

Abstract
Recent studies in yeast have found that processing of DNA-double strand breaks (DSB) for recombination repair involves Sgs1 helicase. Human cells have five Sgs1 homologues with unknown selectivity and significance for repair of different DSB types. Here we examined the importance of WRN helicase in repair of G2-specific DSB caused by abnormal mismatch repair (MMR) of ternary Cr-DNA adducts. We found that Cr(VI) induced a rapid dispersal of WRN from the nucleolus resulting in its prolonged retention in the nucleoplasm. The loss of MSH2 or MLH1 MMR proteins abolished the long-term but not the initial WRN relocalization. WRN-deficient fibroblasts were hypersensitive to Cr(VI)-induced clonogenic death and contained high levels of persistent DSB detected by γ-H2AX/53BP1 foci and pulsed-field gel electrophoresis. WRN was involved in recombination repair of Cr-induced DNA damage, as evidenced by WRN-RAD51 colocalization and defective formation of RAD51 foci in the absence of WRN. The accumulation of unrepaired DSB in WRN-depleted cells was rescued by the inactivation of MMR, indicating that MMR-generated DSB were a key substrate for WRN action in Cr(VI)-treated cells. Competition for the limited amounts of WRN in primary cells between G2 processes of telomere rebuilding and recombinational repair is expected to increase persistence of Cr-induced DSB and may cause telomeric abnormalities in tissues of chronically chromate-exposed workers. Our work provides the first demonstration of the major importance of WRN in repair of the specific class of DSB in human cells.
Introduction
Werner syndrome (WS) is a rare genetic disorder of premature ageing that is manifested by the early appearance of gray hair, cataracts, scleroderma, diabetes and increased incidence of cancer.1–3 WRN, the protein mutated in Werner syndrome, is unique among the five-member human RecQ helicase family as it contains both helicase and exonuclease activities.1,3 Normal localization of WRN is nucleolar4–6 but in afflicted individuals, this protein is often missing the nuclear localization signal resulting in its exclusion from the nucleus.1,2 WS cells display a prolonged S-phase,7 low proliferative capacity3 and increased sensitivity to drugs with S-phase dependent toxicity.8–10 The preferred substrates for WRN action include various types of 3- and 4-way DNA junctions, bubbles, extrahelical loops and DNA overhangs.3,11,12 These abnormal DNA structures can arise spontaneously in the areas of DNA repeats and they can be induced by incomplete repair or replication. WRN is capable of resolving a broad range of alternative DNA structures due to its ability to recognize these structures and process them via unwinding and cleavage reactions.1–3
WRN interacts with several proteins involved in repair of DNA double-strand breaks (DSB)13–18 and localizes to the sites of laser-induced DSB in live cells.19 However, WS cells exhibited only a mild sensitivity toward ionizing radiation in clonogenic survival experiments,2,16 which was indicative of a minor if any role of WRN in the overall DSB repair. Recent studies have found that the yeast RecQ helicase Sgs1 stimulated recombination repair of DSB through its essential function in one of the two redundant DNA ends resection pathways.20,21 The RecQ helicase BLM appears to play a role analogous to that of Sgs1 in campthothecin-treated human cells where it acts in parallel with EXO1 to promote recombination-associated foci and cell survival.22 The presence of five RecQ members in human cells potentially indicates that individual helicases may participate in repair of different classes of DSB. It is currently unknown whether repair of specific DSB in human cells is particularly dependent on WRN or even involves this helicase.
In this work, we investigated the importance of WRN in cellular survival and repair of DSB induced by the potent human carcinogen chromium(VI). The main source of DSB in Cr(VI)-treated human cells is misprocessing of Cr-DNA damage by mismatch repair (MMR).23–25 The majority of Cr-induced DSB are formed in G2 phase following replication of Cr-adducted DNA.23,24 Ternary Cr-DNA adducts, such as cysteine-Cr-DNA or ascorbate-Cr-DNA crosslinks, were particularly good substrates for recognition by MMR proteins.24,25 These adducts were also a primary cause of the mutagenic responses generated in shuttle-vector plasmids damaged during Cr(VI) reduction in vitro with ascorbate26,27 or cysteine.28 Chronic formation of toxic DSB by MMR activity during repetitive exposures to Cr(VI) has been proposed to promote the selection of transformation-prone, Cr-resistant cells lacking MMR, providing an explanation for the unusually high frequency of microsatellite instability (marker of inactive MMR) in lung cancers among chromate workers.29 Unlike other agents with MMR-dependent mechanisms of cytotoxicity,30–32 the formation of DSB in Cr-exposed cells is unusually rapid which results from the unprecedented activation of both the MSH6 and MSH3 branches of MMR.25 MSH3 specifically recognizes and repairs mismatches containing insertion/deletion loops,33,34 pointing to a potential formation of these structures in Cr-damaged DNA. Extrahelical loops are also high affinity substrates for WRN helicase1–3 but they can interfere with the activity of other DNA helicases. Therefore, it is possible that repair of MMR-generated DSB in Cr-damaged DNA may engage WRN. Cr-induced DSB are also well suited for testing the involvement of WRN in homologous recombination because it is a principal DSB repair process in S/G2 cells and the main target for the activity of RecQ helicases in lower organisms.20,21
Materials and Methods
Cell cultures and drug treatments
Human SV40 transformed fibroblast lines GM00637H (NF cells) and GM00847 (LNS cells) were purchased from Coriell Cell Repository. SV40 immortalized Werner syndrome fibroblasts AG11395 (WS cells) were obtained from NIA Aging Cell Culture Repository. Primary human lung IMR90 fibroblasts and colon HCT116 (MLH1−/−) cells were purchased from the American Type Culture Collection. HCT116+ch3 (MLH1+) cell line was a gift from T. Kunkel. NF, WS and IMR90 fibroblasts were cultured in DMEM supplemented with penicillin-streptomycin and serum (15% for NF and WS, 10% for IMR90). LNS cells were grown in MEM with 10% serum. HCT116 cells were grown in DMEM-F12 medium containing 10% serum and penicillin-streptomycin. Media for HCT116+ch3 cells additionally included 600 μg/ml geneticin. All cell lines were grown at 37°C in a humidified atmosphere containing 95% air-5% CO2. Cells were exposed to K2CrO4 [Cr(VI)] for 3 hr in serum-free medium. To increase cellular ascorbate concentrations, cells were incubated for 90 min with 1 mM dehydroascorbic acid in Krebs-HEPES buffer supplemented with 0.5 mM glucose.35 Cellular ascorbate was measured by a modified HPLC procedure based on the fluorescent detection of a specific conjugate with 1,2-diamino-4,5-dimethoxybenzene dihydrochloride.36
Clonogenic survival
A total of 2000 cells were seeded onto 100-mm dishes one day prior to treatments with Cr(VI). Three dishes were used for each dose. Ten to 14 days after exposure, colonies were fixed with 100% methanol and stained with Giemsa solution and counted. Clonogenic experiments were repeated at least twice.
