

Abstract
Background
Timothy Syndrome (TS) is a disease of excessive cellular Ca2+ entry and life-threatening arrhythmias due to a mutation in the primary cardiac L-type Ca2+ channel (CaV1.2). The TS mutation causes loss of normal voltage-dependent inactivation (VDI) of CaV1.2 current (ICa). During cellular Ca2+ overload the calmodulin-dependent protein kinase II (CaMKII) causes arrhythmias. We hypothesized that CaMKII is a part of the proarrhythmic mechanism in TS.
Methods and Results
We developed an adult rat ventricular myocyte model of TS (G406R) by lenti virus-mediated transfer of wild type (WT) and TS CaV1.2. The exogenous CaV1.2 contained a mutation (T1066Y) conferring dihydropyridine resistance, so we could silence endogenous CaV1.2 with nifedipine and maintain peak ICa at control levels in infected cells. TS CaV1.2 infected ventricular myocytes exhibited the signature VDI loss under Ca2+ buffering conditions, not permissive for CaMKII activation. In physiological Ca2+ solutions, TS CaV1.2 expressing ventricular myocytes exhibited increased CaMKII activity and a proarrhythmic phenotype that included action potential prolongation, increased ICa facilitation and afterdepolarizations. Intracellular dialysis of a CaMKII inhibitory peptide, but not a control peptide, reversed increases in ICa facilitation, normalized the action potential and prevented afterdepolarizations. We developed a revised mathematical model that accounts for CaMKII-dependent and CaMKII-independent effects of the TS mutation.
Conclusions
In TS the loss of VDI is an upstream initiating event for arrhythmia phenotypes that are ultimately dependent on CaMKII activation.
Keywords: action potentials, calcium, ion channels, myocytes
Introduction
Timothy Syndrome (TS) is an autosomal genetic disease of the primary voltage-gated cardiac Ca2+ channel (CaV1.2) consisting of a missense mutation in the pore forming α1c subunit protein1. TS is associated with three individual mutations that include G406R on exon 8a2, G406R on exon 81 and G402S1. The G406R mutation on exon 8 used for our model of TS is believed to be the most severe form of TS1. TS patients have an average life expectancy of only 2.5 years due to severe cardiac disease. TS is also known as long QT syndrome 8 (LQT8) and the prolonged QT intervals in TS patients are thought to cause cardiac arrhythmias and sudden death. TS disease phenotypes are apparently initiated by excessive Ca2+ entry, at least in part, due to impaired voltage dependence of inactivation (VDI) of CaV1.2 current (ICa)1, 2 Mathematical modeling predicts that intracellular Ca2+ overload and action potential prolongation stimulate afterdepolarizations that are the cellular mechanism for triggering ventricular arrhythmias in TS1, 2. However, these predictions have not been directly tested in ventricular myocytes.
In ventricular myocytes multiple signaling pathways are activated by increased intracellular Ca2+ entry, including the multifunctional Ca2+ and calmodulin dependent kinase II (CaMKII)3, a procardiomyopathic and proarrhythmic signaling molecule4. Increased CaMKII activity causes action potential prolongation and arrhythmias, similar to the observed phenotypes in TS patients, in part by increasing sarcoplasmic reticulum (SR) Ca2+ leak and ICa facilitation 5, 6. On the other hand, CaMKII inhibition restores normal intracellular Ca2+ homeostasis and suppresses arrhythmias4, 5. Based upon these concepts, we hypothesized that increased Ca2+ entry in TS ventricular myocytes enhances CaMKII actions and that activated CaMKII recruitment is important for the proarrhythmic cellular phenotype in TS. To test this hypothesis, we created an adult rat ventricular myocyte model of TS by lenti viral infection of a dihydropyridine-resistant CaV1.2 α1c subunit7 harboring the TS mutation. Our studies show that TS mutation requires CaMKII activity to cause important proarrhythmic phenotypes in adult ventricular myocytes.
