

Abstract
In this issue of Molecular Cell, Lee et al. (2011) report a novel mechanism for oxygen-sensing in S. pombe, whereby the 2-OG-Fe(II) dioxygenase Ofd protein regulates both the DNA binding activity and the degradation of the hypoxia regulated transcription factor, Sre1p.
In 2005, Hughes et al. described a novel mechanism coupling the regulation of sterol production with hypoxia through the sterol regulatory element binding protein (SREBP) pathway in the yeast S. pombe. SREBPs were first shown to regulate expression of genes involved in the uptake and production of lipids in mammals (Osborne and Espenshade, 2009). The work of Hughes et al. (2005) demonstrated that the S. pombe SREBP homologue, Sre1p, activates genes involved in sterol synthesis under hypoxic conditions. This is congruent with recent studies in mammals that suggest SREBPs evolved to connect lipid metabolism to cellular adaptive processes that require lipids to accomplish their physiological task (Osborne and Espenshade, 2009). The conversion of the committed sterol pathway metabolite squalene into ergosterol, which is the major yeast sterol, requires 12 equivalents of molecular oxygen (Todd et al., 2006). Thus, an increase in production of the key oxygen-sensitive enzymes of sterol biosynthesis would be predicted to retain pathway flux during times of limiting oxygen. The sterol-dependent coupling of Sre1p activation in response to hypoxia only provided an indirect connection between Sre1p and oxygen. However, powerful genetic tools available in yeast coupled with insightful biochemistry, uncovered a direct role for molecular oxygen regulation of Sre1p function (Lee et al., 2009). Further work by Lee et al. (2009), amongst others, showed that the Ofd protein enhanced degradation of Sre1p under normoxic conditions but repression was inactive during hypoxia (Fig. 1). The Ofd protein is predicted to be an 2-OG-Fe(II) dioxygenase dependent hydroxylase enzyme and the amino-terminal oxygen binding hydroxylase domain is linked to a carboxyl-terminal domain that interacts with the Nro1 protein, but only under hypoxic conditions (Lee et al., 2009). During normoxia, oxygen binds to N-terminal Ofd, which in turn alters the Ofd carboxyl-terminal domain so that it no longer interacts with Nro1. When not bound to Ofd, Nro1 binds nuclear Sre1p and accelerates its proteasomal degradation. In studies to decipher how the Nro1 degradation mechanism communicates with the proteasome, Lee et al. (2011) report in this issue of Molecular Cell that when proteasomal degradation of Sre1p was blocked due to an inactivating mutation in Ubr1, the E3 ubiquitin ligase that targets SRE1p to the proteasome, Ofd still regulated Sre1p activity even though its degradation was blocked (Fig. 1). Further studies on the mechanism revealed that the carboxyl terminal domain of Ofd directly inhibited DNA binding by Sre1p. Thus, Ofd regulates the Sre1p hypoxic transcription factor at two different levels.
Fig. 1. Oxygen-dependent regulation of Sre1p by Ofd.
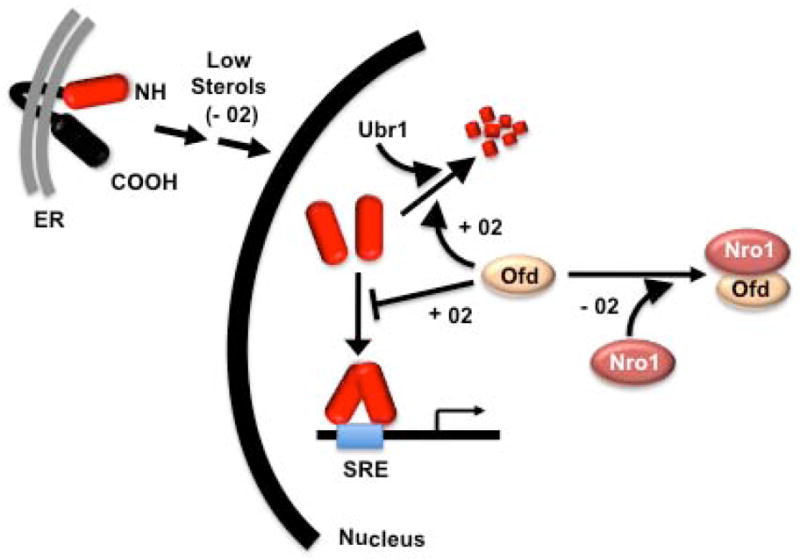
Sre1p is embedded in the ER membrane until sterol levels fall due to decreased synthesis under low oxygen. After a multi-step processing pathway described in more detail in (Osborne and Espenshade, 2009), the mature Sre1p transcription factor (red) enters the nucleus and has different fates depending on the oxygen regulated activity of Ofd. At limiting oxygen, the carboxyl terminal domain of Apo-Ofd binds to Nro1 and Sre1p is free to bind SRE containing promoters to activate its target genes. As oxygen levels rise, the amino-terminal dioxygenase domain of Ofd binds oxygen and it dissociates from Nro1 and inhibits Sre1p at two levels: 1) by stimulating Ubr1 dependent proteasomal degradation; and 2) directly inhibiting Sre1p DNA binding.