Western blotting
Cells were collected by scraping, washed twice with cold phosphate-buffered saline (PBS) and resuspended in a buffer containing 50 mM Tris pH 8.0, 250 mM NaCl, 1% NP40, 0.1% SDS, 5 mM EDTA, 2 mM Na3VO4, 10 mM Na2P2O7, 10 mM NaF, 1 mM PMSF and protease inhibitor cocktail. Lysates were incubated on ice for 10 min and then cell debris was spun down at a speed of 10,000 × g for 5 min at 4°C. Samples were denatured by 2X loading buffer with 10% 2-mercaptoethanol and boiled for 10 min. Proteins were separated by SDS-polyacrylamide gel electrophoresis and electrotransferred to the ImmunoBlot PVDF membrane (Bio-Rad). Primary antibodies were rabbit polyclonal anti-WRN (Santa Cruz Biotechnology), mouse anti-MLH1, anti-MSH6 and anti-MSH2 (BD Biosciences). Protein bands were visualized using horseradish peroxidase-conjugated secondary antibodies (Upstate) and an enhanced chemiluminescence kit (Amersham).
Stable shRNA knockdowns
Stable depletion of protein levels was achieved by expression of short hairpin RNA (shRNA) from pSUPER-RETRO retroviral vector (Oligoengine). The sequence for WRN-targeting shRNA was 5′-GTGTATAGTTACGATGCTA-3′ and for MSH6 was 5′-GGTGATCCCTCTGAGAACT-3′. The control GFP-targeting sequence was 5′-GCAAGCTGACCCTGAAGTT-3′. The luciferase-targeting sequence was 5′-GCGACCAACGCCTTGATTG-3′. Double-stranded oligonucleotides were ligated into the pSUPER-RETRO vector that was linearized with BamHI and HindIII. The pSUPER-RETRO vectors for depletion of MLH1 and MSH224 and details of retroviral infections37 have been described previously. For double knockdown experiments, NF cells were first infected with pSUPER-puro vectors, selected with puromycin for one week and then re-infected with pSUPER-neo vectors and selected with 400 μg/ml geneticin for two weeks. To avoid loss of shRNA efficacy in long-term cultures, new stocks of infected cells were used every 4 weeks.
Immunofluorescence
Cells were grown on Superfrost Plus slides and exposed to Cr(VI) for 3 hr, then returned to complete medium. At the selected times, cells were washed twice with PBS and fixed with 2% paraformaldehyde in PBS for 20 min at room temperature. Cells were then permeabilized for 20 min with 1% Triton X-100, followed by incubation with 3% fetal bovine serum in PBS for 1 hr. Primary antibodies against WRN (rabbit polyclonal antibodies, Santa Cruz) and B23 (mouse monoclonal antibody from Chemicon) were used at 1:200 dilution and incubated overnight at 4°C. For primary cells and Rad51 staining (anti-Rad51 mouse monoclonal antibody from Abcam, 1:400 dilution), cells were incubated at 37°C for 3 hr before and after overnight incubation at 4°C. DSB repair sites were labeled by simultaneous incubation of primary antibodies for γ-H2AX at 1:1000 dilution (Upstate) and anti-53BP1 at 1:500 dilution (Santa Cruz) for 2 hr at 37°C in a humidified chamber. The secondary antibodies, Alexa Fluor 488-conjugated anti-mouse immunoglobulin G, Alexa Fluor 594-conjugated anti-mouse immunoglobulin G, Alexa Fluor 488-conjugated anti-human immunoglobulin G and Alexa Fluor 594-conjugated anti-rabbit immunoglobulin G (Molecular Probes) were incubated for 1 hr at room temperature. Antibodies were diluted in PBS containing 1% bovine serum albumin and 0.5% Tween 20, except for detection of γ-H2AX where the antibodies were diluted in a 2% albumin-PBS solution. Slides were mounted with Vectashield hard set mounting medium with DAPI (Vector Laboratories). Fluorescence images were recorded with a Zeiss Axiovert 100 confocal microscope and analyzed by Phoenix software. Experiments were repeated with 3–6 slides and at least 100 cells were analyzed on each slide.
Pulsed-field gel electrophoresis (PFGE)
Cells were exposed to Cr(VI) for 3 hr and returned to complete medium for 6 hr or 24 hr. PFGE plugs were prepared using BioRad Mammalian CHEF Genomic Plug Kit at 106 cells/plug and incubated in a proteinase K buffer [100 mM EDTA, 1% N-laurylsarcosyl, 10 mM Tris, pH 8.0, and proteinase K 1 mg/ml] at 20°C for 24 hr. The plugs were washed 5 times in a wash buffer [20 mM Tris, pH 8.0 and 50 mM EDTA] with agitation prior to loading. PFGE was performed using a 1% gel (Pulsefield Certified Megabase Agarose, BioRad) in 0.5X TBE buffer on a CHEF Mapper XA Pulsed Field Electrophoresis System (BioRad) for 18 hr at 14°C under the following current conditions: 120° field angle, 240 s switch time, 4 V/cm. DNA was visualized by staining with ethidium bromide.
Results
Increased sensitivity of WRN-deficient cells to Cr(VI) toxicity
Expression of telomerase is known to compensate for the absence of WRN in human WS cells.38,39 The presence of active telomerase is also responsible for suppression of premature aging phenotype in Wrn-knockout mice.40,41 Introduction of active hTERT into BJ human fibroblasts increased resistance and strongly inhibited the induction of chromosomal instability by Cr(VI),42 indicating that telomerase-immortalized cells have altered processing of Cr-induced damage. Therefore, we chose to perform our investigation in telomerase-negative human cells. A potential role of WRN helicase in cellular responses to Cr-DNA damage was first examined by clonogenic survival of Cr-treated WRN-expressing NF and WRN-null WS fibroblasts (Fig. 1A). WS cells displayed high sensitivity to clonogenic lethality by Cr(VI) with only a few colonies surviving at 10 μM Cr while approximately 50% NF cells were clonogenically viable at this dose. Since NF and WS cells are not isogenic, we created a stable knockdown of WRN protein in NF cells by expressing a targeting shRNA from a chromosomally integrated pSUPER-RETRO vector (Fig. 1B, top panel). The selected shRNA decreased WRN levels about 10-fold as compared to NF cells expressing nonspecific shRNA (GFP). Using this isogenic model of WRN deficiency, we found again that the absence of WRN caused hypersensitivity to Cr(VI) lethality (Fig. 1C). To further confirm our findings, we constructed a stable WRN knockdown in telomerase-negative LNS fibroblasts (Fig. 1B, bottom panel). Although LNS cells were more resistant to Cr(VI) relative to NF cells, WRN knockdown in these cells resulted in essentially the same degree of sensitization to Cr(VI) toxicity (Fig. 1D).
