation (as shown in figure 2(C)) or destroyed through a saddle-node on invariant circle bifurcation (see left bifurcation diagram in figure 5(A)). Repetitive spiking begins after either of those types of bifurcations occurs in the V–w system. The precise nature of that bifurcation is not important for our purposes. What is important is that slow changes in z can cause that bifurcation and thereby trigger repetitive spiking.
Figure 5.

Termination of afterdischarge through different mechanisms. We incorporated intracellular Na+ accumulation into our simplified model to terminate afterdischarge. Different combinations of gNa and gNaP (indicated in mS cm−2) were tested. (A) For gNa = 30 mS cm−2 and gNaP = 1.0 mS cm−2, intracellular Na+ accumulation did not cause repetitive spiking to stop, but therein provides a useful starting point from which to consider the effects of Na+ accumulation. The time series illustrates the hyperpolarizing shift in sodium reversal potential (ENa) and the resultant shift in the saddle point. The saddle point was found by re-plotting the bifurcation diagrams (V versus z) at time points corresponding to different ENa values. Reduction of ENa shifts the bifurcation to higher z, and although the z-nullcline remains unchanged, the change in the bifurcation diagram affects where that bifurcation diagram intersects the z-nullcline, which amounts to a shift in the saddle point. Despite shifting the saddle point, the V–z trajectory remains on the far side of the saddle point and repetitive spiking continues once ENa stabilizes at 23 mV. Bifurcation diagram on the right shows z as [Na+]i was systematically varied. The middle branch of this bifurcation diagram, labeled sp, constitutes the saddle point that the z trajectory must cross in order to enter its stable limit cycle. Note how the z-value of the saddle point increases with [Na+]i. (B) With less gNaP, ENa undergoes a smaller reduction but that shift is sufficient to move the saddle point very close to the bifurcation. Eventually, the V–z trajectory crosses back to the near side of the rightward creeping saddle point, causing afterdischarge to stop. Bifurcation diagram on the right illustrates that afterdischarge stops at the [Na+]i-value where the limit cycle collides with the ‘saddle point’ branch. (C) A different set of parameters confirmed that crossing back over the saddle point is the critical termination step. The pattern of intermittent spikes and subthreshold oscillations shown here (orange arrowheads) is evidence of the system drifting back and forth across a subcritical Hopf bifurcation. But although the V–z trajectory moves to the left of the bifurcation, this alone does not terminate the afterdischarge; as long as the trajectory remains on the far side of the saddle point, z will increase to produce more spikes. (D) However, if the bifurcation point shifts beyond an achievable value of z (i.e. z >1), the system will ‘lock up’ in a depolarized state as the z-nullcline forms another intersection with the stable branch of the bifurcation diagram. In this condition, the system stops spiking because the active attractor has converted from a stable limit cycle to a stable fixed point, not because the system has returned to its quiet ‘rest’ attractor. This stable fixed point is also evident near the top right knee of the z-[Na+]i bifurcation diagram. The same stable fixed point also existed for conditions in A and C but, in those cases, [Na+]i did not rise high enough for the full 4D system to stabilize there.
We therefore asked how high z must rise to cause a bifurcation in the V–w system. To answer that, we treated z as a bifurcation parameter (thus converting our model from 3D to 2D) to determine at what z-value the V–w system undergoes a bifurcation. To investigate the effects of persistent inward current, we compared models with different values of gNaP. As gNaP was increased, the bifurcation occurred at smaller and smaller values of z (figures 3(A)–(D)); in fact, there was no bifurcation for low gNaP levels (figure 3(A)). The next question was whether z increases high enough, when treated as a variable, to actually produce this bifurcation. To answer that, we projected the V–z trajectory from the 3D model (black) onto the bifurcation diagram generated from the corresponding 2D model. In all cases, we also projected the z-nullcline (red) onto the bifurcation diagram since its intersection with the bifurcation diagram reveals important fixed points, most notably the saddle point. Figure 3(B) shows an example where the evoked spike did not cause a large enough increase in z to force the system across the saddle point so that z could continue to increase high enough to produce the bifurcation, hence no afterdischarge was evoked. In figure 3(C), the evoked spike did cause z to traverse the saddle point and thereafter increase high enough to cause the bifurcation, thus initiating afterdischarge. In figure 3(D), gNaP was so high that the stable attractor and saddle point were destroyed through a saddle-node bifurcation; in the absence of that stable attractor, z increased even in the absence of any perturbation and caused spontaneous spiking.