Methods
Cloning
The plasmids pLentiNB CaV1.2 DHPR HA WT and pLentiNB CaV1.2 DHPR HA TS are described in Supplementary Methods.
Lenti virus
Lenti virus was prepared using the manufacturer’s protocol (Invitrogen) and as described in Supplementary Methods.
Ventricular myocyte isolation, culturing and viral transduction
Adult male Sprague-Dawley rat (250-300g) ventricular myocytes were isolated as previously published8 and cultured as described in Supplementary Methods. Procedures were in accordance with the Institutional Animal Care and Use Committee of the University of Iowa. Lenti virus was added to cells at a multiplicity of infection (MOI) of 1-3, and cultures were maintained for 24-36 hours.
Electrophysiology
Electrophysiology for HEK293 cells and myocytes is detailed in the Supplementary Methods including pipette solutions, bath solutions, voltage clamp protocols, current clamp protocols and data analysis.
Immunoflourescence
HEK293 and myocytes were fixed, permeabilized, incubated with primary antibody Ig and a fluorescent secondary antibody Ig. Confocal images were collect on a Zeiss 510 Meta confocal microscope (Carl Zeiss). Detailed information on cell preparation, antibodies and acquisition of confocal images is available in Supplementary Methods.
Calcium imaging
Ca2+ transients, SR content and sparks were acquired by confocal laser scanning from myocytes loaded with Flou-3. Supplementary methods contain details on cell preparation and confocal Ca2+ imaging.
Mathematical modeling
Mathematical models of the WT and TS myocytes are based on the Luo-Rudy dynamic model of the mammalian ventricular action potential9, 10. See Supplementary Methods for equations that differ from the published model.
Statistics
Data presented as means with SEM. Sigma Stat was used to compare two groups with a Student T-test and multiple groups with an ANOVA. Significance was set at a P value < 0.05. Categorical data between two groups was compared using a 2-tailed Fisher Exact Test with significance set at P<0.05.
The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
Results
An adult ventricular myocyte TS model
We marked exogenous CaV1.2 by the addition of an extracellular hemaglutanin (HA) epitope11 (Figure 1A, green circle) and introduced a validated dihydropyridine-insensitivity mutation 7 (Figure 1A, black circle). The dihydropyridine-insensitivity mutation allows the virally introduced CaV1.2 to remain functional while using nifedipine to inhibit endogenous CaV1.2 7. Exogenous CaV1.2 expression was confirmed by immunoblot (Figure 1B) and immunofluorescence (Figure 1C) in transduced HEK293T cells. The function of CaV1.2 wild type (WT) and TS (G406R exon 8) were confirmed by recording ICa using whole cell voltage clamp in HEK293T cells. ICa recorded from TS expressing HEK293T cells exhibited a significant loss of VDI (Figure 1D), as previously published1, 2, 12.
Figure 1.