Figure 1. Loss of WRN decreases survival of cells treated with Cr(VI).
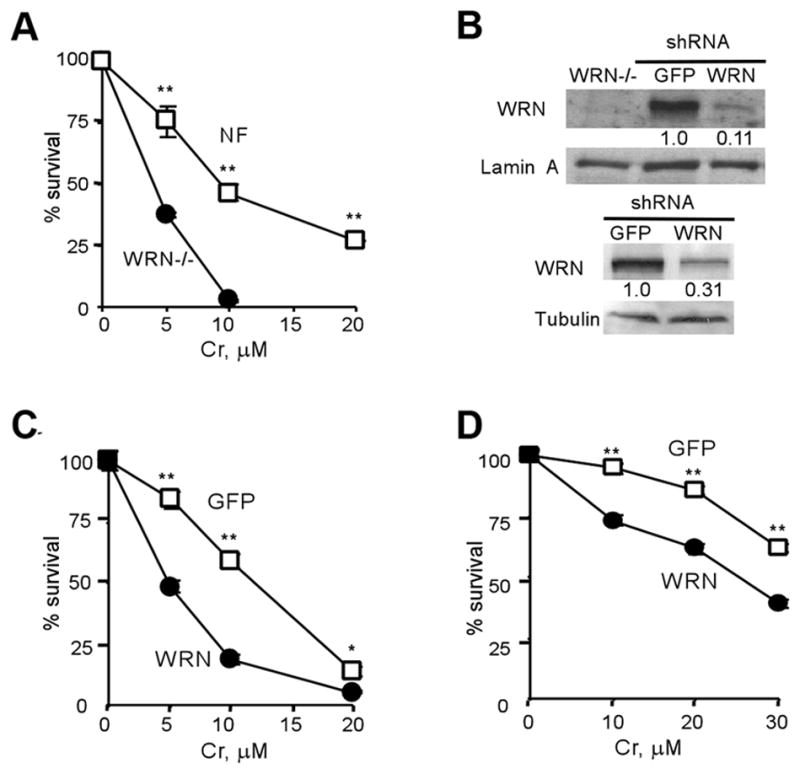
Cell survival was determined by clonogenic assay. Results are means±SD of 2–3 independent experiments, each with triplicate dishes per dose. Where not seen, error bars were smaller than the symbol. Statistics (Student’s t-test): *- p<0.05, **- p<0.01 relative to WRN-deficient cells. (A) Clonogenic viability of WS and NF fibroblasts treated with Cr(VI). (B) WRN levels in NF (top panel) and LNS (lower panel) cells expressing targeting and nonspecific (GFP) shRNA. (C) Cr(VI) toxicity in NF cells expressing WRN-targeting or nonspecific (GFP) shRNA. (D) Survival of LNS cells expressing WRN or GFP-targeting shRNA.
Altered intranuclear localization of WRN after Cr(VI) damage
WRN is typically found in the nucleolus of primary and transformed human cells.5 Costaining for WRN and the nucleolar marker B23 showed that the majority of NF cells also exhibited nucleolar localization of WRN (Fig. 2A). In response to Cr(VI) exposure, WRN relocalized from the nucleolus into the nucleoplasm. The nucleoplasmic localization was observed immediately after 3 hr-long exposures to 10–20 μM Cr(VI) and it was retained in treated cells up to 24 hr post-treatment. The majority of relocalized WRN produced a largely uniform diffuse staining pattern. Costaining of Cr(VI)-treated cells with antibodies for WRN and a marker of DSB, γ–H2AX,43 showed that most DSB-positive cells displayed the diffuse WRN nuclear distribution and many γ–H2AX foci colocalized with WRN (Fig. 2B,C).
Figure 2. Cr(VI) causes dispersion of WRN from nucleolus.

(A) Confocal images of control and 20 μM Cr(VI)-treated NF cells immunostained for WRN and the nucleolar marker B23. (B) A representative confocal image of Cr-treated cells displaying colocalization WRN with γ-H2AX foci. (C) Presence of γ-H2AX foci in NF cells with different patterns of WRN staining. Cells were treated with 20μM Cr(VI) and fixed for immunostaining at indicated times. Data are means±SD from three slides with >100 cells scored per slide.
Persistence of DSB in the absence of WRN
The observed colocalization of WRN with γ–H2AX foci suggested that WRN could be involved in repair of Cr(VI)-induced DSB. To test this possibility, we examined the effects of WRN deficiency on the persistence of DSB after Cr(VI) removal. The presence of DSB was detected by the appearance of 53BP1 and γ–H2AX foci and only cells exhibiting focal staining for both proteins were scored as DSB-positive (Fig. 3A). 53BP1 is recruited following remodeling of chromatin regions flanking the break44 and serves as a second marker of DSB. Levels of DSB induced by moderate doses of Cr(VI) typically approached their peak at about 6 hr post-exposure.23,24 Treatment of NF and LNS cells with moderately toxic Cr(VI) doses (~60% clonogenic survival) also resulted in a strong production of DSB foci at 6 hr post-Cr and WRN depletion had no significant effect on this initial induction of DSB (Fig. 3B,C). However, at 24 hr post-exposure, there were approximately twice as many DSB foci-positive cells with WRN knockdown as compared to their shGFP controls. Cr(VI) requires reductive activation inside cells in order to cause DNA damage.45 Human cells in culture contain low levels of ascorbate,25,35 which is the most rapid cellular reducer of Cr(VI).46,47 Restoration of physiological levels of ascorbate in primary human cells strongly potentiated the formation of DSB by Cr(VI).24 Our HPLC analyses detected approximately 25 μM ascorbate in NF cells, which is much lower than physiological levels of this vitamin in tissues.47 Therefore, we preloaded NF cells with ascorbate to 0.25 mM and then examined DSB accumulation at 24 hr post-Cr (Fig. 3D,E). As in primary cells,24 increasing cellular ascorbate levels enhanced DSB-producing activity of Cr(VI), and WRN depletion resulted in the presence of persistent DSB even after exposure to as low as 2 μM Cr(VI), which is the current EPA standard for chromium in drinking water.47 Analysis of chromosomal DNA by PFGE further confirmed the importance of WRN in repair of Cr-induced DSB as evidenced by 2.7-times (p<0.01) higher levels of remaining DNA breaks in WRN-depleted LNS cells at 24 hr after Cr(VI) exposure (Fig. 3F). Control LNS cells with normal WRN levels were also capable of repairing the majority of DSB generated even by 50 μM Cr(VI) while WRN-depleted cells were unable to show significant repair of much fewer DSB induced by 30 μM Cr(VI) (Fig. 3G).