Figure 3.

Initiation of afterdischarge by a single perturbation. The question of whether afterdischarge is initiated for a given parameter set was addressed by breaking the questions into two parts: (1) How strongly must gNaP be activated in order to sustain repetitive spiking? (2) How strongly is gNaP actually activated? The first question was addressed by converting z from a variable to a parameter (thus reducing the model to two dimensions) and systematically varying z as a bifurcation parameter to determine at what point the V–w system undergoes a bifurcation. The second question was addressed by projecting the V–z trajectory from the original 3D model onto the bifurcation diagram from the 2D model. The z-nullcline (red) is also projected onto the bifurcation diagram in order to identify the saddle point (sp). Different values of gNaP (indicated in mS cm−2) were tested in A–D; in all cases, gNa = 20 mS cm−2. (A) For gNaP = 0.1 mS cm−2, the bifurcation diagram shows that no amount of activation can produce repetitive spiking, i.e. there is no bifurcation for z ≤ 1. (B) For gNaP = 0.8 mS cm−2, the bifurcation diagram indicates that repetitive spiking occurs for z > 0.57, but the superimposed trajectory shows that z does not rise that high following a single evoked spike; specifically, the V–z trajectory approaches the stable branch of the bifurcation diagram on the near side of the saddle point (arrowhead), whereupon the trajectory is directed back to the stable fixed point. Compare with trajectory in C. (C) For gNaP = 1 mS cm−2, the bifurcation diagram indicates that repetitive spiking occurs for z > 0.45. The superimposed trajectory shows that z does indeed rise high enough to produce that bifurcation in the 3D model. Sustained activation of gNaP is predicted by the V–z trajectory approaching the stable branch of the bifurcation diagram on the far side of the saddle point (arrowhead), which allows z to continue increasing. (D) For gNaP = 4 mS cm−2, the bifurcation diagram shows that the stable fixed point defining the ‘quiet’ attractor is destroyed, which allows the system to move to its other (now sole) ‘active’ attractor even in the absence of any perturbation. Modified from Coggan et al (2010).
The analysis described above suggests two conclusions. First, afterdischarge requires the system to be bistable; however, as will be shown in the 4D model, the bistability may exist only transiently, in which case the active attractor is not truly stable on a long timescale. For conditions tested in our 3D model, the system is bistable only if the density of persistent Na+ channels is sufficiently high. The minimum persistent Na+ conductance required to make the system bistable depends on other channel densities (data not shown; see Coggan et al 2010), but at least some persistent Na+ current must be present. Second, if the system is bistable, initiation of afterdischarge requires a perturbation that is large enough to knock the system from one basin of attraction, across the saddle point, into the other basin of attraction. The minimum perturbation depends on the distance from the stable fixed point to the saddle point (figure 3). We investigated the perturbation requirements further by testing whether we could replicate the multispike-induced afterdischarge observed in the realistic axon model (see figure 1(E)). For this, we used the parameters from figure 3(B) since our analysis revealed that the model was bistable but that a single evoked spike did not trigger afterdischarge. We evoked spikes at 15 ms intervals until the V–z trajectory had crossed the saddle point, at which point we predicted that afterdischarge should be initiated. As predicted, continuing the simulation (without further evoked spikes) confirmed that afterdischarge was inevitable once the saddle point had been crossed (figure 4).
Figure 4.
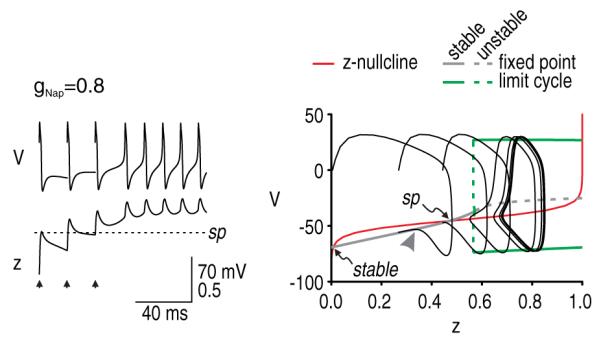
Initiation of afterdischarge by multiple perturbations. Bifurcation analysis in figure 3(B) showed that a single spike may be insufficient to provoke afterdischarge despite the system being bistable. We predicted that a stronger perturbation, such as multiple evoked spikes, would trigger afterdischarge if that cumulative perturbation was sufficient to force the V–z trajectory across the saddle point. As predicted, three spikes evoked at 15 ms intervals were sufficient to initiate afterdischarge. On the time series, note that z did not fall below the saddle point after the third evoked spike. Similarly, the V–z trajectory approached the stable branch of the bifurcation diagram from the near side of the saddle point after the first two evoked spikes, but approached from the far side after the third evoked spike, after which point afterdischarge was inevitable.