In multicellular eukaryotes from worms to mammals, the heterodimeric Hif (hypoxia-inducible factor) family of proteins are responsible for major hypoxia regulated transcriptional responses (Semenza 2011). The Hifs are composed of an α and β, with α being the key regulated subunit. The mechanism for hypoxic regulation of Hifs also involves 2-OG-Fe(II) dioxygenase dependent hydroxylation enzymes that, similar to Sre1p, control Hif at two different levels (Semenza 2011). Whereas it is clear that the hydroxylase activity of Ofd is required for Sre1p regulation it is not clear whether Ofd directly hydroxylates itself or Sre1p. In contrast, the proline dependent PHD domain enzymes directly hydroxylate critical proline residues of Hifα proteins, targeting them for proteasomal degradation through the VHL E3 ubiquitin ligase. A distinct FIH arginine hydroxylase modifies critical arginine residues within the HIFα transactivation domain that inhibit its interaction with the P300/CBP family of transcriptional coactivators and decreased Hif dependent transcriptional activation.
SREBPs, including Sre1p, are expressed as ER membrane targeted precursors that are tethered through two closely spaced membrane spanning helices such that both the amino-terminal half corresponding to the mature transcription factor, and the carboxyl terminal regulatory domain project into the cytoplasm (Osborne and Espenshade, 2009). The oxygen dependent Sre1p turnover occurs only after sterol dependent cleavage of the precursor from the membrane (Fig. 1). Proteolytic cleavage from the membrane occurs only after the precursor SREBPs are escorted from the ER to the Golgi apparatus. In mammals, the regulated translocation requires several additional proteins and comparative sequencing coupled with key biochemical studies have demonstrated that although the fundamental mechanism seems to be conserved from yeast to man, the molecular itinerary has species specific components that likely reflect the unique role of SREBPs in different organisms.
Sre1p protein processing is regulated by oxygen dependent sterol availability and its nuclear protein level is regulated directly by the oxygen sensing Ofd protein. There is also evidence that mammalian SREBPs are also regulated at both levels. After the sterol dependent protein maturation, nuclear mammalian SREBPs are regulated by additional nutrient sensitive mechanisms. Once in the nucleus, mammalian SREBPs are acetylated by CBP/P300 transcriptional coactivators, which prevent the mature SREBPs from being phosphorylated and targeted for degradation (Sundqvist et al., 2005). After SIRT1 deacetylation, SREBP becomes a substrate for phosphorylation by GSK3 and possibly other kinases and phosphorylated SREBPs are targeted to the proteasome for degradation through interaction with Fbx7 ubiquitin ligase. SIRT1 activity is induced in the mammalian liver by fasting and repressed by refeeding providing a mechanism to rapidly modulate hepatic nuclear SREBP-1 levels in accordance with its key role in feeding dependent hepatic lipogenesis (Walker et al., 2010). GSK3 activity is inhibited by AKT signaling which is another major pathway for growth dependent regulation of SREBPs (Sundqvist et al., 2005). In fact, Duvel et al. (2010) showed that SREBPs are essential for the high growth factor-independent proliferation of mouse MEFs that have constitutive TORC1 signaling owing to an inactivation of the negative regulatory TSC1/TSC2 complex. Signaling through AKT results in the inhibition of TSC1/TSC2 and de-repression of TORC1. In studies using the TOR inhibitor rapamycin coupled with mouse embryo fibroblasts that lack either TSC1 or 2, Duvel et al. (2010) showed that SREBP-1 and Hif are the two major transcriptional activation programs that are activated in response to constitutive TORC1. The mechanism for SREBP regulation by PI3 kinase signaling is complex and beyond the scope of this preview.