Figure 3. Persistence of Cr-induced DSB in WRN-depleted cells.

(A) Confocal images of GFP and WRN-shRNA expressing NF cells stained with DAPI and antibodies for 53BP1 and γ-H2AX. Shown are representative images of untreated and 10 μM Cr-treated cells at 6 hr post-exposure. (B) Frequency of DSB-positive NF cells at 6 and 24 hr after exposure to 10 μM Cr(VI). Cells with ≥5 foci of both 53BP1 and γ-H2AX were counted as DSB-positive. WRN -cells expressing WRN-targeting shRNA, GFP - cells expressing nonspecific shRNA. Data are means ±SD for 5 slides with >100 cells analyzed per slide. Statistics: *- p<0.05, **- p<0.01 relative to shGFP cells. (C) Frequency of DSB-positive LNS cells following exposure to 30 μM Cr(VI). Definitions of cells are as in panel B. Results are means±SD for 3 slides with >100 cells counted per slide. (D) DSB levels in normally cultured NF cells at 24 hr post-exposure to low Cr(VI) doses. Data are means ±SD for 4 slides with >100 cells analyzed per slide. (E) Elevated DSB levels in NF cells preloaded with 0.25 mM ascorbic acid prior to Cr(VI) exposure. DSB foci were counted at 24 hr post-Cr. Data are means±SD for 4 slides with >100 cells counted per slide. (F) Determination of DSB repair by PFGE in LNS cells treated with 30 μM Cr(VI) (means±SD). (G) PFGE of chromosomal DNA from shGFP- and shWRN-LNS cells treated with 50 and 30 μM Cr(VI), respectively.
Attenuated formation of RAD51 foci in WRN-deficient cells
The majority of Cr-induced DSB are present in G2 cells.23,24 Since DSB repair in G2 cells occurs primarily via homologous recombination,48,49 we examined the effect of WRN on this repair process. The activity of recombination repair was assessed by scoring foci of RAD51, which is one of the essential recombination repair proteinsand a specific marker of recombination repair sites.50 Treatment of NF cells with either moderate 10 μM or highly toxic 20 μM doses of Cr(VI) did not increase the production of RAD51 foci above background levels at 6 hr post-exposure (Fig. 4A,B). At 24 hr post-Cr, NF cells with nonspecific GFP-shRNA displayed a significant induction of RAD51 foci at both Cr(VI) doses while WRN-depleted cells showed no change relative to untreated controls. Thus, activation of recombination repair in NF cells significantly lagged the induction of DSB in the presence of WRN (Fig. 3B,C) or was absent in cells with WRN knockdown. To further confirm these findings, we investigated the formation of RAD51 foci in LNS cells (Fig. 4C). Control LNS cells showed a strong induction of RAD51 foci already at 6 hr post-Cr but WRN depletion completely abrogated recombination foci at this time. At 24 hr post-Cr, WRN-depleted LNS cells displayed a significant increase in the number of RAD51 foci, which was still modestly lower than in control cells. Thus, both cell lines showed a strong positive effect of WRN on the formation of RAD51 foci. A more rapid activation of recombination repair in LNS cells may at least be partially responsible for their higher resistance to Cr(VI) lethality (Fig. 1D). We also analyzed RAD51 foci in WRN-null WS cells treated with 10 and 20 μM Cr(VI), doses that were clearly genotoxic to these cells (Fig. 1A). Neither Cr(VI) dose caused even a marginal increase in the induction of RAD51 foci at 24 hr post-exposure (Fig. 4D), further confirming the importance of WRN protein for the initiation of recombination repair of Cr-DNA damage. A large fraction of Cr-induced RAD51 focus-positive NF and LNS cells also showed a partial colocalization (ranging from 20 to 80%) of RAD51 foci with WRN (Fig. 4E,F).
Figure 4. WRN depletion inhibits the formation of RAD51 foci in Cr-treated cells.

(A) Representative confocal images of NF and LNS cells stained with DAPI and anti-RAD51 antibody. Cells were treated with 10 μM (NF cells) or 30 μM Cr(VI) (LNS cells) for 3 hr and fixed 6 and 24 hr later. (B) Induction of RAD51 foci Cr(VI) in NF cells. Cells with 3 or more foci were scored as focus-positive. Data are means±SD for 4 slides with >100 cells scored per slide. (C) Formation of RAD51 foci in LNS cells treated with 30 μM Cr(VI). Data are means±SD for 4 slides with >100 cells counted per slide. Statistics for panels B and C: *-p<0.05, **- p<0.01 relative to shGFP cells. (D) Number of RAD51 foci-positive WS cells at 24 hr post-Cr. Data are means±SD for 3 slides with at least 100 cells scored per slide. (E) Colocalization of WRN and RAD51 foci in LNS cells at 6 and 24 hr after treatment with 30 μM Cr(VI). (F) WRN and RAD51 colocalization in NF cells at 24 hr following exposure to 10 μM Cr(VI). For panels E and F, nuclei containing at least one clear RAD51 focus colocalizing with WRN were scored as positive. Typical colocalization for positive cells was 20–80% of all RAD51 foci. “−” Frequency of RAD51-positive cells without WRN colocalization, “+” –frequency of cells with RAD51 and WRN colocalization. Results are means±SD for 3 slides with >30 RAD51 foci-containing cells scored for colocalization. Statistics for panels E and F: *-p<0.05, **- p<0.01 relative to untreated cells.
