

Abstract
This review focuses on current developments in the field of nanostructured bulk polymers and their application in bioengineering and therapeutic sciences. In contrast to well-established nanoscale materials, such as nanoparticles and nanofibers, bulk nanostructured polymers combine nanoscale structure in a macroscopic construct, which enables unique application of these materials. Contemporary fabrication and processing techniques capable of producing nanoporous polymer films are reviewed. Focus is placed on techniques capable of sub-100 nm features since this range approaches the size scale of biological components, such as proteins and viruses. The attributes of these techniques are compared, with an emphasis on the characteristic advantages and limitations of each method. Finally, application of these materials to biofiltration, immunoisolation, and drug delivery are reviewed.
1. Introduction
In the past several decades, nanoscale materials have received substantial interest due to the distinct properties that can be achieved, and a broad range of applications have emerged as a result.1 Among these, the field of bioengineering and therapeutic sciences has seen great advances in the development and application of nanostructured materials to medicine. With advent of National Institutes of Health (NIH) nanotechnology programs, such as the NIH Nano Task Force,2 the burgeoning field of nanobiotechnology has received considerable attention in recent years. Much of the interest in nanostructured materials is due to their properties at the nanoscale – specifically their interactions with biological molecules and the structures within living cells.3–10 Furthermore, the wide range of materials and potential applications makes this field one that is full of opportunities. Among these materials, nanoparticles and nanofibers have received significant attention, while nanostructured bulk and thin films have received comparably less attention. To date, the majority of investigated bulk nanostructured materials have been inorganic, which has limited the range of possible chemical and mechanical properties. Regardless, nanostructured inorganics have exhibited compelling attributes, such as improved immunogenicity compared with equivalent materials that lack structure.3, 4 Furthermore, nanostructured inorganics have been employed for immunoisolation11, 12 and controlled release applications,13–15 opening novel therapeutics avenues.
Naturally, the successes of inorganic materials have prompted advances in the development of analogous polymeric films. Chemical synthesis has allowed an extensive range of chemical functionality and mechanical properties for polymeric materials, providing a means to cater polymers to their intended application. Relative to the wide range of existing porous polymers,16–18 nanoporous membranes have unique properties that can be exploited advantageously in biomedical sciences. For instance, nanoscale porosity enables biofiltration as the size of many important filtrates is less than 10 nm. Similarly, immunoisolation membranes employ size exclusion to reject proteins (a few nm) while maintaining transport of small molecule nutrients and waste (nm or less) to and from isolated cells. Controlled drug delivery can benefit likewise, where nanoporous membranes can control release when therapeutic and pore size are comparable. In all these examples, nanostructure in a polymer film facilitates a therapeutic application. While the advent of nanostructured bulk polymers in biomedical engineering has been slow, an increasing number of methods have been developed in recent years to fabricate and apply nanostructured polymers.
In this review, an overview of contemporary fabrication of nanostructured polymers is presented, with a focus on bioengineering applications, such as biofiltration, immunoisolation, and drug delivery. Because significant attention has been paid to the development and characterization of nanoparticles and nanofiber materials elsewhere,19–23 the scope of this review focuses on bulk and thin film polymers with nanostructured features.
2. Fabrication Techniques
A prime advantage associated with polymeric materials is the wide range of properties and fabrication techniques available. Advances in polymer synthesis and novel processing techniques have led to a variety of nanoporous polymers. Table 1 summarizes existing fabrication techniques that produce nanoporous polymers along with their characteristic pore size and density, film thickness, and structural uniformity. Among these techniques are lithography, pattern-transfer, track etching, solvent-based formation, layer-by-layer growth, block copolymer self-assembly, and various biologically-derived materials: lithographic and pattern-transfer approaches utilize pre-defined patterns that are transfer into a polymer film; polymers radiated with high energy particles can form nanoporous membranes with the track etch technique; various solvent-based procedures take advantage of natural formation of nanostructured polymers induced via precipitation; layer-by-layer assembly forms layered structures of polyelectrolytes that can become nanoporous with an appropriate treatment sequence; self-assembly of block copolymers employ a combination of polymer design and processing to allow formation of nanostructures (often dense and highly ordered); finally, a range of biologically-derived structures can form nanostructures, including widely available biomaterials such as cellulose, naturally forming structures like bacterial-derived crystalline cellular layers (S-layers), or synthetically engineered polypeptides. In the following sections, each of these fabrication techniques is reviewed.
Table 1.
Summary of various fabrication approaches for the formation of nanoporous polymeric materials.
| Approach | Polymer Used | Pore Diameter (nm) | Pore Density (cm−2) | Film Thickness | Film Uniformity | Ref. |
|---|---|---|---|---|---|---|
| Lithography | ||||||
| Optical Lithography | various photoresists | ~100 | – | – | High | 24 |
|
| ||||||
| Electron Beam Lithography | PMMA | 10 | – | – | High | 31, 32 |
|
| ||||||
| Aperture Array Lithography | PMMA | 200–350 A | 2.4×107–4×108 | 160 nm | High | 33 |
|
| ||||||
| Colloidal Lithography | PES | 230 | 109 | 0.5 μm | High | 34 |
|
| ||||||
| PIP | 55 | 5×109 | 40 nm | High | 35 | |
|
| ||||||
| Pattern Transfer | ||||||
| Nanowire Templated | PCL | 20–30 | 5×109 | 0.5 μm | High | 41 |
|
| ||||||
| Molding/Imprint | PMMA | 25 | 7×109 | 100 nm | High | 42 |
|
|
||||||
| PS | 25–30 | 5×1010 | – | High | 116 | |
|
| ||||||
| Track Etch | PC, PE | 15 | <109 | 5–20 μm | High | 46, 47 |
|
| ||||||
| Solvent-based Techniques | ||||||
| Immersion Precipitation | PVDF, PSF | 20–120 | – | 200 μm | Moderate | 52 |
|
| ||||||
| Phase Separation | PCL | 5–200 | 2×108 | 50 μm | Low | 53 |
|
| ||||||
| CO2 Foaming | PIM, PSF | 20–50 | ~1010 | – | Moderate-High | 54 |
|
|
||||||
| PPQ | 8–24 | – | 2–50 μm | Moderate | 55 | |
|
| ||||||
| Light Induced Polymerization | complex monomer mixture | 20–40 | – | – | – | 58 |
|
| ||||||
| Block Copolymer | ||||||
| PS-co-PMMA | 15 | ~5×1010 B | 80 nm C | Moderate-High | 77 | |
|
|
||||||
| PS-co-PL | 16 | ~1011 | 300 μm | High D(h) | 78 | |
|
|
||||||
| PS-co-PIP | 20–30 | ~2×1010 | 100–300 μm | High | 76 | |
|
| ||||||
| Reactive Pore Formation | PS-co-PMMA | 3–8 | 1011 | 30–300 nm | High | 80, 81 |
|
| ||||||
| Substrate Aligned Pores | PS-co-PMMA | ~25 | ~1011 | <0.1 μm | High | 86 |
|
| ||||||
| Electric Field Aligned Pores | PS-co-PMMA | 14–50 | 1010–1011 | ~1 μm | Moderate-High | 85 |