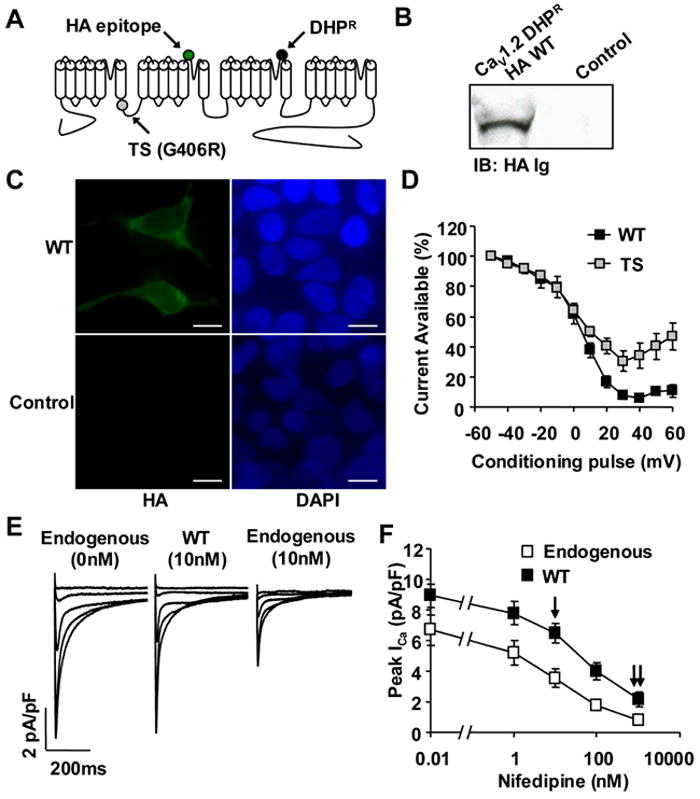
Dihydropyridine-resistant CaV1.2 α subunit Timothy Syndrome (TS) model. (A) A topology diagram of CaV1.2 depicting dihydropyridine resistance mutation (DHPR, black circle), extracellular hemaglutanin epitope (HA, green circle) and the TS mutation (G406R, gray circle) on the I-II intracellular loop. (B) Immunoblot (HA Ig) of HEK293T cells expressing the modified CaV1.2 or empty vector control. (C) FITC immunofluorescence (HA Ig) of HEk293T cells expressing the modified CaV1.2 with corresponding nuclear stain by DAPI (Scale bar, 10μm). (D) CaV1.2 TS expressing HEK293T cells show a reduction in VDI as compared to HEK293T cells transfected with CaV1.2 WT (N=5 cells/point). (E) Exogenous CaV1.2 with dihydropyridine mutation is resistant to nifedipine. (F) Preserved ICa during exposure to nifedipine. The single arrow indicates the nifedipine concentration (10nM) used to study the cellular consequences of the TS mutation, and the double arrow indicates the nifedipine concentration (1μM) to overcome dihydropyridine resistance and block the majority of ICa (N=5-8 cells/point, P<0.05 at each nifedipine concentration).
Over-expression of CaV1.2 in ventricular myocytes yielded 33.7% increase in peak ICa (Figure 1F) and an average 31.9% increase in total CaV1.2 protein (Supplemental Figure 1). Due to the dihydropyridine-resistance mutation7, peak ICa in CaV1.2 infected ventricular myocytes was significantly resistant to nifedipine, as compared to uninfected cells (Figure 1E,F). In TS and WT infected ventricular myocytes 10nM nifedipine resulted in a peak ICa (WT 6.6±0.7 pA/pF N=5, TS 6.9±0.7 pA/pF N=6) that was similar to the peak ICa (6.7±1.0 pA/pF N=8) measured in non-infected myocytes recorded without nifedipine (Figure 1E,F). This nifedipine engineered balance of endogenous and exogenous CaV1.2 allowed us to determine the effects of the TS mutation on cardiac electrophysiology independent of over-expression induced changes in peak ICa.
TS ventricular myocytes exhibit increased CaMKII autophosphorylation
We confirmed expression of exogenous CaV1.2 in cultured adult ventricular myocytes by immuno-staining for the HA epitope (Figure 2D,G). Virally introduced CaV1.2 was properly targeted to the transverse-tubule (T-tubule) network, based upon the punctate appearance and 1.8 μm spacing of the HA immunofluorescence that is consistent with known distances between T-tubules in a resting sarcomere13. No HA immuno-staining was detected in uninfected ventricular myocytes (Figure 2A).
Figure 2.