Mismatch repair (MMR)-mediated DNA lesions and WRN
The main cause of DSB in human cells treated with low to moderately toxic doses of Cr(VI) is abnormal processing of Cr-DNA crosslinks by MMR.23–25 We have also observed that ascorbate, which enhances DSB production by aberrant MMR activity,24 increased the levels of DNA breaks in WRN-depleted cells at Cr(VI) concentrations that were otherwise nongenotoxic (Fig. 3D,E). Therefore, we tested whether WRN was responding to secondary DNA lesions generated by MMR. We first examined the dynamics of WRN localization in Cr(VI)-treated human HCT116 cells lacking or expressing MLH1, which is one of the essential MMR proteins.33,34 The majority of control cells in both lines showed costaining of WRN with the nucleolar marker B23, demonstrating that MLH1 has no effect on the retention of WRN in nucleoli of unstressed cells (Fig. 5A). Immediately after Cr(VI) exposure, WRN dispersed from the nucleolus into the nucleoplasm in the majority of cells irrespective of their MLH1 status. Similarly to NF cells (Fig. 2), MLH1+ HCT116 cells also retained nucleoplasmic WRN at 24 hr post-Cr. In contrast, MLH1−/− HCT116 cells restored the normal nucleolar localization of WRN at 3–6 hr post-exposure. To further investigate the MMR-dependence of WRN dynamics, we created stable knockdowns of MLH1 or MSH2 MMR proteins in primary human IMR90 fibroblasts (Fig. 5B). We found that WRN relocalized in all IMR90 constructs immediately after Cr(VI) exposure but while control Luc-shRNA cells retained WRN in the nucleoplasm, cells with MLH1 or MSH2 knockdowns restored nucleolar WRN staining already at 3 hr post-Cr (Fig. 5C). Thus, the initial relocalization of WRN occurred directly in response to Cr(VI)-induced stress but the long-term retention in the nucleoplasm required the presence of MMR proteins. The nucleoplasmic retention of WRN was not due to binding to MMR proteins, as co-immunoprecipitation experiments found no detectable association of WRN with MSH6 or MSH2 in control or Cr(VI)-treated NF cells (Fig. 5D). MSH6 immunoprecipitates contained large amounts of its binding partner MSH2,33,34 validating the effectiveness of the employed procedure. Sensitization of WRN-depleted cells to Cr(VI) toxicity did not appear to result from the alterations in the MMR system as expression of three major MMR proteins MSH6, MSH2 and MLH1 did not vary between GFP-shRNA and WRN-shRNA cells (Fig. 5E). Cr(VI) exposure also had no significant effect on the protein levels of WRN itself (Fig. 5E).
Figure 5. Nucleoplasmic retention of WRN in Cr(VI)-treated cells requires intact MMR.

(A) Frequency of MLH1−/− and MLH1+ HCT116 cells with nucleoplasmic WRN following treatment with 20 μM Cr(VI) (means±SD for 3 slides with >100 cells analyzed/slide). Statistics: *- p<0.05, **- p<0.01 relative to MLH1−/− cells. (B) Western blot of protein extracts from IMR90 cells expressing nonspecific (Luc), MSH2 or MLH1-targeting shRNA. (C) Quantitation of nuclear WRN dispersion in IMR90 cells treated with 10 μM Cr(VI) (means±SD for 3 slides with >100 cells analyzed/slide). Statistics: *- p<0.05, **- p<0.01 relative to MLH1- or MSH2-shRNA. (D) Coimmunoprecipitation of WRN with MSH6 or MSH2 in control or 20 μM Cr-treated NF cells. (E) Expression of MMR proteins in GFP-shRNA and WRN-shRNA NF cells after exposure to 20μM Cr(VI).
The observed MMR-dependent dynamics of WRN in the absence of direct interactions with MMR proteins suggested that WRN could be responding to MMR-induced DSB. To test this possibility, we constructed NF cells with double knockdowns of MSH6 and WRN (Fig. 6A). We found that depending on Cr(VI) dose, knockdown of MSH6 in WRN-depleted cells either completely abrogated or significantly suppressed the levels of persistent DSB foci (Fig. 6B). The presence of ascorbate strongly potentiated the formation of DSB by Cr(VI) (Fig. 3E) and at low doses essentially all ascorbate-promoted DSB are MMR-generated.24 We found that depletion of MSH6 completely eliminated the presence of persistent DSB in Cr-treated/ascorbate-preloaded cells with WRN knockdown (Fig. 6C). This effect was quite striking, as even cells with normal WRN levels (GFP/GFP controls) were unable to fully repair DSB. As with DSB induction, the formation of RAD51 foci by Cr(VI) was enhanced by preloading of control cells with ascorbate and MSH6 depletion abrogated the appearance of the recombination-active cells (Fig. 6D,E).
Figure 6. Rescue of WRN hypersensitivity by MSH6 depletion.

(A) Western blot for MSH6 and WRN in NF cells after double shRNA infections. WRN-shRNA was expressed from pSUPER-puro vector, MSH6 – from pSUPER-neo and nonspecific GFP-shRNA was expressed from either pSUPER-puro or pSUPER-neo depending on the resistance marker in the targeting vector. (B) DSB foci (53BP1 + γ-H2AX) in WRN-depleted NF cells with and without MSH6 knockdown (● - WRN-GFP shRNAs, □ - GFP-GFP shRNAs, ◇- GFP-MSH6 shRNAs, ▲-WRN-MSH6 shRNAs). Cells were treated with Cr(VI) for 3 hr and fixed for immunofluorescence 24 hr later. Data are means±SD for 3 slides with >100 cells counted per slide (*- p<0.05 for both GFP/GFP and WRN/MSH6 relative to WRN/GFP cells). (C) Frequency of DSB-positive double knockdown NF cells preloaded with 0.25 mM ascorbate prior to 5 μM Cr(VI) exposure. Cells were fixed for DSB foci scoring (53BP1 + γ-H2AX foci) at 24 hr post-Cr. Shown are means±SD for 3 slides with >100 cells counted per slide (*- p<0.05, **-p<0.01 relative to untreated cells). (D) Frequency of RAD51 foci-positive NF cells expressing nonspecific (GFP) and MSH6-targeting shRNA following exposure to Cr(VI) under normal culture conditions. RAD51 foci were scored at 24 hr post-treatment. Data are means±SD for 3 slides with >100 cells counted per slide. (E) As panel D except that cells were preloaded with 0.25 mM ascorbate prior to treatments with Cr(VI). Data are means±SD for 3 slides. Statistics for panels D and E: *- p<0.05, **- p<0.01 relative to shGFP cells.