|
| ||||||
| Triblock Copolymer | PL-co-PDMA-co-PS | 19 | 1011 | –E | High | 87 |
|
| ||||||
| Graft Copolymer | PVDF-graft-POEM | ~2 | >1011 | ~3.5 μm F | Moderate-High | 82 |
|
| ||||||
| Asymmetric Membranes | PS-co-PDMAEMA | 20–80 | – | 50 nm – 1 μm 85 μm total |
Moderate | 84 |
|
| ||||||
| PS-co-P4VP | 40 | 2.4×1010 | 200–300 nm 100 μm total |
High | 83 | |
|
| ||||||
| Polyelectrolyte Multilayers | ||||||
| PAA/PAH | 50–200 | – | 50–80 nm | High | 66 | |
|
|
||||||
| PAA/PAH/P4VP | 10–50 | – | ~25nm | Moderate | 71 | |
|
|
||||||
| PAA/PAH | 30–40 | ~1010 | 0.2–0.7 μm | Moderate-High | 67 | |
|
| ||||||
| Spin Assisted Deposition | PAH/PSS | ~40 | ~5×1010 | 15–30 nm | Moderate-High | 70 |
|
| ||||||
| Nanoparticle Templated Pores | PAA/PAH/silica nanoparticles | 20–30 | ~2×1010 | <100 nm | Moderate | 72 |
|
| ||||||
| Spatially Patterned Films | PAArVBA/ PAH/PSS | ~10 | – | ~150 nm | – | 68 |
|
| ||||||
| Asymmetric Membranes | PEI/PAA | ~100 | – | ~10 μm | Low-Moderate | 69 |
|
| ||||||
| Biologically-Derived Materials | ||||||
| Cellulose-based | Cellulose esters | < 10 G | ~1011 | >100 μm | Moderate | 89, 117 |
|
| ||||||
| Cellular Layers (S-layers) | bacterium-based | 4–5 | >1011 | 10’s of nm H | High | 92, 93, 104, 118 |
|
| ||||||
| Peptide-based Multilayers | polypeptides | 50–150 | – | – | – | 73 |
|
| ||||||
| Engineered polypeptides | P(GPV) | ~70 | ~5×109 | 8 nm I | Moderate-High | 97 |
Abbreviated polymers are as follows: PC - poly(carbonate), PE - poly(ester), PS - poly(styrene), PMMA - poly(methyl methacrylate), PL – poly(lactide), PIP -poly(isoprene), PAA= poly(acrylic acid), PAH - poly(allylamine hydrochloride), PCL - poly(caprolactone), PSS - poly(sodium-4-styrene sulfonate), POEM -poly(oxyethylene methacrylate), PVDF - poly(vinylidene fluoride), PDMA - poly(N,N-dimethylacrylamide), PDMAEMA - poly(N,N-dimethylaminoethyl methacrylate), P4VP - poly(4-vinylpyridine), PAArVBA - poly(acrylic acid-ran-vinylbenzylacrylate), PEI - poly(ethylene imine), PES - poly(ether sulfone), PSF - polysulfone, PIM -Polyimide, P(GPV) - peptide polymer with gylcine, L-proline, and L-valine monomer base units, PPQ – poly(phenylquinoxaline)
2.1. Lithographic Techniques
From its inception, the field of nanotechnology has been largely driven by semiconductor processing, which primarily utilizes photolithographic techniques to generate complex structures.24 While patterning robust nanoscale devices with inorganic materials is well established, polymers are less frequently used as active materials, despite their central role in the patterning process. Limited use is primarily due to high process temperatures or exposure to harsh chemicals during conventional semiconductor processing; modified techniques and improved materials have recently been developed to pattern arbitrary polymers using photolithography, but these have not yet been demonstrated at the nanoscale.25, 26 The primary advantage of lithographic techniques is the ability to produce user-defined patterns. While a variety of lithographic, printing, and machining techniques are capable of patterning polymeric materials,27 these are often non-ideal for producing regular high-density features in polymers compared to existing alternatives.
Optical lithography is the most widely used lithographic technique. In this approach, a thin coating of photoresist is deposited on a host substrate, typically by spin casting. When the photoresist is exposed to light through a shadow mask, the mask pattern is transferred to the resist (Figure 1). Depending on the type of photoresist, exposed (positive-tone resist; Figure 1D) or unexposed (negative-tone resist; Figure 1E) regions may be removed to produce a patterned polymer layer. Many popular photoresists are two-component mixtures consisting of a photo sensitizer and a matrix resin. A common two-component positive-tone photoresist is a mixture of diazoquinone ester and phenolic novolak resin (often referred to as DQN), which becomes base-soluble upon exposure to UV light. A common two-component negative-tone photoresist is a mixture of bis(aryl)azide and cyclized poly(cis-isoprene), which becomes crosslinked upon exposure to UV light. Recent advances have resulted in chemically amplified resists, where resolutions on the order of 100 nm can be obtained for production-quality photolithography using deep ultraviolet light.24 An in depth review of photolithography is beyond the scope of this review and can be found elsewhere.24 Direct use of lithographic techniques requires a photocrosslinkable or photodegradable polymer, which restricts potential materials selection. While biologically-relevant materials compatible with these techniques are available,28–30 simpler, higher-throughput alternatives typically make lithography disadvantageous for generating nanoporous materials.
Figure 1.
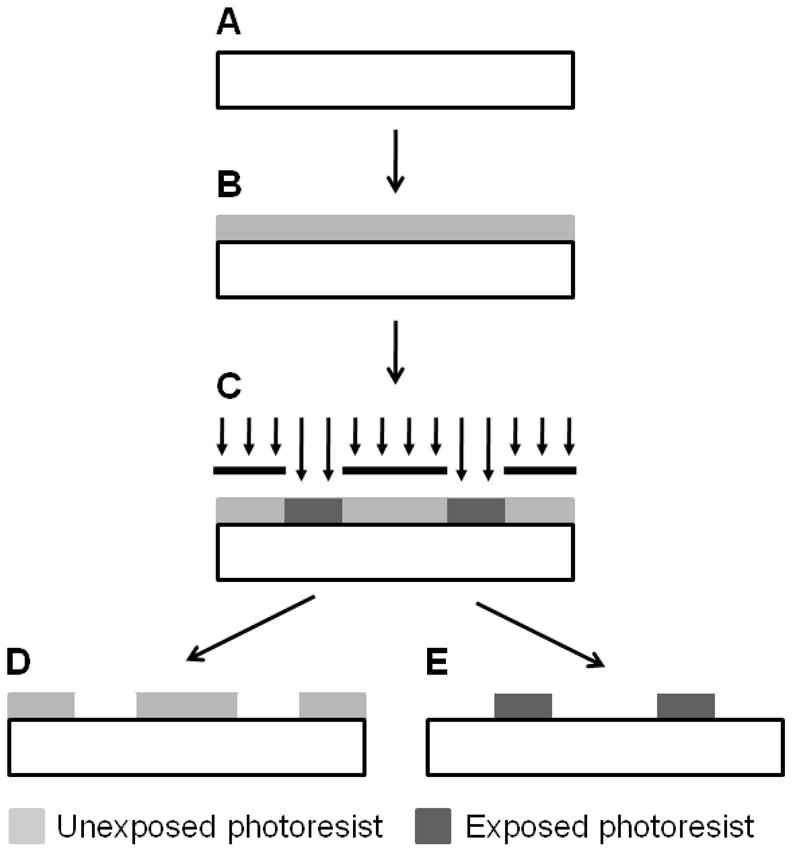
Lithographic patterning of nanostructures. Patterns can be generated on a target substrate (A) by depositing a photosensitive polymer (photoresist), typically by spin casting (B) and selectively exposing regions to ultraviolet light (C). Upon exposure to ultraviolet light, positive-tone photoresists can be removed (D), generating patterns in the unexposed photoresist. Likewise, unexposed regions of negative-tone photoresist can be removed (E), generating patterns in the exposed photoresist.
Similarly, electron-beam lithography (EBL) and ion-beam lithography (IBL) utilize electrons and charged particles, respectively, to pattern polymeric materials. By scanning an electron- or ion-beam across a target polymer, it is possible to generate spatially defined patterns, where typically exposed material can be removed as with a positive-tone photoresist. The most common charged particle beam resist is poly(methylmethacrylate) (PMMA), where incident charge particles result in chain scission within the polymer and render it soluble. For EBL, patterns with critical dimensions of 10 nm or less can be obtained with commercial EBL systems.31, 32 Due to charged-particle scattering within the resist polymer, ultimate pattern resolution and thickness are intimately coupled: it is difficult to pattern features considerably smaller than the resist thickness. Because patterns must be scanned spatially, a major limitation of EBL/IBL techniques is throughput for this serial process. Alternatively, aperture-array lithography combines the parallel nature of optical lithography with the serial nature of EBL/IBL by utilizing an array of focusing apertures that replicates the scanned pattern. With a sufficiently high number of apertures, patterns can be produced over large areas with increased throughput, which has resulted in regular features patterned with He+ ions down to 200 nm.33 Further information regarding charged particle beam lithography can be found elsewhere.24
In addition to user-defined geometries, self-assembling materials can be utilized as lithographic masks. For instance, colloidal silica has been used to fabricate nanoporous membranes.34, 35 Colloidal silica used as a sputter mask can produce relatively thick films (500 nm) with 200 nm pores34 or used as a mask for optical crosslinking can produce smaller pores (55 nm) in considerably thinner films (40 nm).35 In these examples, pore density is largely determined by particle size, which allows for densities of 109 cm−2 and 5×109 cm−2, respectively. Self-assembled lithography masks benefit from simplified pattern generation, but they lack the ability to generate user-defined structures, a normal advantage of lithography.