Termination of afterdischarge
As in our initial multicompartment axon model, afterdischarge continued indefinitely in our initial simplified 3D models. Therefore, like we did for the multicompartment model, we added intracellular Na+ accumulation to our simplified model to implement ultra-slow negative feedback that terminates afterdischarge, thus making our model 4D. According to the Nernst equation, an increase in intracellular Na+ causes a reduction in the sodium reversal potential ENa. Since INaP = gNaP z (V–ENa), changes in ENa will change the driving force and alter the magnitude of the inward current. Importantly, ENa changes on a much slower timescale than each of the other variables.
Sodium accumulation does not necessarily terminate afterdischarge (figure 5(A)). Nonetheless, reduction of ENa invariably shifts the V–z bifurcation diagram, which (1) increases the z required for a bifurcation in the V–w system and (2) shifts the saddle point. As long as the V–z trajectory remains on the far side of the shifted saddle point once afterdischarge is initiated, afterdischarge will continue (figure 5(A)). However, for different parameter values (cf figures 5(A)–(C)), the saddle point can shift close enough to the bifurcation that the V–z trajectory eventually dips below the saddle point, whereupon the V–z trajectory is directed to the quiet attractor (figure 5(B)); this is also evident on the z-[Na+]i bifurcation diagrams described below. The V–z trajectory passing leftward through the bifurcation does not predict the termination of the afterdischarge since the trajectory will be forced back through the bifurcation so long as the system remains on the far side of the saddle point. This is verified in figure 5(C), where the V–z trajectory is seen to clearly dip below the minimum value of z required to maintain repetitive spiking (this is evidenced by the subthreshold oscillations highlighted with orange arrowheads) but, since the trajectory remains on the far side of the saddle point, the system continues spiking, albeit falteringly. Note that because of the slow Na+ dynamics, the spiking oscillation is not strictly a stable limit cycle; however, on a fast timescale (such as that of individual spikes), ENa is practically constant like in the 3D system described above, and we have a quasi-stable limit cycle. As for initiation, the termination of afterdischarge is thus dictated by the V–z trajectory relative to the saddle point although, in the case of termination, we must also consider how that saddle point moves (although this can also be a factor in initiation; see figure 7).
Figure 7.

Intracellular Na+ accumulation affects afterdischarge initiation when initiation depends on repeated perturbations. In all panels, gNa = 20 mS cm−2 and gNaP = 0.8 mS cm−2 and stimuli were repeated at 15 ms intervals, as indicated by the arrowheads. (A) When ENa is constant, the only requirement for initiating afterdischarge is that repeated perturbations cause cumulative activation of z (red dotted line on time series) such that the V–z trajectory eventually crosses the stationary saddle point. Bifurcation diagram on right shows how z increases with each evoked spike without a concomitant increase in [Na+]i; arrows point to values to which z falls after each spike (compare with time series). (B) However, if the saddle point shifts on the same time scale as cumulative activation of z, then the Na+ accumulation caused by each evoked spike can push the saddle point away faster than each spike can increase z, resulting in the V–z trajectory never overtaking the shifting saddle point, as seen here. (C) Increasing the surface area-to-volume ratio of our simulated compartment by increasing the radius from 0.5 to 2 μm reduced the rate of Na+ accumulation. Under these conditions, spikes evoked at 15 ms intervals were capable of initiating afterdischarge. Note that z-[Na+]i bifurcation diagrams are the same in parts A–C and that the only difference, therefore, is in the trajectory. These results highlight that it is the relative rates of g activation and Na+ NaP accumulation that are important, as opposed to the absolute rates.