Discussion
In this work, we found that WRN was actively engaged in cellular responses to Cr(VI). Immediately after short treatments with this carcinogen, WRN relocalized from the nucleolus into the nucleoplasm where it remained even at 24 hr post-exposure. The loss of WRN resulted in increased clonogenic toxicity of Cr(VI), demonstrating the importance of this RecQ-type helicase in cell survival. Cellular reduction of Cr(VI) generates a transient oxidative stress due to the formation of redox-active CrV/IV intermediates51,52 and yields stable Cr-DNA adducts.45 The prolonged nucleoplasmic retention of WRN in Cr-treated cells, but not its initial relocalization, was dependent on the presence of intact MMR. Thus, the early WRN movement was likely caused by the cellular stress during Cr(VI) metabolism but its subsequent nucleoplasmic presence reflected other events involving MMR. Processing of Cr-DNA adducts by human MMR generates DSB as secondary, more toxic DNA lesions.23–25 We found that WRN played a major role in repair of MMR-induced DSB in Cr(VI)-treated human fibroblasts without significant effects on the initial production of DNA breaks. The presence of unrepaired DSB was likely responsible for the decreased survival of WRN-depleted cells in the clonogenic assay. Poor repair of DSB in WRN-lacking cells was associated with their deficiency in activation of early steps in homologous recombination, as revealed by the absence or severely delayed formation of RAD51 foci after Cr-DNA damage. Functional recombination repair is known to play an important role in cellular resistance to Cr(VI) toxicity.53,54
Induction of DSB by MMR typically begins within 1–3 hr post-Cr exposure and occurs in G2 cells following their replication of Cr-adducted DNA.23,24 The late steps in MMR-dependent processing of Cr-DNA damage prior to DNA breakage are dependent on the activity of MSH3-MSH2 heterodimer (MutSβ complex).25 MutSβ dimer exhibits high specificity for DNA loops of various sizes,33,34 suggesting that MMR-induced DSB could be associated with the presence of loops or hairpins. While the presence of loops or hairpins can interfere with the activity of many DNA-processing enzymes, DNA ends with adjacent loops are high affinity substrates for WRN.2 Additionally, a strong requirement of WRN for the initiation of RAD51-dependent recombination in Cr-treated cells could have also reflected a limited availability of EXO1 that operates in a parallel pathway leading to the activation of recombination repair of DSB.20–22 The role of the RecQ helicases in DSB processing was most evident when cells were made deficient in EXO1/Exo1. Because EXO1 is also an essential component of human MMR,33,34 a massive formation of ternary Cr-DNA adducts recruiting MMR complexes and stimulating their activity is expected to sequester a major fraction of this low abundance exonuclease.22
Both MSH2-MSH3 and MSH2-MSH6 mismatch-sensing complexes were capable of stimulating DNA duplex unwinding by WRN in vitro.55 The biological relevance of these findings was further supported by physical interactions between WRN and MSH2 in yeast two-hybrid studies. High duplex unwinding activity should promote DSB repair, as helicase activity is essential for 5′→3′ exonuclease degradation of DSB ends in the RecQ/Sgs1-dependent recombination process.20–22 Since MMR proteins are present at the sites of Cr-induced DSB,25 they could recruit WRN and/or stimulate its strand unwinding activity and thereby contribute to its prominent role in DSB ends processing that is necessary for RAR51 loading. However, we were so far unable to detect interactions of WRN with MSH2 or MSH6 by co-IP in either control or Cr(VI)-treated cells. It is possible that association of WRN with MMR proteins predominantly occurs in chromatin-bound DSB repair complexes that remained insoluble during the protein extraction procedures or our IP procedure employed too stringent lysis/washing conditions. Alternatively, WRN can be recruited to the sites of Cr-induced DSB by other components of MMR, such as the upstream-acting MLH1-PMS2 heterodimer.55
Implications for Cr(VI) genotoxicity
Long DSB persistence and defective formation of RAD51 recombination foci in the absence of WRN indicate that repair of MMR-induced DSB in Cr(VI)-treated fibroblasts is dependent on this helicase. Thus, unlike other types of DSB that are processed for recombination repair by both RecQ-dependent and independent (EXO1) pathways,20–22 a reliance on a WRN-dependent process for repair of Cr/MMR-induced breaks can result in more persistent DSB. Cr(VI)-treated cells did display a relatively slow DSB repair and this phenomenon was particularly noticeable in primary human cells which showed a steady accumulation of DSB over many hours even after exposure to subtoxic environmental doses of Cr(VI).24,25 High promoter-repressive activity of Rb complexes in primary cells results in low levels of WRN expression,56 which could limit repair rates for Cr-induced DSB in tissues. Persistent DSB can cause a variety of gross genetic rearrangements, including chromosomal deletions and translocations,57,58 that are likely contributing to the high carcinogenicity of even moderate chromate exposures found in the modern workplace environment.47,59 Consequently, individuals with low WRN levels could be at a greater risk for Cr(VI)-associated genotoxicity and carcinogenesis. Since Cr(VI)-induced DSB are predominantly found in G2 cells and their repair requires WRN, this may restrict WRN availability for its other biological functions such as rebuilding of telomere structures which also occurs in G2 phase.60 Thus, one of the potential consequences of competition for WRN between telomeric functions and DSB repair could be slower rates for both processes and, possibly, accelerated attrition of telomeres in cells of individuals chronically exposed to Cr(VI).
Acknowledgments
This work was supported by R01 grant ES008786, training grant T32 ES007272 and training core of grant P42 ES013660 from the National Institute of Environmental Health Sciences.
Abbreviations
- DSB
double-strand break
- MMR
mismatch repair
- PBS
phosphate-buffered saline
- shRNA
short hairpin RNA
- PFGE
pulsed-field gel electrophoresis
References
- 1.Hickson ID. RecQ helicases: caretakers of the genome. Nat Rev Cancer. 2003;3:169–78. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- 2.Opresko PL, Cheng H, von Kobbe C, Harrigan JA, Bohr VA. Werner syndrome and the function of the Werner protein; what they can teach us about the molecular aging process. Carcinogenesis. 2003;24:791–802. doi: 10.1093/carcin/bgg034. [DOI] [PubMed] [Google Scholar]