2.2. Pattern Transfer
A closely related process is template fabrication, where a template structure is directly transferred into a polymer film (Figure 2). Templating is a process that has been applied to a variety of applications and has been used to fabricate nanostructured materials over the past several decades.36, 37 While templating has predominantly features inorganic materials, a few recent examples have fabricated nanostructured polymers using templates, such as nanostructured poly(caprolactone) (PCL) rods from anodize alumina membranes.38–40 Applying similar processing techniques, zinc oxide nanorod templates have been used to fabricate nanoporous (PCL) thin films.41 This approach effectively transferred nanostructured zinc oxide rods and generated 20–30 nm pores in PCL with modest pore densities (5×109 cm−2), as shown in Figure 2B. Because this approach does not rely on the properties of the polymer to generate nanostructures, this approach is widely applicable to many biologically-relevant and biodegradable polymers. In general, the primary constraint for possible patterns is the quality and structure of the template material.
Figure 2.

Template fabrication of nanostructures. (A) Physical templates (i) can be used for patterning by deposition of a target polymer (ii). Through chemical or mechanical removal of the template material, a nanoporous polymer can be generated (iii). (B) A characteristic SEM image of biodegradable poly(caprolactone) fabricated from a zinc oxide nanorod template.
Nanopores can also be fabricated in polymers utilizing imprint techniques, where a master pattern is physically transferred to a polymer target. Typically, an external pressure is applied to a polymer above its glass transition temperature and a rigid master such that the master pattern is transferred to the polymer. For instance, imprinting a silicon master onto a PMMA film produced regular porous structures with this approach. The resultant PMMA film had 25 nm pores and pore densities as high as 7×109 cm−2.42 Similarly, tri-block copolymers have been used as an imprint mold and generated a nanoporous poly(styrene) (PS) layer with 25–30 nm pores with greater densities (5×1010 cm−2) than EBL molds.43 While transfer to PS was effective, existing block copolymer techniques (see Section 2.6) can achieve similar structures in PS with less complexity, so this approach will need to be utilized with a wider range of polymers to demonstrate expansive applicability.
2.3. Track Etch
Track etch processing takes advantage of natural damage that occurs when high energy particles bombard a surface. Porous structures can be generated by etching the linear paths of travel associated with incident particles within the polymer film (Figure 3). Initially this approach was utilized for simple particle detectors44 but was later adapted to reproducibly fabricate membranes with highly uniform pore size (down to 15 nm) and low defect density.45, 46 Since each pore is the result of a spatially-random incident particle, agglomeration of pores limits maximum pore density in order to maintain a low dispersion in pore size. This results in an upper limit for pore density on the order of 108 cm−2.47 While this approach is useful for commercial membrane fabrication (prominent examples include poly(carbonate) and poly(ethylene terephthalate)), it is infrequently used to generate membranes from arbitrary materials. In depth review of track etch methods is beyond the scope of this review and are covered in depth elsewhere.48
Figure 3.

Track etch fabrication of nanostructures. (A) Radiation incident on a target polymer (i) results in physical damage (ii) that can be etched to form nanoporous polymers (iii). (B) A characteristic SEM image of poly(ethylene terephthalate) fabricated using track etch methods. B – Reprinted from Nuclear Instruments and Methods in Physics Research Section B, 209, P.Y. Apel et al., Effect of nanosized surfactant molecules on the etching of ion tracks: New degrees of freedom in design of pore shape, 329–334 Copyright (2003), with permission from Elsevier.115
2.4. Solvent-based Techniques
In contrast to lithographic and pattern-transfer techniques that produce features defined by a predetermined pattern or template, other methods can be used to produce porous structures intrinsic to the materials and/or processing techniques. The most prevalent of these are solvent-based precipitation techniques, which exploits solubility variations of a target polymer depending on concentration, solvent, or process conditions. When initially dissolved in a good solvent, nanostructures can be induced from a polymer solution by solvent evaporation, cooling, or exposure to non-solvents (often water).18 This has produced porosity in a variety of polymers, including commercial examples,49–51 and can generate sub-100 nm pores.52, 53 For instance, PCL dissolved in a mixture of dioxane and 2-methoxyethanol can generate a nanostructured film when cast and subsequently submerged in water.53 Control of pore size and distribution is highly variable with this technique, but the ease and simplicity of this approach make it an attractive option. Along similar lines, supercritical carbon dioxide can be used to form nanostructured films in a process often referred to as carbon dioxide foaming. For this a polymer film is saturated with carbon dioxide, and upon quick release of pressure, porous structures are generated. Recent examples have achieved pore size as low as 8 nm with relatively high pore densities (~1010 cm−2) and thick films (2–50 μm).54, 55 This process is compatible with a variety of polymers but generally produces irregular structures.54–57 In addition, solvent-based techniques are not limited to post-synthesis polymers: direct polymerization can also generate nanoporous structures. With an appropriate mixture of monomer, initiator, and solvent, exposure to ultraviolet light can induce polymerization and fix a nanoporous structure. For example, 20–40 nm pores were produced, but flexibility in material selection, pore size, density, and uniformity is severely limited and must be investigated on a case-by-case basis.58
2.5. Layer-by-Layer
Layer-by-layer (LbL) assembly forms layered structures of polyelectrolytes by the sequential deposition of cationic and anionic polymers, which takes advantage of attractive and repulsive electrostatic forces.59 During each deposition step, ionic polymers in solutions are electrostatically attracted to oppositely charged polymers on the deposition surface, which results in deposition of a monolayer (Figure 4A). A common polylelectrolyte combination is poly(allylamine hydrochloride) (PAH) and poly(acrylic acid) (PAA), which act as a polycation and polyanion, respectively. Because only either PAH or PAA is deposited during each growth step, deposition beyond a monolayer is prevented by electrostatic repulsion of the like-charge present on the surface. This allows deposition of an arbitrary number of layers with excellent control over film thickness; however, because this is a serial process, increased film thickness requires increased deposition time. An attractive consequence of LbL assembly is the resultant film conformity, which assembles coatings of consistent thickness and composition regardless of an object’s geometry. Some level of intermixing between layers is expected with most LbL films, but this is not a significant concern for the majority of applications. The primary limitation of this approach is available materials, which require a combination of polyelectrolytes: commercially available options are limited, so custom polymer synthesis is often favored in order to cater LbL materials to specific applications. LbL techniques have been reviewed extensively elsewhere59–61 and have been deployed in a variety of therapeutic applications.62–65
Figure 4.

Layer-by-layer assembly of nanostructures. (A) Sequential deposition of oppositely charged polylelectrolytes produces layered structures. Nanostructures can be induced in such films by (B) solution-based film reorganization or (C) with selectively removable polymers or (D) particles. (E) A characteristic AFM image of a poly(allylamine hydrochloride)/poly(acrylic acid) layer-by-layer film with solution-induced nanostructures. E – Reprinted in part with permission from reference 67. Copyright 2006 American Chemical Society).
Porous LbL films have emerged in recent years as an interesting adaptation of this approach. Initial examples obtained pore sizes in the range of 50–200 nm for a two component system that was exposed to a conditioning solution (Figure 4B).66 Rearrangement of the polymer constituents generated a porous structure, and the porous structure was stabilized with a heat treatment that crosslinked the polymer. To date, the smallest pore size demonstrated with this approach is 30–40 nm, which is shown in Figure 4E.67 Exposure to ultraviolet light has been used to photopattern such nanoporous films, providing versatility in future fabrication schemes.68 It is also possible to obtain asymmetric membranes through post-deposition treatments (frequently involving exposure to aqueous solution of a particular pH), which have produced relatively thick films (~10 μm) with moderate pore size (100 nm).69 As an alternative to solution assembly, spin casting a LbL solution has been used to generate porous structures by polymer dewetting at the coating surface.70 With an increasing number of layers, the pore size decreased for this approach such that the porous structure became substantially less prevalent. Pore size down to 40 nm were observed with transition electron microscopy, and smaller pores may be possible, but artifacts associated with atomic force microscopy (the primary characterization technique utilized) make accurate determination of pore size difficult at this size scale.