Bifurcation diagrams on the right of figure 5 use [Na+]i as the bifurcation parameter and provide another way of explaining the initiation and termination of afterdischarge. The middle branch of these bifurcation diagrams correspond to the saddle point; therefore, afterdischarge initiation can be understood from the z-[Na+]i trajectory staying above this branch and entering the stable limit cycle rather than returning to the stable fixed point. Afterdischarge termination can be understood from where the limit cycle collides with one of the fixed point branches. Repetitive spiking stops when Na+ accumulation brings the system to the end of the stable limit cycle branch. In some respects, these dynamics resemble those associated with parabolic, type 2, or circle/circle bursting (Rinzel and Lee 1987, Rinzel et al 1998, Izhikevich 2007). Parabolic bursting involves interaction between two slow variables, which in our case correspond to z and [Na+]i. Afterdischarge terminates through the same mechanism as a parabolic burst of spikes, but afterdischarge does not initiate spontaneously. For our model to initiate repetitive spiking through the same mechanism as in parabolic bursting, [Na+]i would need to fall to <0.1 mM to allow the system to escape around the bottom-left knee of the bifurcation diagram, which is physiologically impossible. Instead, repetitive spiking is initiated only when a perturbation forces the system across a saddle point threshold. An important observation is that z can exist in two steady states (one being a limit cycle) across a reasonably broad range of [Na+]i.
Notably, there is another mechanism whereby the system can stop spiking repetitively without returning to its resting state (figure 5(D)). In effect, the active attractor can switch from a limit cycle to a fixed point, as will be explained. In this case, termination occurs because the bifurcation point (on the V–z bifurcation diagram) moves beyond an attainable value of z, where 0 ≤ z ≤ 1. Under those conditions (but unlike in figure 3(A), in which there was also no possibility of repetitive spiking), the z-nullcline and the stable branch of the bifurcation diagram intersect at three points. The new fixed point represents a stable fixed point at which the system can settle rather than spike repetitively. Although spiking stops, the system does not return to its resting membrane potential. Information is not relayed to downstream neurons in the absence of spiking (hence, there should be no positive symptoms), but remaining in a depolarized state risks causing severe excitotoxicity (Stys et al 1992). This might be a contributing factor to the gray matter pathology associated with MS (Geurts and Barkhof 2008). This second stable fixed point is also evident in the z-[Na+]i bifurcation diagrams in figures 5(A), (C) and (D); evidently, [Na+]i rises high enough for the system to reach this fixed point only in the case of figure 5(D).
Based on insight gleaned from figure 5, we predicted that the kinetics of gNaP activation are important for termination. Specifically, when the kinetics of gNaP activation are relatively fast, z will increase rapidly during the spike but will also tend to decrease rapidly after the spike (figure 6(A)). Large post-spike dips in z make the V–z trajectory more prone to crossing back over the saddle point. We therefore predicted that slower gNaP activation kinetics would discourage afterdischarge termination. This was tested in figure 6(B). Note that multiple, rapidly evoked spikes were necessary to drive the V–z trajectory across the saddle point because the incremental increase in z with each evoked spike was less than that in figure 6(A), consistent with the change in kinetics. But although harder to initiate, once initiated, afterdischarge in the model with slower kinetics did not stop, unlike the model with faster kinetics (cf figures 6(A) and (B)), thus confirming our prediction.
Figure 6.

Kinetics of gNaP affect traversal of the saddle point. (A) When the kinetics of gNaP are relatively fast, z increases rapidly during each spike but so too will it decrease rapidly after each spike. As the saddle point creeps rightward as ENa decreases, large post-spike dips in z increase the likelihood of the V–z trajectory crossing back over the saddle point. Compare with B. (B) When the kinetics of gNaP activation are slowed down, z changes more slowly. Consequently, initiation of afterdischarge required three spikes evoked in quick succession (5 ms intervals) to drive z across the saddle point. However, once initiated, afterdischarge did not terminate (despite an even larger reduction in ENa than in A), consistent with the smaller dips in z. For both A and B, gNa = 30 mS cm−2 and gNaP = 0.8 mS cm−2. Note that peak gNaP activation was equivalent in both cases (dotted orange line). Bifurcation diagrams on the right show that [Na+]i can rise higher without terminating afterdischarge for conditions in B. Note the spacing between the maximum and minimum of the limit cycle, as shown on these bifurcation diagrams.