- 3.Shen JC, Loeb LA. Unwinding the molecular basis of the Werner syndrome. Mech Ageing Dev. 2001;122:921–44. doi: 10.1016/s0047-6374(01)00248-2. [DOI] [PubMed] [Google Scholar]
- 4.Karmakar P, Bohr VA. Cellular dynamics and modulation of WRN protein is DNA damage specific. Mech Ageing Dev. 2005;126:1146–58. doi: 10.1016/j.mad.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 5.Marciniak A, Lombard DB, Johnson FB, Guarantee L. Nucleolar localization of the WRN syndrome protein in human cells. Proc Natl Acad Sci USA. 1998;95:6686–92. doi: 10.1073/pnas.95.12.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Von Kobbe C, Borh VA. A nucleolar targeting sequence in the Werner syndrome protein resides within residues 949–1092. J Cell Sci. 2002;115:3901–07. doi: 10.1242/jcs.00076. [DOI] [PubMed] [Google Scholar]
- 7.Poot M, Hoehn H, Runger TM, Martin GM. Impaired S-phase transit of Werner syndrome cells expressed in lymphoblastoid cell lines. Exp Cell Res. 1992;202:267–73. doi: 10.1016/0014-4827(92)90074-i. [DOI] [PubMed] [Google Scholar]
- 8.Sidorova JM, Li N, Folch A, Monnat RJ., Jr The RecQ helicase WRN is required for normal replication fork progression after DNA damage or replication fork arrest. Cell Cycle. 2008;7:796–807. doi: 10.4161/cc.7.6.5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dhillon KK, Sidorova J, Saintigny Y, Poot M, Gollahon K, Rabinovitch PS, et al. Functional role of the Werner syndrome RecQ helicase in human fibroblasts. Aging Cell. 2007;6:53–61. doi: 10.1111/j.1474-9726.2006.00260.x. [DOI] [PubMed] [Google Scholar]
- 10.Zhang N, Kaur R, Lu X, Shen X, Li L, Legerski RJ. The PSO4 mRNA splicing and DNA repair complex interacts with WRN for processing of DNA interstrand cross-links. J Biol Chem. 2005;280:40559–67. doi: 10.1074/jbc.M508453200. [DOI] [PubMed] [Google Scholar]
- 11.Machwe A, Xiao L, Orren DK. Length-dependent degradation of single-stranded 3′ ends by the Werner syndrome protein (WRN): implications for spatial orientation and coordinated 3′ to 5′ movement of its ATPase/helicase and exonuclease domains. BMC Mol Biol. 2006;7:6. doi: 10.1186/1471-2199-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen JC, Loeb LA. Werner syndrome exonuclease catalyzes structure-dependent degradation of DNA. Nucleic Acids Res. 2000;28:3260–8. doi: 10.1093/nar/28.17.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baynton K, Otterlei M, Bjoras M, von Kobbe C, Bohr VA, Seeberg E. WRN interacts physically and functionally with the recombination mediator protein RAD52. J Biol Chem. 2003;278:36476–86. doi: 10.1074/jbc.M303885200. [DOI] [PubMed] [Google Scholar]
- 14.Li B, Comai L. Requirements for the nucleolytic processing of DNA ends by the WRN syndrome protein-Ku70/80 complex. J Biol Chem. 2001;276:9896–02. doi: 10.1074/jbc.M008575200. [DOI] [PubMed] [Google Scholar]
- 15.Otterlei M, Bruheim P, Ahn B, Bussen W, Karmakar P, Baynton K, et al. Werner syndrome protein participates in a complex with RAD51, RAD54, RAD54B and ATR in response to ICL-induced replication arrest. J Cell Sci. 2006;119:5137–46. doi: 10.1242/jcs.03291. [DOI] [PubMed] [Google Scholar]
- 16.Yannone SM, Roy S, Chan DW, Murphy MB, Huang S, Campisi J, et al. Werner syndrome protein is regulated and phosphorylated by DNA-dependent protein kinase. J Biol Chem. 2001;276:38242–8. doi: 10.1074/jbc.M101913200. [DOI] [PubMed] [Google Scholar]
- 17.Constantinou A, Tarasounas M, Karow JK, Brosh RM, Bolu VA, Hickson ID, et al. Werner’s syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO Rep. 2000;1:80–4. doi: 10.1093/embo-reports/kvd004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liao S, Toczylowski T, Yan H. Identification of the Xenopus DNA2 protein as a major nuclease for the 5′->3′ strand-specific processing of DNA ends. Nucleic Acids Res. 2008;36:6091–00. doi: 10.1093/nar/gkn616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lan L, Nakajima S, Komatsu K, Nussenzweig A, Shimamoto A, Oshima J, et al. Accumulation of Werner protein at DNA double strand-breaks in human cells. J Cell Sci. 2005;118:4153–62. doi: 10.1242/jcs.02544. [DOI] [PubMed] [Google Scholar]
- 20.Zhu Z, Chung WH, Shim EY, Lee E, Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–94. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–4. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gravel S, Chapman JR, Magill C, Jackson SP. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22:2767–72. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peterson-Roth E, Reynolds M, Quievryn G, Zhitkovich A. Mismatch repair proteins are activators or toxic responses to chromium-DNA damage. Mol Cell Biol. 2005;25:3596–607. doi: 10.1128/MCB.25.9.3596-3607.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reynolds M, Stoddard L, Bespalov I, Zhitkovich A. Ascorbate acts as a highly potent inducer of chromate mutagenesis and clastogenesis: linkage to DNA breaks in G2 phase by mismatch repair. Nucleic Acids Res. 2007;35:465–76. doi: 10.1093/nar/gkl1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reynolds MF, Peterson-Roth EC, Johnston T, Gurel VM, Menard HL, Zhitkovich A. Rapid DNA double-strand breaks resulting from processing of Cr-DNA crosslinks by both MutS dimers. Cancer Res. 2009;69:1071–9. doi: 10.1158/0008-5472.CAN-08-2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quievryn G, Peterson E, Messer J, Zhitkovich A. Genotoxicity and mutagenicity of chromium(VI)/ascorbate-generated DNA adducts in human and bacterial cells. Biochemistry. 2003;42:1062–70. doi: 10.1021/bi0271547. [DOI] [PubMed] [Google Scholar]
- 27.Quievryn G, Messer J, Zhitkovich A. Lower mutagenicity but higher stability of Cr-DNA adducts formed during gradual chromate activation with ascorbate. Carcinogenesis. 2006;27:2316–21. doi: 10.1093/carcin/bgl076. [DOI] [PubMed] [Google Scholar]
- 28.Zhitkovich A, Song Y, Quievryn G, Voitkun V. Non-oxidative mechanisms are responsible for the induction of mutagenesis by reduction of Cr(VI) with cysteine: role of ternary DNA adducts in Cr(III)-dependent mutagenesis. Biochemistry. 2001;40:549–60. doi: 10.1021/bi0015459. [DOI] [PubMed] [Google Scholar]
- 29.Zhitkovich A, Peterson-Roth E, Reynolds M. Killing of chromium-damaged cells by mismatch repair and its relevance to carcinogenesis. Cell Cycle. 2005;4:1050–2. [PubMed] [Google Scholar]