In addition to LbL films that naturally form porous structures, porosity can also be obtained through a selectively removable component. For instance, incorporation of a hydrogen-bonded polymer, such as poly(4-vinylpyridine), during the polycation deposition allowed selective removal of the non-ionically-bound constituent without disrupting the bulk LbL film (Figure 4C).71 This resulted in pores ranging from 10–50 nm but was only demonstrated with thin films (25 nm). In addition, silica nanoparticles can be used similarly as a removable component. Silica particles 25 nm and greater were incorporated into LbL films and subsequently dissolved with hydrofluoric acid to generated a porous structure (Figure 4D). Pores down to 20–30 nm were produced and pore size closely mimicked the particle sizes utilized.72
One drawback cited for LbL films is the lack of biocompatible materials. To improve upon this, peptide-based polymers have been developed for and deployed in LbL films, which were combinations of cysteine, glycine, glutamic acid, lysine, tyrosine, and valine subunits.73 Depending on the specific polypeptides utilized, nanotopography with surface roughness ranging from 4–20 nm was obtained. While small molecule release is observed from these films, it is difficult to establish whether this was the result of solid-state diffusion or an inherently porous structure.
2.6. Block Copolymers
Block copolymer (BCP) techniques take advantage of phase separation in polymers with two or more distinct chemical blocks. A wide range of structures have been demonstrated74, 75 and depend highly on the chemical functionality and block lengths. A common route to generate porous structures is through cross-linking and subsequent solvent removal of a soluble block (Figure 5A,C). The most prominent BCP is poly(styrene)-block-poly(methylmethacrylte) (PS-b-PMMA), where cylindrical domains of PMMA form with hexagonal symmetry with a PS matrix; when exposed to UV ozone treatment, the PS phase is crosslinked and the PMMA domain can be selectively removed. Self-assembly of these materials often generate patterns with high two-dimensional symmetry, where hexagonal patterns are the most prevalent (as shown in Figure 5C). Patterns of high symmetry typically require a layer to orient domains perpendicular to the substrate and is frequently a random copolymer of the constituent blocks. With simple combinations of hydrophilic and hydrophobic blocks, pores on the order of 15–30 nm can be achieved.76–78 Because of the high degree of symmetry and small pore size, it is common to observe pore densities of 1011 cm−2 or greater. Depending on the conditions employed, possible film thickness spans a wide range from tens of nanometers to hundreds of microns. Both processing conditions and the specific BCP combinations synthesized have an impact on porous structures. Other sources have extensively reviewed BCP techniques, theory, and practice, and further details can be found elsewhere.18, 74, 79
Figure 5.

Block copolymer self-assembly of nanostructures. A) Deposition of di-block copolymers (i) can result in the formation of nanoscale regions consisting of hydrophilic and hydrophobic segments (ii). Pores can be generated with cross-linking and solvent removal (iii) or with ozone exposure(iv). In addition, hydrophilic segments in ozone exposed films can be removed to generate larger pores (iii). (B) Deposition of tri-block copolymers (i) results in similar nanostructures (ii), and upon removal of one block, a hydrophilic surface character can be acheived (iii). (C) A characteristic SEM image of a poly(styrene)-block-poly(4-vinylpyridine) BCP film showing typical hexagonal ordering or pores. C – Reprinted by permission from Macmillan Publishers Ltd: Nature Materials (Reference 83), copyright (2007).
Given the attractive properties of these materials, many techniques have been developed to influence BCP structure formation. The conventional approach to induce pores is through solvent dissolution, but it is also possible to do so with ozone treatment, which selectively degrades the center of hydrophilic regions. In particular, much smaller pores have been demonstrated (down to 3 nm): although the hydrophilic block remains sensitive to solvent removal (in acetic acid for instance), where approximately 25 nm pores would result (Figure 5A-iv). 80, 81 Another approach to achieve similar porosity is the use of graft copolymers (where one extended block has many smaller blocks pendant from the main block). This is capable of very small pores (~2 nm),82 but it is unclear what film thickness can be achieved with this type of polymer. For thinner films, the structural integrity of copolymer membranes can be improved by mounting nanoporous films on thicker macroporous supports, which has allowed their use in filtration.77 In addition, uniform BCP films can also be obtained with relatively thick films,76, 78 but fabrication of BCP monoliths depends on the specific copolymer of interest. Furthermore, with the appropriate casting conditions, it is also possible to obtain asymmetric membranes that are composed of a thin nanoporous region and a thicker region with coarser pores.83, 84
As a means to lower defect densities and improve alignment with underlying substrates, electric field and surface patterning have been used. With electric-field alignment, it is possible to obtain micron thick films with nanoscale pores oriented perpendicular to the substrate.85 By patterning the underlying substrate using e-beam lithography, it is possible to guide self-assembly.86 Such guided assembly utilized lower density surface patterns, which improved defect density and pore uniformity when compared with conventional BCP self-assembly.
In general, the largest drawback of BCP approaches is the need for two dissimilar blocks in order for self-assembly to occur. This restricts polymer selection and consequently the resulting porous polymer. For instance, the hydrophobic block is typically crosslinked and the hydrophilic block removed, so most porous films are characteristically hydrophobic, which limits the versatility of this approach. Alternatively, an approach to alter BCP surface chemistry is with triblock copolymers. While typical pore formation removes the hydrophilic block, in the case of triblock copolymers, it is possible to remove one block and leave a hydrophilic and a hydrophobic segment (Figure 5B). When designed correctly, this places a hydrophilic segment at the surface, and the subsequent porous structure has hydrophilic surface properties.87
2.7. Biologically-Derived Materials
Many important biological functionalities operate at the nanoscale, so it is sensible to consider biology in the development of nanoscale structures. Cellulose membranes are one of the most prolific and well established biologically-derived nanoscale materials used in medicine today. These membranes are the corner stone of biofiltration, particularly dialysis treatments, and represent a >$1 billion market.17, 18 Interest in cellulose was originally due to its natural abundance along with low solubility in water and many common organic solvents. Derivative forms of cellulose, such as cellulose nitrate and cellulose acetate, allowed the use of solvent casting as a preparation technique (see section 2.4).88 These membranes are often used in the derivative form, but conversion back to the unfunctionalized cellulose chemical structure (termed regenerated cellulose) restores its chemical robustness. Commercial examples of cellulose-based membranes are abundant and available in a wide range of pore sizes and molecular weight cut-offs (including MW cut-offs down to 1 kDa).51, 89 Given its lengthy history, cellulose-based membranes have previously been covered in other resources.88, 90
In addition to cellulose-based membranes, nanostructures can also be found in the outermost cell envelope of certain bacteria. In some cases, this can form crystalline surface layers (termed S-layers), which form a variety of two-dimensional symmetries including oblique (p2), square (p4), and hexagonal (p6).91 When deposited on supporting structures, S-layers can form highly uniform pores that range from 2–8 nm, with a pore-to-pore spacing from 3 to 35 nm depending on the particular bacterial strain.92 Furthermore, surface modifications can be performed to influence protein interactions with these biologically-derived membranes, which are particularly useful for filtration (see Section 3.1).92, 93 In-depth detail regarding Slayers can be found elsewhere,94–96 and examples of relevant prokaryotes are presented in the supplementary materials.
Finally, engineered polypeptides provide a synthetic route whereby biologically-relevant materials can be generated. For example, synthetic polypeptides (primarily consisting of glycine, L-glutamic acid, L-valine, and proline subunits) were able to produce elastomeric films with a pore size of approximately 70 nm, and these films are expected to exhibit improved biocompatibility.97 In addition, peptide-based polymers have been used with LbL techniques (see section 2.5).73 Finer nanostructures were shown for polypeptides deposited with LbL, yet it is unclear whether the demonstrated nanotopography constitutes a nanoporous film.
3. Applications of Nanoporous Polymers
While a variety of applications in bioengineering and therapeutic sciences utilize porous membranes,10, 16–18 nanoporous membranes have unique advantages that can be exploited in biomedical sciences. In particular, a wide range of fabrication techniques and versatility in chemical and mechanical properties make nanostructured polymers attractive for a variety of applications. In the area of biofiltration and immunoisolation, nanoporous membranes are valuable given the membrane’s pore size relative to the size of potential filtrates. For membranes with high uniformity, nanopores act as an effective means to prevent passage of larger molecules and allow passage of small molecules: in addition to filtration, this can be used to isolate implanted cells from the immune system while allowing passage of nutrients and waste. Similarly, size selectivity can be utilized in drug delivery, where nanoscale pores can control diffusive release of potential therapeutics when pore and therapeutic size are comparable. In this section, applications of nanoporous polymers, including biofiltration, immunoisolation, and drug delivery, are considered.