Other effects of intracellular sodium accumulation
Analysis in figure 5 clearly established that intracellular Na+ accumulation can cause a shift in the saddle point, which, if it collided with the stable limit cycle, would force the model to stop spiking. Here, we asked how changes in ENa might also affect afterdischarge initiation. The Na+ accumulation that occurs following a single evoked spike causes a small shift in the saddle point such that the precise threshold for single-spike-evoked afterdischarge is raised slightly compared to that in the same model with constant ENa (data not shown). However, we predicted that the effect would be much larger for multi-spike-evoked afterdischarge, especially if the time-scale of cumulative increase in z (see dotted red lines on figure 7) is comparable to the time-scale of Na+ accumulation. To test this, we evoked spikes at 15 ms intervals in a model with constant Na+ (figure 7(A)) and in a model with dynamic Na+ (figure 7(B)). As predicted, afterdischarge was readily evoked in the former but not in the latter case (cf figures 7(A) and (B)). Hence, afterdischarge initiation in a model with Na+ dynamics relies on cumulative changes in z overtaking a shifting saddle point. This is clearly evident from trajectories on the z–[Na+]i bifurcation diagrams since the middle branch of the bifurcation diagram depicts the saddle point across a range of [Na+]i values. These results are consistent with those in figure 6(B), where rapidly evoked spikes were necessary to initiate afterdischarge not only because cumulative activation of gNaP had been slowed down but also because the saddle point shifted because of the Na+ accumulation caused by each evoked spike. Overall, these results suggest that it is the relative kinetics of g activation and Na+ NaP accumulation that are important. To test this, we increased the axon radius fourfold, thus reducing the surface area-to-volume ratio of our model and reducing the rate of Na+ accumulation. As expected, ENa decreased more slowly, thereby causing the saddle point to shift more slowly and increasing the likelihood of cumulative changes in z overtaking that saddle point (figure 7(C)). As shown, ENa continued to decrease after cumulative activation of z overtook the saddle point but activation of z accelerates once z crosses the saddle point. These results emphasize the importance of the relative rates of different processes, and highlights that the same processes may interact differently in cell compartments with different dimensions.
In addition to considering how rapidly Na+ accumulates during afterdischarge, one must also consider how quickly intracellular Na+ levels recover upon termination of afterdischarge. We modeled Na+ extrusion as a simple exponential decay rather than using a more realistic Na–K pump, but our basic results were nonetheless revealing. We predicted that lingering changes in ENa would produce a post-afterdischarge refractory period. Figure 8 illustrates that V and z returned to baseline faster than ENa following termination of an afterdischarge as expected given that Na+ dynamics were relatively slow. If additional spikes are evoked before ENa recovers, it is more difficult to elicit a second afterdischarge although the model is nonetheless capable of generating individual spikes (figures 8(A) and (B)). In a real axon, the demyelinated region would propagate spikes without initiating afterdischarge; in other words, the axon would return to comparatively normal operation. It is also notable that the Na+ accumulation caused by multiple evoked spikes may preclude afterdischarge (figure 8(C)).
Figure 8.

Slow Na+ kinetics cause a refractory period. All simulations are with gNa = 30 mS cm−2 and gNaP = 0.8 mS cm−2. (A) A burst of six spikes evoked at 5 ms intervals could not elicit a second afterdischarge when that burst arrived 200 ms after the start of the first afterdischarge. Response to second stimulus set is projected onto the z versus [Na+]i bifurcation diagram. For these conditions, the z-trajectory fails to stay above the saddle point branch and enter the limit cycle. (B) When delivered 300 ms after the first start of the first afterdischarge, only two spikes evoked at 5 ms intervals were required to elicit afterdischarge, although this is still more than the single spike required to elicit the first afterdischarge. Patterns in A and B are indicative of a refractory period. Notably, the model is refractory to afterdischarge but can support spike generation, as evidenced by the evoked spikes. (C) Evoking multiple spikes may, by causing a large rise in [Na+]i, mask an afterdischarge that would have been elicited by fewer evoked spikes.