- 30.Stojic L, Mojas N, Cejka P, Di Pietro M, Ferrari S, Marra G, et al. Mismatch repair-dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes Dev. 2004;18:1331–44. doi: 10.1101/gad.294404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan T, Berry SE, Desai AB, Kinsella TJ. DNA mismatch repair (MMR) mediates 6-thioguanine genotoxicity by introducing single-strand breaks to signal a G2-M arrest in MMR-proficient RKO cells. Clin Cancer Res. 2003;9:2327–34. [PubMed] [Google Scholar]
- 32.Meyers M, Wagner MW, Mazurek A, Schmutte C, Fishel R, Boothman DA. DNA mismatch repair-dependent response to fluoropyrimidine-generated damage. J Biol Chem. 2005;280:5516–26. doi: 10.1074/jbc.M412105200. [DOI] [PubMed] [Google Scholar]
- 33.Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7:335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 34.Modrich P. Mechanisms in eukaryotic mismatch repair. J Biol Chem. 2006;281:30305–30309. doi: 10.1074/jbc.R600022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quievryn G, Messer J, Zhitkovich A. Carcinogenic chromium(VI) induces cross-linking of vitamin C to DNA in vitro and in human lung A549 cells. Biochemistry. 2002;41:1062–70. doi: 10.1021/bi011942z. [DOI] [PubMed] [Google Scholar]
- 36.Reynolds M, Zhitkovich A. Cellular vitamin C increases chromate toxicity via a death program requiring mismatch repair but not p53. Carcinogenesis. 2007;28:1613–20. doi: 10.1093/carcin/bgm031. [DOI] [PubMed] [Google Scholar]
- 37.Reynolds M, Peterson E, Quievryn G, Zhitkovich A. Human nucleotide excision repair efficiently removes chromium-DNA phosphate adducts and protects cells against chromate toxicity. J Biol Chem. 2004;279:30419–24. doi: 10.1074/jbc.M402486200. [DOI] [PubMed] [Google Scholar]
- 38.Hisama FM, Chen YH, Meyn MS, Oshima J, Weissman SM. WRN or telomerase constructs reverse 4-nitroquinoline 1-oxide sensitivity in transformed Werner syndrome fibroblasts. Cancer Res. 2000;60:2372–6. [PubMed] [Google Scholar]
- 39.Wyllie FS, Jones CJ, Skinner JW, Haughton MF, Wallis C, Wynford-Thomas D, et al. Telomerase prevents the accelerated cell ageing of Werner syndrome fibroblasts. Nat Genet. 2000;24:16–17. doi: 10.1038/71630. [DOI] [PubMed] [Google Scholar]
- 40.Chang S, Multani AS, Cabrera NG, Naylor ML, Laud P, Lombard D, et al. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat Genet. 2004;36:877–82. doi: 10.1038/ng1389. [DOI] [PubMed] [Google Scholar]
- 41.Du X, Shen J, Kugan N, Furth EE, Lombard DB, Cheung C, et al. Telomere shortening exposes functions for the Werner and Bloom syndrome genes in mice. Mol Cell Biol. 2004;24:8437–46. doi: 10.1128/MCB.24.19.8437-8446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Glaviano A, Nayak V, Cabuy E, Baird DM, Yin Z, Newson R, et al. Effects on hTERT on metal ion-induced genomic instability. Oncogene. 2006;25:3424–35. doi: 10.1038/sj.onc.1209399. [DOI] [PubMed] [Google Scholar]
- 43.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 44.Stewart GS. Solving the RIDDLE of 53BP1 recruitment to sites of damage. Cell Cycle. 2009;8:1532–8. doi: 10.4161/cc.8.10.8351. [DOI] [PubMed] [Google Scholar]
- 45.Zhitkovich A. Importance of chromium-DNA adducts in mutagenicity and toxicity of chromium(VI) Chem Res Toxicol. 2005;18:3–11. doi: 10.1021/tx049774+. [DOI] [PubMed] [Google Scholar]
- 46.Standeven AM, Wetterhahn KE. Ascorbate is the principal reductant of chromium(VI) in rat lung ultrafiltrates and cytosols, and mediates chromium-DNA binding in vitro. Carcinogenesis. 1992;13:1319–24. doi: 10.1093/carcin/13.8.1319. [DOI] [PubMed] [Google Scholar]
- 47.Salnikow K, Zhitkovich A. Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic and chromium. Chem Res Toxicol. 2008;21:28–44. doi: 10.1021/tx700198a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rodrigue A, Lafrance M, Gauthier MC, McDonald D, Hendzel M, West SC, et al. Interplay between human DNA repair proteins at a unique double-strand break in vivo. EMBO J. 2006;25:222–31. doi: 10.1038/sj.emboj.7600914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takata M, Sasaki MS, Sonoda E, Fukushima T, Morrison C, Albala JS, et al. The Rad51 paralog Rad51B promotes homologous recombinational repair. Mol Cell Biol. 2000;20:6476–82. doi: 10.1128/mcb.20.17.6476-6482.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.West SC. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol. 2003;4:435–45. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- 51.Martin BD, Schoenhard JA, Hwang JM, Sugden KD. Ascorbate is a pro-oxidant in chromium-treated human lung cells. Mutat Res. 2006;610:74–84. doi: 10.1016/j.mrgentox.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 52.Messer J, Reynolds M, Stoddard L, Zhitkovich A. Causes of DNA single-strand breaks during reduction of chromate by glutathione in vitro and in cells. Free Radic Biol Med. 2006;40:1981–92. doi: 10.1016/j.freeradbiomed.2006.01.028. [DOI] [PubMed] [Google Scholar]
- 53.O’Brien TJ, Fornsaglio JL, Cervak S, Patierno SR. Effects of hexavalent chromium on the survival and cell cycle distribution of DNA repair-deficient S. cerevisiae. DNA Repair (Amst) 2002;1:617–627. doi: 10.1016/s1568-7864(02)00078-2. [DOI] [PubMed] [Google Scholar]
- 54.Bryant HE, Ying S, Helleday T. Homologous recombination is involved in repair of chromium-induced DNA damage in mammalian cells. Mutat Res. 2006;599:116–23. doi: 10.1016/j.mrfmmm.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 55.Saydam N, Kanagaraj R, Dietschy T, Garcia PL, Peña-Diaz J, Shevelev I, et al. Physical and functional interactions between Werner syndrome helicase and mismatch-repair initiation factors. Nucleic Acids Res. 2007;35:5706–16. doi: 10.1093/nar/gkm500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Y, El-Naggar S, Clem B, Chesney J, Dean DC. The Rb/E2F pathway and Ras activation regulate RecQ helicase gene expression. Biochem J. 2008;412:299–306. doi: 10.1042/BJ20070975. [DOI] [PubMed] [Google Scholar]
- 57.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27:247–54. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 58.Povirk LF. Biochemical mechanisms of chromosomal translocations resulting from DNA double-stranded breaks. DNA Repair. 2006;5:1199–212. doi: 10.1016/j.dnarep.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 59.Costa M, Klein CB. Toxicity and carcinogenicity of chromium compounds in humans. Crit Rev Toxicol. 2006;36:155–63. doi: 10.1080/10408440500534032. [DOI] [PubMed] [Google Scholar]
- 60.Verdun RE, Karlseder J. The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell. 2006;127:709–20. doi: 10.1016/j.cell.2006.09.034. [DOI] [PubMed] [Google Scholar]