3.1 Biofiltration and Immunoisolation
Membranes have historically been the foundation of filtration processes, and the development of nanoporous membranes has resulted in enhanced performance and broader applicability. In particular, dialysis has benefited substantially from improved membranes for this vital procedure. Led by cellulose-based membranes89 and various solvent-cast synthetic membranes,98–101 a wide range of size- or MW-based restrictions are currently available. Table 2 summarizes contemporary nanoporous membranes that have been developed for biofiltration and immunoisolation, including rejection/passage properties and relevant size cutoffs. However, the focus of this review is nanoporous membranes and consideration of all membranes utilized for biofiltration and immunoisolation is beyond the scope of this review and excellent auxillary resources can be found elsewhere.16–18
Table 2.
Summary of biofiltration/immunoisolation membranes.
| Approach | Material | Rejects A | Passes | Cut-off | Ref | |
|---|---|---|---|---|---|---|
| MW (kDa) | Size (nm) | |||||
| Cellulose (Spectra/Por®) | Cellulose Esters | N/A B | 1–50 | – | 89 | |
| Track Etch (Nucleopore™) | PC | N/A B | – | 15 - 1.2×104 | 46 | |
| Nanosphere Lithography | PES | 300 nm SiO2 particles | 60 nm SiO2 particles | – | < 300 | 34 |
| S-layers |
B. stearothermophilus C. thermohydrosulfuricum |
Ovalbumin, BSA, Ferritin | Myoglobin, CA | <67 | 1–8 C | 104 |
| B. stearothermophilus | Ovalbumin, BSA | Myoglobin D, CA E | <67 | – | 93 | |
| B. spaericus | CA F, Ovalbumin, BSA, Ferritin | Myoglobin | – | 4–5 | 92 | |
| Block Copolymer | PS-b-PMMA | Human Rhinovirus Type 14 | – | – | >30 | 77 |
| PS-b-P4VP | Albumin G | – | – | ~8 H | 83 | |
| Graft copolymer | PVDF-g-POEM | Alcian blue+ | Rhodamine B+ | 0.4–1.3 | ~ 10 | 82 |
| Brilliant blue− | Congo red− | 0.7–0.85 | ||||
| Phase Separation | PCL | IgG | Glucose I | <150 | – | 53 |
Abbreviations are as follows: PC - poly(carbonate), PCL - poly(caprolactone), PS - poly(styrene), PMMA - poly(methyl methacrylate), P4VP - poly(4-vinylpyridine), PVDF - poly(vinylidene fluoride), POEM - poly(oxyethylene methacrylate), PES - poly(ether sulfone), CA – Carbonic anhydrase
Commercial examples of filtration membranes are plentiful and provide a wide range of size selectivity and chemical compatibility. Both track etch and cellulose-based membranes are well known examples that can be found in a wide range of pore sizes (15 nm to 12 μm) and MW-cut-offs (1–50 kDa).46, 51, 89, 102, 103 Synthetic nanostructured membranes are also available, where membrane resistance to solvent exposure is often a primary advantage. One basic test of filtration performance is the passage of nanoparticles. For poly(ether sulfone) membranes fabricated using nanosphere lithography, rejection of large (300 nm) silica particles and passage of smaller (60 nm) silica particles has been demonstrated.34 While biological applications are limited at this size scale, smaller colloids have been demonstrated with this approach,35 which may improve the performance of this processing scheme.
More biologically relevant is the filtration of proteins. To date, S-layers have received the most extensive investigation of their filtration properties. Analytes investigated included ferritin, bovine serum albumin (BSA), ovalbumin, and the enzyme carbonic anhydrase.92, 93, 104 Various rejection capabilities were demonstrated depending on the origin of the S-layer and the characteristic pore size. Overall, larger proteins (ferritin and BSA) consistently exhibited good rejection due to their size relative to the pore size. In addition, chemical functionalization has been used as an avenue to control permeability and has been shown to influence passage of various species.92, 93
Copolymer membranes have also been used in a variety of applications to selectively filter small molecules,82 biomolecules,83 and viruses.77 PS-b-PMMA BCP membranes with 15 nm pores were effective at preventing the passage of human rhinovirus (with approximate size of 30 nm), which is excepted based on pore size and high degree of uniformity of these BCP membranes.77 Similarly, thick BCP membranes were capable of rejecting albumin, indicating a cut-off size on the order of ~7 nm;83 based on their structure, passage of small molecules is expected to be efficient, making these membranes candidates for immunoisolation. The effectiveness of graft copolymers for filtration of charged small molecules has also been investigated. These materials were shown to differentially filter positive molecules Alican Blue and Rhodamine B and negative molecules Brilliant Blue and Congo Red, indicating these membranes may be valuable for fine-scale molecular separations.82
In contrast to polymer membranes used for filtration, membranes targeted to immunoisolation are comparatively rare, and most isolation examples have focused on inorganic membranes or capsules;12, 105 however, PCL has been used as a immunoisolation membrane for mouse embryonic stem cells.53 This work demonstrated that cellular viability was unaffected by the addition of immunoglobulin G (IgG), indicating that IgG was prevented from transversing the nanoporous membrane, while nutrient availability was not significantly affected.
3.2 Drug Delivery
Recent advances in drug delivery have resulted in improved control over dose and localized release, which has improved treatment outcomes and led to innovative therapies.106, 107 Nanoscale materials have been consistent contributors to emergent delivery strategies and continue to have significant impacts.108 Various technologies have utilized membranes for drug delivery,17, 27 but typically these membranes are not nanoporous in nature. Many nanoscale drug delivery devices utilize nanoscale geometries to increase surface area for release;17, 58, 109 however, nanoporous membranes also have the potential to constrain diffusion physically. Developed theoretically110 and later demonstrated in zeolites111 and other materials,11, 15 non-first order diffusion can be achieved with porous materials when the size of the diffusing species is comparable to the pore size. This process is often referred to as “single-file” diffusion and can lead to zero-order kinetics. For many therapies constant zero-order release is an ideal way to maintain stable therapeutic concentrations over extended periods while avoiding side effects, wasted therapeutic, or sub-therapeutic troughs: for this reason, nanoporous materials of particular interest to drug delivery applications. Nanoporous degradable polymers loaded with a molecule for release are omitted here since release is dominated by polymer degradation and porosity primarily increases surface area for release.
Table 3 summarizes diffusion/release of small molecules and proteins from nanoporous polymers. Early examples utilized commercial nanoporous cellulose membranes (Millipore), and release of glucose and BSA was studied over the course of hours.11 Glucose transport was first-order and BSA transport was linear over the course of 6 hours. Unfortunately, because less than 10% of the loaded BSA diffused across the membrane, it difficult to establish the characteristic concentration dependence since zero- and first-order release both appear linear over this time course.
Table 3.
Summary of loading and release of therapeutics and model molecules released from/through nanoporous materials
| Approach | Material | Molecule(s) | Molecule Solubility | Loading Type | Kinetics | Time course | Ref |
|---|---|---|---|---|---|---|---|
| Commercial (Millipore) | Cellulose Esters | Glucose0 | Moderate | Reservior | Exponential A | hours | 11 |
| BSA−119, 120 | Moderate | Linear A | days B | ||||
| Layer-by-layer | PAA/PAH/PSS | Ketoprofen0 | Low (0.24 mg/ml)113 | Immersion | Linear A | days | 67 |
| Cytochalasin D0 | Essentially insoluble112 | ||||||
| Phase separation | PCL | Lysozyme+121 | Moderate122 | Reservior | Sublinear A | days | 53 |
| Light induced polymerization | Acrylate-based | Rhodamine BZ | High | Immersion | Sublinear C,D | a few hours | 58 |
| Template | PCL | Fluorescein− | High | Reservior | Exponential E | days | 41 |
| BSA−119, 120 | Moderate | Linear E |
The net charge of molecules investigated is indicated with the following markers:
positive,
negative,
neutral/isoelectric point,
zwitterionic.