Discussion
Our goal was to explain the initiation and termination of ectopic spiking thought to underlie paroxysmal neurological symptoms. To this end, we replicated afterdischarge in as simple a model as possible and then applied nonlinear dynamical analysis to reveal the responsible mechanisms. Specifically, using the same biophysical mechanisms in each model, we replicated phenomena observed in our multicompartment Hodgin–Huxley model in a single compartment Morris–Lecar model; however, the latter model is not formally derived from the former, and even the former model may not reproduce the true biological mechanisms used by real axons. Nonetheless, our results suggest that afterdischarge has two requirements, one relating to the system (i.e. the axon) and the other relating to the perturbation (i.e. evoked spikes). First, the axon is susceptible to afterdischarge only if it is (at least transiently) bistable. Second, the axon will only exhibit afterdischarge if a sufficiently strong perturbation knocks the system from its quiet attractor to its active attractor. In an axon, that perturbation is dictated by the number and timing of ‘evoked’ spikes arriving in the hyperexcitable region. The critical transition from one attractor to the other is controlled by changes in a slow activation variable z relative to a saddle point that defines threshold. However, if the saddle point is not static (like in our 4D model where it shifts according to changes in [Na+]i), the active attractor can be destroyed if the stable limit cycle and saddle point collide, whereupon afterdischarge stops.
How do these general principles inform our biological understanding of afterdischarge? For one, they pinpoint factors that may be necessary and sufficient to explain afterdischarge. The dual requirements of (1) bistability and (2) sufficient perturbation reconcile how an axon may continue to function normally despite pathological changes in channel expression—those changes may not manifest overt changes in excitability until the appropriate trigger occurs. When the appropriate trigger does occur, the onset and offset of symptoms are dictated by the nonlinear dynamics of the system and can thus be very abrupt. The intrinsic nonlinear dynamics obviate any need to invoke unrealistically fast changes in ion channel expression, phosphorylation, etc to explain the attack. By extension, what constitutes a ‘sufficient’ perturbation depends on quantifiable features of the system’s bistability; specifically, the closer the quiet attractor (i.e. the stable fixed point) is to threshold (i.e. the saddle point), the more likely a perturbation is to knock the system to its active attractor. Similarly, the further the active attractor operates from the threshold, the more robust the afterdischarge will likely be (i.e. more Na+ accumulation will be required for termination). Previous experiments have suggested that motor axons can exist at two stable states on the basis of swings in extracellular K+ concentration combined with effects of the electrogenic Na+ pump (Bostock et al 1991, Baker 2000). Such mechanisms seem to explain spontaneous spiking (and parasthesia) over the time interval during which ion concentrations re-equilibrate following prolonged (15 min) ischemia. Such mechanisms are not sufficient to explain the evoked afterdischarge described here, although they could certainly modulate the initiation and/or termination of afterdischarge.
The strength of the perturbation required to trigger afterdischarge, or whether ectopic spiking can arise spontaneously, depends also on other physiological factors that have not been addressed. For instance, ischemia and hyperventilation increase the excitability of otherwise normal axons, giving rise to spontaneous (i.e. stimulus-independent) parasthesias and less commonly to muscle fasciculations (Mogyoros et al 1997). The greater likelihood of sensory symptoms compared with motor symptoms points to a differential susceptibility of sensory and motor axons to ectopic spiking. Motor axons may be less susceptible to the experimental manipulation (e.g. their ion gradients may be less affected by a certain duration of ischemia) or perhaps they are simply ‘less’ bistable. The same considerations apply when comparing normal and pathologically altered axons. For instance, the observation that normal sensory axons can quite readily be rendered hyperexcitable (see above) suggests that they, more than motor axons, may become ectopically active after small changes in the expression of certain ion channels.
An important question is changes in which channels are most likely responsible for afterdischarge; indeed, expression changes in certain channels may have no effect, or only indirect effects. Figure 3 illustrates that afterdischarge is not possible without sufficient expression of a slow inward current. The reason, explained in figure 2, hinges on the slow inward current implementing a positive feedback loop that encourages repetitive spiking by outlasting the negative feedback responsible for spike repolarization. The fast positive feedback loop responsible for generating individual spikes is clearly incapable of providing that sustained positive feedback. Thus, a large change in fast sodium conductance may be less consequential than a small change in slow sodium conductance. It should also be pointed out that a reduction in slow negative feedback (e.g. M-type or Ca2+-activated K+ channels) is liable to have an effect comparable to an increase in slow positive feedback (e.g. persistent Na+ channels). In short, conductances operating on different time scales interact in complex ways. This is not a new concept even in the context of axonal hyperexcitability (Rizzo et al 1996), but minimal modeling offers the framework to formally characterize and understand those complex interactions. Notably, although our minimal model reproduced all of the paroxysmal phenomena that we considered, and did so using physiologically plausible mechanisms, we cannot be certain that real axons necessarily use the dynamical mechanisms described herein. Nevertheless, identifying putative dynamical mechanisms should help direct experiments toward establishing definitive connections.