Interestingly, similar to the findings of Lee et al. that Ofd is involved in hypoxia, its mammalian orthologue, OGFOD1 appears to be a nuclear protein involved in regulation of oxygen supply, yet there is no apparent alteration of nuclear SREBP-1 levels upon knockdown of OGFOD1 (Saito et al., 2010). Consistent with this result, there is no 5 evidence that SREBPs are directly involved in hypoxic regulation in multi-cellular eukaryotes. However, a connection to Hif factors through TORC1 signaling (Duvel et al., 2010) provides a mechanism whereby SREBPs and the major hypoxic regulatory pathway are coupled in higher eukaryotes. The coordinate regulation of SREBPs and Hif is likely important in the PI3 kinase-dependent growth of tumor cells that occurs along with the shift to aerobic glycolysis also referred to as the “Warburg Effect”. Like the mammalian SREBPs, the S. pombe Sre1p is also activated by cellular stress but whether TOR signaling also targets it remains to be determined.
Epigenetic mechanisms are also likely involved in the hypoxic response because additional 2-OG-Fe(II) dependent dioxygenase targets of Hifa are the JmjC class of histone demethylases that are involved in dynamically regulating histone methylation levels (Xia et al., 2009). Similar to the Sre1p coupling of sterol metabolism to hypoxia, the histone demethylases are likely up-regulated to maintain levels of methylated histones during hypoxia. Whether a similar process is also regulated by Sre1p in S. pombe is not clear but two of the “uncharacterized targets” of Sre1p in S. pombe are predicted to be 2-OG-Fe++ dependent dioxygenases (Todd et al., 2006) and it is tempting to speculate that these enzymes may function in regulating additional processes where oxygen is also required like histone de-methylation.
In summary, SREBPs have evolved in both unicellular and multicellular eukaryotes to couple lipid metabolism to a myriad of physiological adaptations that require communication with the extracellular environment. The acute responses required to adapt to changes in extracellular oxygen levels in single-celled organisms involves a novel mechanism that couples hypoxia to cellular metabolic pathways that require oxygen. A key component of this adaptation involves the bivalent action of the 2-OGFe( II) dioxygenase Ofd on the Sre1p transcription factor described by Lee et al. in this issue.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, et al. Molecular cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hughes AL, Todd BL, Espenshade PJ. Cell. 2005;120:831–842. doi: 10.1016/j.cell.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 3.Lee CY, Stewart EV, Hughes BT, Espenshade PJ. EMBO J. 2009;28:135–143. doi: 10.1038/emboj.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee CY, Yeh TL, Hughes BT, Espenshade PJ. Molecular Cell. 2011 doi: 10.1016/j.molcel.2011.08.031. [CITATION DETAILS TO BE INSERTED WHEN AVAILABLE PLEASE] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Osborne TF, Espenshade PJ. Genes & development. 2009;23:2578–2591. doi: 10.1101/gad.1854309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saito K, Adachi N, Koyama H, Matsushita M. FEBS letters. 2010;584:3340–3347. doi: 10.1016/j.febslet.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 7.Semenza GL. N Engl J Med. 2011;365:537–547. doi: 10.1056/NEJMra1011165. [DOI] [PubMed] [Google Scholar]
- 8.Sundqvist A, Bengoechea-Alonso MT, Ye X, Lukiyanchuk V, Jin J, Harper JW, Ericsson J. Cell metabolism. 2005;1:379–391. doi: 10.1016/j.cmet.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 9.Todd BL, Stewart EV, Burg JS, Hughes AL, Espenshade PJ. Mol Cell Biol. 2006;26:2817–2831. doi: 10.1128/MCB.26.7.2817-2831.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walker AK, Yang F, Jiang K, Ji JY, Watts JL, Purushotham A, Boss O, Hirsch ML, Ribich S, Smith JJ, et al. Genes & development. 2010;24:1403–1417. doi: 10.1101/gad.1901210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xia X, Lemieux ME, Li W, Carroll JS, Brown M, Liu XS, Kung AL. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:4260–4265. doi: 10.1073/pnas.0810067106. [DOI] [PMC free article] [PubMed] [Google Scholar]