More recently, nanoporous layer-by-layer films were loaded with two small molecule therapeutics, ketoprofen and cytochalasin D. Release of both therapeutics from nanoporous films was linear, where the duration of release could be extended for multiple weeks. Given the low aqueous solubility of these molecules112, 113 and the relatively large pore size compared to the molecules of interest, the observed linear dependence may not be the result of constrained release but rather additional complications influencing release. Similarly, nanoporous polymer films formed using light-induced polymerization have been studied for the release of the small molecule Rhodamine B.58 Rhodamine B was released over a few hours, characterized by an initial burst release followed by relatively linear release kinetics. Compared with a non-porous film, release differed by a factor of approximately two and were qualitatively similar. Given the similarity between non-porous and nanoporous films and the short time course of release, the difference observed here may be due to increased surface area in the porous films.
Due to its biodegradability and biocompatibility,114 PCL has been a nanoporous material of particular interest. Nanoporous PCL fabricated for immunoisolation of mouse embryonic stem cells (Section 3.1) was also examined for diffusion of lysozyme.53 Release over a few days was sub-linear and is likely first-order in nature, which is reasonable given a pore size that is large relative to the diffusing species. Additionally, nanoporous PCL films fabricated using templating techniques were characterized with fluorescein and fluorescein-isothiocyanate-labeled BSA (FITC-BSA).41 Diffusion of the small molecule fluorescein was first-order while diffusion of FITC-BSA was zero-order. Since FITC-BSA is similar to the pore size reported here, it is reasonable that the constant-rate was due to physically constrained transport as described above. Cell culture of NIH 3T3 fibroblasts on these nanostructured PCL films demonstrated preliminary biocompatibility, but further work will be required to establish in vivo biocompatibility. While examples of therapeutic delivery controlled by nanoporous films are limited, improved nanostructure fabrication and wider materials availability make this approach a promising avenue for macromolecule delivery in therapeutic applications. Further work will be required to characterize release kinetics and biocompatibility of candidate materials alongside development of novel materials.
4. Concluding Remarks
Nanotechnology, in the form of particles, fibers, and nanostructured films, has made significant strides in recent years to provide solutions in the field of bioengineering and therapeutic sciences. In particular, advantageous use of nanoscale materials has resulted in a variety of filtration, immunoisolation, drug delivery, and tissue engineering applications. In particular, development of nanoporous materials has resulted in improved material selection produced using a wider range of fabrication techniques, making these materials more viable for biomedical applications. While improved fabrication techniques for inorganic materials have been responsible for many of the applications of nanoporous materials, developments in polymer fabrication and processing will allow these materials to compete more effectively with inorganic alternatives. To be competitive, polymers will need to facilitate solutions that make use of their tunable chemical properties, variable mechanical properties, and versatility in form-factor. Emerging nanoporous polymers are poised to make significant inroads with biological applications; however, significant work, both in vitro and in vivo, will be required to verify the compatibility and effectiveness of these materials in biomedical and therapeutic applications.
Acknowledgments
The authors would like to acknowledge Miquella Chavez and Rachel Lowe for valuable review of this manuscript. This work was supported by the National Institutes of Health and the UC Discovery Program. Scanning electron microscopy of nanoporous PCL was performed at the Stanford Nanocharacterization Laboratory through the Stanford CIS grant program.
Biographies
 Daniel A. Bernards
Daniel A. Bernards
Daniel Bernards received his PhD in Materials Science and Engineering from Cornell University for work investigating mixed conduction of ionic and electronic charge in organic semiconductor devices in the group of George Malliaras. In 2007, he joined the group of Tejal Desai at the University of California, San Francisco developing nanostructured biodegradable polymers for drug delivery applications.
 Tejal A. Desai
Tejal A. Desai
Dr. Tejal Desai is currently Professor of Bioengineering and Therapeutic Sciences at the University of California, San Francisco. She is also a member of the California Institute for Quantitative Biomedical Research and chair of the UCSF/UC Berkeley Graduate Group in Bioengineering. Dr. Tejal Desai directs the Laboratory of Therapeutic Micro and Nanotechnology at UCSF. Her research uses micro and nanofabrication techniques to create implantable biohybrid devices for cell encapsulation, targeted drug delivery, and templates for cell and tissue regeneration. In addition to authoring over 130 technical papers and delivering over 150 invited talks, she is the co-editor of an encyclopedia on Therapeutic Microtechnology.
Footnotes
Highly dependent particular processing parameters
Not reported, estimated from best available data
Thickness of nanoporous layer; mounted/attached to a thick membrane
Indicator for symmetry of layer: (h) hexagonal symmetry, (g) double gyroid network, (p) 2, 3, 4, or 6-fold symmetry, (u) user-defined patterns.
Results presented make it difficult to make reasonable estimates of these parameters
Nanoporous region is a fraction of this reported thickness
One kDa dialysis membranes indicate sub 10 nm pores are readily obtainable
Crystalline cell layers are typically supported on polymer membranes
Pore depth as measured with AFM. Likely an underestimate of total pore depth
Defined as >=90% rejection of listed species, unless noted otherwise
Specific rejection/passage depends highly on the particular membranes used
Range of pore sizes observed with different S-layers.
Depending on processing, Myoglobin passage ranges from 45–100%
Depending on processing, Carbonic anhydrase passage ranges from 15–90%
78% rejection
82% rejection, as measured at the isoelectric point for albumin
Effective pore diameter
Nutrients required for cell survival are able to pass membrane
Experiments performed at an unspecified temperature.
This is an estimate of expected release time scales. Transport of BSA was tested over 7 hours at which point less than 10% has pass through the membrane.
Experiments performed at room (20–25 °C) temperature.
Sublinear behavior is preceded by burs time release for the first 5% of the delivery time course
Experiments performed at physiological (37 °C) temperature.
References
- 1.Rejeski D, Lekas D. J Clean Prod. 2008;16:1014–1017. [Google Scholar]
- 2.NIH. Nanotechnology at the the National Institutes of Health. 2009 http://www.nih.gov/science/nanotechnology/sci_NANO_brochure.pdf.