There is, of course, a long history of modeling spike initiation and propagation. With increases in computing power, there is less and less practical need to keep models simple. However, as models become more complicated, they become harder to analyze (in any formal mathematical manner at least) and ultimately harder to understand. Especially when one’s goal is to explain the basis for some well-characterized phenomenon, building a model with the minimally sufficient components may be a good approach. Such models afford the best opportunity to apply tools from dynamical system theory to formally characterize the nonlinear dynamical basis for the phenomenon (Rinzel et al 1998, Izhikevich 2007). As illustrated in this study, clinically relevant changes in excitability can be replicated in surprisingly simple models, and can be explained on the basis of a relatively small number of complex nonlinear interactions.
Acknowledgments
This work was supported by the Howard Hughes Medical Institute and NIH (5R01 MH079076) (TJS) and by start-up funds from the University of Pittsburgh (SAP). SAP is also a Rita Allen Foundation Scholar in Pain and the 53rd Mallinckrodt Scholar.
References
- Baker MD. Axonal flip-flops and oscillators. Trends Neurosci. 2000;23:514–9. doi: 10.1016/s0166-2236(00)01645-3. [DOI] [PubMed] [Google Scholar]
- Baldissera F, Cavallari P, Dworzak F. Motor neuron ‘bistability’. A pathogenetic mechanism for cramps and myokymia. Brain. 1994;117:929–39. doi: 10.1093/brain/117.5.929. [DOI] [PubMed] [Google Scholar]
- Bostock H, Baker M, Reid G. Changes in excitability of human motor axons underlying post-ischaemic fasciculations: evidence for two stable states. J. Physiol. 1991;441:537–57. doi: 10.1113/jphysiol.1991.sp018766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvin WH, Devor M, Howe JF. Can neuralgias arise from minor demyelination? Spontaneous firing, mechanosensitivity, and afterdischarge from conducting axons. Exp. Neurol. 1982;75:755–63. doi: 10.1016/0014-4886(82)90040-1. [DOI] [PubMed] [Google Scholar]
- Coggan JS, Prescott SA, Bartol TM, Sejnowski TJ. Imbalance of ionic conductances contributes to diverse symptoms of demyelination. Proc. Natl. Acad. Sci. USA. 2010;107:20602–9. doi: 10.1073/pnas.1013798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu. Rev. Neurosci. 2009;32:1–32. doi: 10.1146/annurev.neuro.051508.135531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor M, Amir R, Rappaport ZH. Pathophysiology of trigeminal neuralgia: the ignition hypothesis. Clin.J.Pain. 2002;18:4–13. doi: 10.1097/00002508-200201000-00002. [DOI] [PubMed] [Google Scholar]
- Devor M, Wall PD. Cross-excitation in dorsal root ganglia of nerve-injured and intact rats. J. Neurophysiol. 1990;64:1733–46. doi: 10.1152/jn.1990.64.6.1733. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 2010;33:325–47. doi: 10.1146/annurev-neuro-060909-153234. [DOI] [PubMed] [Google Scholar]
- Donaghy M, Compston A, Rossor M, Warlow C. Clinical diagnosis. In: Donaghy M, editor. Brain’s Diseases of the Nervous System. Oxford University Press; New York: 2001. pp. 1–59. [Google Scholar]
- Ermentrout B. Simulating, Analyzing, and Animating Dynamical Systems: A Guide to XPPAUT for Researchers and Students. SIAM; Philadelphia, PA: 2002. [Google Scholar]
- Felts PA, Kapoor R, Smith KJ. A mechanism for ectopic firing in central demyelinated axons. Brain. 1995;118:1225–31. doi: 10.1093/brain/118.5.1225. [DOI] [PubMed] [Google Scholar]
- Geurts JJ, Barkhof F. Grey matter pathology in multiple sclerosis. Lancet Neurol. 2008;7:841–51. doi: 10.1016/S1474-4422(08)70191-1. [DOI] [PubMed] [Google Scholar]