- 3.Ainslie KM, Tao SL, Popat KC, Desai TA. ACS Nano. 2008;2:1076–1084. doi: 10.1021/nn800071k. [DOI] [PubMed] [Google Scholar]
- 4.Ainslie KM, Thakar RG, Bernards DA, Desai TA. In: Biological Interactions on Material Surfaces: Understanding and Controlling Protein, Cell and Tissue Responses. Puleo DM, Bizios R, editors. Springer; New York: 2009. [Google Scholar]
- 5.Roco MC. Curr Opin Biotechnol. 2003;14:337–346. doi: 10.1016/s0958-1669(03)00068-5. [DOI] [PubMed] [Google Scholar]
- 6.Whitesides GM. Nat Biotechnol. 2003;21:1161–1165. doi: 10.1038/nbt872. [DOI] [PubMed] [Google Scholar]
- 7.Seeman NC, Belcher AM. Proc Natl Acad Sci U S A. 2002;99:6451–6455. doi: 10.1073/pnas.221458298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sarikaya M, Tamerler C, Jen AKY, Schulten K, Baneyx F. Nat Mater. 2003;2:577–585. doi: 10.1038/nmat964. [DOI] [PubMed] [Google Scholar]
- 9.N. R. f. M. Research. Nanomedicine Initiative. 2009. [Google Scholar]
- 10.Adiga SP, Jin C, Curtiss LA, Monteiro-Riviere NA, Narayan RJ. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology. 2009;1:568–581. doi: 10.1002/wnan.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desai TA, Hansford DJ, Leoni L, Essenpreis M, Ferrari M. Biosens Bioelectron. 2000;15:453–462. doi: 10.1016/s0956-5663(00)00088-9. [DOI] [PubMed] [Google Scholar]
- 12.Mendelsohn A, Desai T. In: Therapeutic Applications of Cell Microencapsulation. Pedraz JL, Orive G, editors. Landes Bioscience; Austin, Texas: 2008. [Google Scholar]
- 13.Kipke S, Schmid G. Adv Funct Mater. 2004;14:1184–1188. [Google Scholar]
- 14.Paulose M, Prakasam HE, Varghese OK, Peng L, Popat KC, Mor GK, Desai TA, Grimes CA. J Phys Chem C. 2007;111:14992–14997. [Google Scholar]
- 15.Martin F, Walczak R, Boiarski A, Cohen M, West T, Cosentino C, Ferrari M. J Control Release. 2005;102:123–133. doi: 10.1016/j.jconrel.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 16.Adiga SP, Curtiss LA, Elam JW, Pellin MJ, Shih CC, Shih CM, Lin SJ, Su YY, Gittard SA, Zhang J, Narayan RJ. Jom. 2008;60:26–32. [Google Scholar]
- 17.Stamatialis DF, Papenburg BJ, Girones M, Saiful S, Bettahalli SNM, Schmitmeier S, Wessling M. J Membr Sci. 2008;308:1–34. [Google Scholar]
- 18.Ulbricht M. Polymer. 2006;47:2217–2262. [Google Scholar]
- 19.Couvreur P, Barratt G, Fattal E, Legrand P, Vauthier C. Crit Rev Ther Drug Carr Syst. 2002;19:99–134. doi: 10.1615/critrevtherdrugcarriersyst.v19.i2.10. [DOI] [PubMed] [Google Scholar]
- 20.Frenot A, Chronakis IS. Curr Opin Colloid Interface Sci. 2003;8:64–75. [Google Scholar]
- 21.Kumbar SG, James R, Nukavarapu SP, Laurencin CT. Biomed Mater. 2008:3. doi: 10.1088/1748-6041/3/3/034002. [DOI] [PubMed] [Google Scholar]
- 22.Goldberg M, Langer R, Jia XQ. J Biomater Sci-Polym Ed. 2007;18:241–268. doi: 10.1163/156856207779996931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. J Control Release. 2001;70:1. doi: 10.1016/s0168-3659(00)00339-4. [DOI] [PubMed] [Google Scholar]
- 24.Madou MJ. Fundamentals of Microfabrication. 2. CRC; 2002. [Google Scholar]
- 25.DeFranco JA, Schmidt BS, Lipson M, Malliaras GG. Organic Electronics. 2006;7:22. [Google Scholar]
- 26.Taylor PC, Lee JK, Zakhidov AA, Chatzichristidi M, Fong HH, DeFranco JA, Malliaras GC, Ober CK. Adv Mater. 2009;21:2314. [Google Scholar]
- 27.Lu Y, Chen SC. Adv Drug Deliv Rev. 2004;56:1621–1633. doi: 10.1016/j.addr.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 28.Sideridou I, Tserki V, Papanastasiou G. Biomaterials. 2002;23:1819–1829. doi: 10.1016/s0142-9612(01)00308-8. [DOI] [PubMed] [Google Scholar]
- 29.Metters AT, Anseth KS, Bowman CN. Polymer. 2000;41:3993–4004. [Google Scholar]
- 30.Bryant SJ, Cuy JL, Hauch KD, Ratner BD. Biomaterials. 2007;28:2978–2986. doi: 10.1016/j.biomaterials.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elionix Inc. ELS-7000 Ultra-High Precision Electron Beam Lithography System. 2009 [Google Scholar]
- 32.Jeol Ltd. JBX-5500FS Electron Beam Lithography System. 2009 [Google Scholar]
- 33.Han KP, Xu WD, Ruiz A, Ruchhoeft P, Chellam S. J Membr Sci. 2005;249:193–206. [Google Scholar]
- 34.Acikgoz C, Ling XY, Phang IY, Hempenius MA, Reinhoudt DN, Huskens J, Vancso CJ. Adv Mater. 2009;21:2064–2067. [Google Scholar]
- 35.Xu H, Goedel WA. Langmuir. 2002;18:2363–2367. [Google Scholar]
- 36.Martin CR. Science. 1994;266:1961–1966. doi: 10.1126/science.266.5193.1961. [DOI] [PubMed] [Google Scholar]
- 37.Cao GZ, Liu DW. Adv Colloid Interface Sci. 2008;136:45–64. doi: 10.1016/j.cis.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 38.Johnson SA, Ollivier PJ, Mallouk TE. Science. 1999;283:963–965. doi: 10.1126/science.283.5404.963. [DOI] [PubMed] [Google Scholar]
- 39.Cepak VM, Martin CR. Chem Mat. 1999;11:1363–1367. [Google Scholar]
- 40.Tao SL, Desai TA. Nano Lett. 2007;7:1463–1468. doi: 10.1021/nl0700346. [DOI] [PubMed] [Google Scholar]
- 41.Bernards DA, Desai TA. Adv Mater. 2009 doi: 10.1002/adma.200903439. in press. [DOI] [PubMed] [Google Scholar]
- 42.Chou SY, Krauss PR, Renstrom PJ. Appl Phys Lett. 1995;67:3114–3116. [Google Scholar]
- 43.Chuang VP, Ross CA, Gwyther J, Manners I. Adv Mater. 2009;21:3789–3793. [Google Scholar]
- 44.Price PB, Walker RM. Physics Letters. 1962;3:113. [Google Scholar]
- 45.OxyPhen AG. RoTrac(R) Membranes. 2009 [Google Scholar]
- 46.Whatman Whatman Nucleopore Track Etch Membranes. 2009 [Google Scholar]
- 47.Spohr R. Radiat Meas. 2005;40:191–202. [Google Scholar]
- 48.Fleischer RL. Tracks to Innovation: Nuclear Tracks in Science and Technology. Springer; New York: 1998. [Google Scholar]
- 49.Pall Corporation. Polysulfone Membranes. 2009. [Google Scholar]
- 50.Whatman Whatman Polysulfone Membranes. 2009 [Google Scholar]
- 51.Whatman Whatman Cellulose Membranes. 2009 [Google Scholar]
- 52.Akthakul A, McDonald WF, Mayes AM. J Membr Sci. 2002;208:147–155. [Google Scholar]
- 53.Zhang XL, He HY, Yen C, Ho W, Lee LJ. Biomaterials. 2008;29:4253–4259. doi: 10.1016/j.biomaterials.2008.07.032. [DOI] [PubMed] [Google Scholar]
- 54.Krause B, Diekmann K, van der Vegt NFA, Wessling M. Macromolecules. 2002;35:1738–1745. [Google Scholar]
- 55.Merlet S, Marestin C, Schiets F, Romeyer O, Mercier R. Macromolecules. 2007;40:2070–2078. [Google Scholar]
- 56.Reverchon E, Cleofe Volpe M, Caputo G. Current Opinion in Solid State and Materials Science. 7:391. [Google Scholar]
- 57.Tomasko DL, Han X, Liu D, Gao W. Current Opinion in Solid State and Materials Science. 7:407. [Google Scholar]
- 58.Yan W, Hsiao SVK, Zheng YB, Shariff YM, Gao TY, Huang TJ. Thin Solid Films. 2009;517:1794–1798. [Google Scholar]
- 59.Decher G. Science. 1997;277:1232–1237. [Google Scholar]
- 60.Zhang X, Chen H, Zhang HY. Chem Commun. 2007:1395–1405. doi: 10.1039/b615590a. [DOI] [PubMed] [Google Scholar]
- 61.Dobrynin AV, Rubinstein M. Prog Polym Sci. 2005;30:1049–1118. [Google Scholar]