- Henderson WR. Trigeminal neuralgia: the pain and its treatment. Br. Med. J. 1967;1:7–15. doi: 10.1136/bmj.1.5531.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izhikevich EM. Dynamical Systems in Neuroscience. MIT Press; Cambridge, MA: 2007. [Google Scholar]
- Mogyoros I, Kiernan MC, Burke D, Bostock H. Excitability changes in human sensory and motor axons during hyperventilation and ischaemia. Brain. 1997;120:317–25. doi: 10.1093/brain/120.2.317. [DOI] [PubMed] [Google Scholar]
- Morris C, Lecar H. Voltage oscillations in the barnacle giant muscle fiber. Biophys. J. 1981;35:193–213. doi: 10.1016/S0006-3495(81)84782-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochoa J, Torebjork E. Sensations evoked by intraneural microstimulation of single mechanoreceptor units innervating the human hand. J. Physiol. 1983;342:633–54. doi: 10.1113/jphysiol.1983.sp014873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochoa JL, Torebjork HE. Paraesthesiae from ectopic impulse generation in human sensory nerves. Brain. 1980;103:835–53. doi: 10.1093/brain/103.4.835. [DOI] [PubMed] [Google Scholar]
- Ostermann PO, Westerberg CE. Paroxysmal attacks in multiple sclerosis. Brain. 1975;98:189–202. doi: 10.1093/brain/98.2.189. [DOI] [PubMed] [Google Scholar]
- Prescott SA, De Koninck Y, Sejnowski TJ. Biophysical basis for three distinct dynamical mechanisms of action potential initiation. PLoS Comput. Biol. 2008;4:e1000198. doi: 10.1371/journal.pcbi.1000198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinzel J, Ermentrout GB. Analysis of neural excitability and oscillations. In: Koch C, Segev I, editors. Methods in Neuronal Modeling: From Ions to Networks. MIT Press; Cambridge, MA: 1998. pp. 251–91. [Google Scholar]
- Rinzel J, Lee YS. Dissection of a model for neuronal parabolic bursting. J. Math. Biol. 1987;25:653–75. doi: 10.1007/BF00275501. [DOI] [PubMed] [Google Scholar]
- Rizzo MA, Kocsis JD, Waxman SG. Mechanisms of paresthesiae, dysesthesiae, and hyperesthesiae: role of Na+ channel heterogeneity. Eur. Neurol. 1996;36:3–12. doi: 10.1159/000117192. [DOI] [PubMed] [Google Scholar]
- Sakurai M, Kanazawa I. Positive symptoms in multiple sclerosis: their treatment with sodium channel blockers, lidocaine and mexiletine. J. Neurol. Sci. 1999;162:162–8. doi: 10.1016/s0022-510x(98)00322-0. [DOI] [PubMed] [Google Scholar]
- Seltzer Z, Devor M. Ephaptic transmission in chronically damaged peripheral nerves. Neurology. 1979;29:1061–4. doi: 10.1212/wnl.29.7.1061. [DOI] [PubMed] [Google Scholar]
- Strogatz SH. Nonlinear Dynamics and Chaos: With Applications to Physics, Biology, Chemistry, and Engineering. Addison-Wesley; Don Mills, ON: 1998. [Google Scholar]
- Stys PK, Waxman SG, Ransom BR. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na+-Ca2+ exchanger. J. Neurosci. 1992;12:430–9. doi: 10.1523/JNEUROSCI.12-02-00430.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey JA, Espir ML. Paroxysmal symptoms as the first manifestations of multiple sclerosis. J. Neurol. Neurosurg. Psychiatry. 1980;43:296–304. doi: 10.1136/jnnp.43.4.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG. Membranes, myelin, and the pathophysiology of multiple sclerosis. N. Engl. J. Med. 1982;306:1529–33. doi: 10.1056/NEJM198206243062505. [DOI] [PubMed] [Google Scholar]
- Waxman SG. Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nat. Rev. Neurosci. 2006;7:932–41. doi: 10.1038/nrn2023. [DOI] [PubMed] [Google Scholar]
- Waxman SG, Kocsis JD, Black JA. Pathophysiology of demyelinated axons. In: Waxman SG, Kocsis JD, Stys PK, editors. The Axon: Structure, Function and Pathophysiology. Oxford University Press; New York: 1995. pp. 438–61. [Google Scholar]
- Wolfe J, Houweling AR, Brecht M. Sparse and powerful cortical spikes. Curr. Opin. Neurobiol. 2010;20:306–12. doi: 10.1016/j.conb.2010.03.006. [DOI] [PubMed] [Google Scholar]