- 62.Tang ZY, Wang Y, Podsiadlo P, Kotov NA. Adv Mater. 2006;18:3203–3224. [Google Scholar]
- 63.De Geest BG, Vandenbroucke RE, Guenther AM, Sukhorukov GB, Hennink WE, Sanders NN, Demeester J, De Smedt SC. Adv Mater. 2006;18:1005. [Google Scholar]
- 64.Zhang JT, Chua LS, Lynn DM. Langmuir. 2004;20:8015–8021. doi: 10.1021/la048888i. [DOI] [PubMed] [Google Scholar]
- 65.Wood KC, Boedicker JQ, Lynn DM, Hammon PT. Langmuir. 2005;21:1603–1609. doi: 10.1021/la0476480. [DOI] [PubMed] [Google Scholar]
- 66.Mendelsohn JD, Barrett CJ, Chan VV, Pal AJ, Mayes AM, Rubner MF. Langmuir. 2000;16:5017–5023. [Google Scholar]
- 67.Berg MC, Zhai L, Cohen RE, Rubner MF. Biomacromolecules. 2006;7:357–364. doi: 10.1021/bm050174e. [DOI] [PubMed] [Google Scholar]
- 68.Olugebefola SC, Kuhlman WA, Rubner MF, Mayes AM. Langmuir. 2008;24:5172–5178. doi: 10.1021/la703936p. [DOI] [PubMed] [Google Scholar]
- 69.Lutkenhaus JL, McEnnis K, Hammond PT. Macromolecules. 2008;41:6047–6054. [Google Scholar]
- 70.Zimnitsky D, Shevchenko VV, Tsukruk VV. Langmuir. 2008;24:5996–6006. doi: 10.1021/la7038575. [DOI] [PubMed] [Google Scholar]
- 71.Li Q, Quinn JF, Caruso F. Adv Mater. 2005;17:2058. [Google Scholar]
- 72.Li Q, Quinn JF, Wang YJ, Caruso F. Chem Mat. 2006;18:5480–5485. [Google Scholar]
- 73.Zhong Y, Whittington CF, Zhang L, Haynie DT. Nanomed-Nanotechnol Biol Med. 2007;3:154–160. doi: 10.1016/j.nano.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 74.Park C, Yoon J, Thomas EL. Polymer. 2003;44:6725–6760. [Google Scholar]
- 75.Li MQ, Coenjarts CA, Ober CK. Block Copolymers Ii. Vol. 190. Springer-Verlag Berlin; Berlin: 2005. pp. 183–226. [Google Scholar]
- 76.Hashimoto T, Tsutsumi K, Funaki Y. Langmuir. 1997;13:6869–6872. [Google Scholar]
- 77.Yang SY, Ryu I, Kim HY, Kim JK, Jang SK, Russell TP. Adv Mater. 2006;18:709. [Google Scholar]
- 78.Zalusky AS, Olayo-Valles R, Taylor CJ, Hillmyer MA. J Am Chem Soc. 2001;123:1519–1520. [Google Scholar]
- 79.Bockstaller MR, Mickiewicz RA, Thomas EL. Adv Mater. 2005;17:1331–1349. doi: 10.1002/adma.200500167. [DOI] [PubMed] [Google Scholar]
- 80.Jeong UY, Ryu DY, Kim JK, Kim DH, Russell TP, Hawker CJ. Adv Mater. 2003;15:1247. [Google Scholar]
- 81.Jeong UY, Ryu DY, Kim JK, Kim DH, Wu XD, Russell TP. Macromolecules. 2003;36:10126–10129. [Google Scholar]
- 82.Akthakul A, Salinaro RF, Mayes AM. Macromolecules. 2004;37:7663–7668. [Google Scholar]
- 83.Peinemann KV, Abetz V, Simon PFW. Nat Mater. 2007;6:992–996. doi: 10.1038/nmat2038. [DOI] [PubMed] [Google Scholar]
- 84.Schacher F, Ulbricht M, Muller AHE. Adv Funct Mater. 2009;19:1040–1045. [Google Scholar]
- 85.Xu T, Kim HC, DeRouchey J, Seney C, Levesque C, Martin P, Stafford CM, Russell TP. Polymer. 2001;42:9091–9095. [Google Scholar]
- 86.Ruiz R, Kang HM, Detcheverry FA, Dobisz E, Kercher DS, Albrecht TR, de Pablo JJ, Nealey PF. Science. 2008;321:936–939. doi: 10.1126/science.1157626. [DOI] [PubMed] [Google Scholar]
- 87.Rzayev J, Hillmyer MA. Macromolecules. 2005;38:3–5. [Google Scholar]
- 88.Kamide K. Cellulose and Cellulose Derivatives. Elsevier Science; Amsterdam: 2005. [Google Scholar]
- 89.Spectrum Laboratories. Spectra/POR Dialysis Membranes. 2009 [Google Scholar]
- 90.Zugenmaier P. Crystalline Cellulose and Derivatives. Springer; Berlin: 2008. [Google Scholar]
- 91.Sara M, Sleytr UB. J Bacteriol. 2000;182:859–868. doi: 10.1128/jb.182.4.859-868.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Weigert S, Sara M. J Membr Sci. 1995;106:147–159. [Google Scholar]
- 93.Kupcu S, Sara M, Sleytr UB. J Membr Sci. 1991;61:167–175. [Google Scholar]
- 94.Sara M, Sleytr UB. J Bacteriol. 2000;182:859–868. doi: 10.1128/jb.182.4.859-868.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sleytr UB, Beveridge TJ. Trends in Microbiology. 1999;7:253. doi: 10.1016/s0966-842x(99)01513-9. [DOI] [PubMed] [Google Scholar]
- 96.Sleytr UB, Sara M. Trends Biotechnol. 1997;15:20–26. doi: 10.1016/S0167-7799(96)10063-9. [DOI] [PubMed] [Google Scholar]
- 97.Reguera J, Fahmi A, Moriarty P, Girotti A, Rodriguez-Cabello JC. J Am Chem Soc. 2004;126:13212–13213. doi: 10.1021/ja047417f. [DOI] [PubMed] [Google Scholar]
- 98.Fresenius Medical Care. F60 (Fresenius Polysulfone) 2009 [Google Scholar]
- 99.Minntech Corporation. Primus 1350 Membranes (Polyphen) 2009 [Google Scholar]
- 100.WR Grace Incorporated. National Medical Care BioCare 130 (Biosulfane) 2009 [Google Scholar]
- 101.Hoenich NA, Woffindin C, Brennan A, Cox PJ, Matthews JN, Goldfinch M. J Am Soc Nephrol. 1996;7:871–876. doi: 10.1681/ASN.V76871. [DOI] [PubMed] [Google Scholar]
- 102.Millipore Millipore BioMax Membranes. 2009 [Google Scholar]
- 103.Thermo Scientific. SnakeSkin Dialysis Tubing. 2009 [Google Scholar]
- 104.Sara M, Sleytr UB. J Membr Sci. 1987;33:27–49. [Google Scholar]
- 105.Desai TA, Hansford DJ, Ferrari M. Biomol Eng. 2000;17:23–36. doi: 10.1016/s1389-0344(00)00063-0. [DOI] [PubMed] [Google Scholar]
- 106.Langer R. Science. 1990;249:1527–1533. doi: 10.1126/science.2218494. [DOI] [PubMed] [Google Scholar]
- 107.Allen TM, Cullis PR. Science. 2004;303:1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 108.Tong R, Christian DA, Tang L, Cabral H, Baker JR, Kataoka K, Discher DE, Cheng J. MRS Bull. 2009;34:422–431. [Google Scholar]
- 109.Hughes GA. Dm Disease-A-Month. 2005;51:342–361. doi: 10.1016/j.disamonth.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 110.Chemistry JOP, Levitt DG. Phys Rev A. 1973;8:3050–3054. [Google Scholar]
- 111.Hahn K, Karger J, Kukla V. Phys Rev Lett. 1996;76:2762–2765. doi: 10.1103/PhysRevLett.76.2762. [DOI] [PubMed] [Google Scholar]
- 112.Aldrich Website. C8273 - Product Information Sheet. 2009 [Google Scholar]
- 113.Fujii M, Hori N, Shiozawa K, Wakabayashi K, Kawahara E, Matsumoto M. Int J Pharm. 2000;205:117. doi: 10.1016/s0378-5173(00)00496-8. [DOI] [PubMed] [Google Scholar]
- 114.Sun HF, Mei L, Song CX, Cui XM, Wang PY. Biomaterials. 2006;27:1735–1740. doi: 10.1016/j.biomaterials.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 115.Apel PY, Blonskaya IV, Orelovitch OL, Root D, Vutsadakis V, Dmitriev SN. Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms. 2003;209:329. [Google Scholar]
- 116.Vivian PC, Caroline AR, Jessica G, Ian M. Adv Mater. 2009;21:3789–3793. [Google Scholar]
- 117.Millipore MF-Millipore Membrane Filters. 2009 [Google Scholar]
- 118.Schuster B, Pum D, Sara M, Braha O, Bayley H, Sleytr UB. Langmuir. 2001;17:499–503. [Google Scholar]
- 119.Böhme U, Scheler U. Chemical Physics Letters. 2007;435:342. [Google Scholar]
- 120.Foghandersen N, Bjerrum PJ, Siggaardandersen O. Clin Chem. 1993;39:48–52. [PubMed] [Google Scholar]
- 121.Kuehner DE, Engmann J, Fergg F, Wernick M, Blanch HW, Prausnitz JM. The Journal of Physical Chemistry B. 1999;103:1368. [Google Scholar]
- 122.Rieskautt MM, Ducruix AF. J Biol Chem. 1989;264:745–748. [PubMed] [Google Scholar]