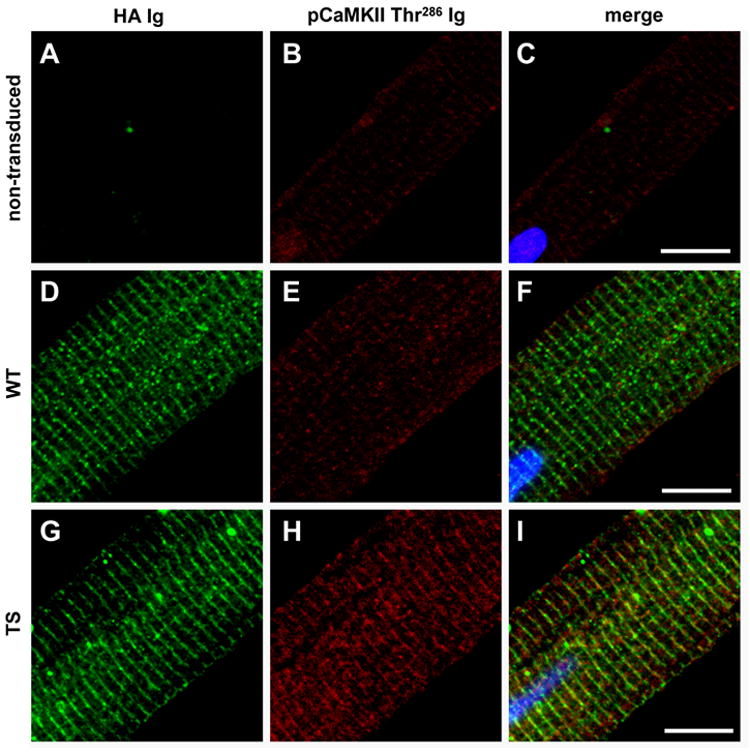
CaMKII recruitment in the TS adult ventricular myocyte model. (A-C) Non-transduced, (D-F) WT and (G-I) TS adult ventricular myocytes (field stimulated 1Hz for 5 minutes in Tyrodes with 1.8mM CaCl2). (A,D,G) Exogenous CaV1.2 channels are expressed in regularly distributed punctae across ventricular myocytes as shown by HA immunostaining. Both WT and TS CaV1.2 show spacing consistent with T-tubule network localization. HA immunofluorescence section of CaV1.2 WT, TS mutation, and uninfected negative control. (H) More activated CaMKII (pCaMKII Thr286) immuno-stained with (I) TS ventricular myocytes as compared to WT (E and F) and non-transduced (B and C) ventricular myocytes. (Scale bar, 10μm)
We immuno-stained for the CaMKII autophosphorylation site, Thr 286, which is a marker of CaMKII activation14. TS ventricular myocytes (Figure 2H,I) exhibited greater levels of CaMKII autophosphorylation compared to both WT (Figure 2E,F) and uninfected ventricular myocytes (Figure 2B,C). Total CaMKII immuno-staining revealed no changes in CaMKII protein levels between WT, TS and uninfected ventricular myocytes (Supplemental Figure 2). These data show that activated CaMKII is recruited in TS CaV1.2 expressing ventricular myocytes and suggest that CaMKII activity may contribute to the cellular arrhythmia phenotypes in TS.
Action potential prolongation in TS ventricular myocytes is reversed by CaMKII inhibition
Stimulated action potentials (arrow head, Figure 3A) were recorded in nifedipine treated (10nM) WT and TS ventricular myocytes. Compared to WT, the TS mutation significantly prolonged the action potential duration (Figure 3A,B) as determined by the time to 90% repolarization (APD90). Excessive action potential prolongation favors the generation of afterdepolarizations15. We observed afterdepolarizations from TS ventricular myocytes (5 out of 10 cells, Figure 3A,C), whereas none were observed in any of the WT cells (0 out of 10 cells, Figure 3A,C). Most afterdepolarizations were delayed afterdepolarizations (DADs), but early afterdepolarizations (EADs) were also recorded from TS ventricular myocytes. DADs are favored by increased diastolic Ca2+ leak from the sarcoplasmic reticulum (SR)16, 17 and EADs are caused by increased ICa facilitation 18. The action potential prolongation and the tendency for afterdepolarizations in TS ventricular myocytes are consistent with predictions from computational modeling1, 2.
Figure 3.
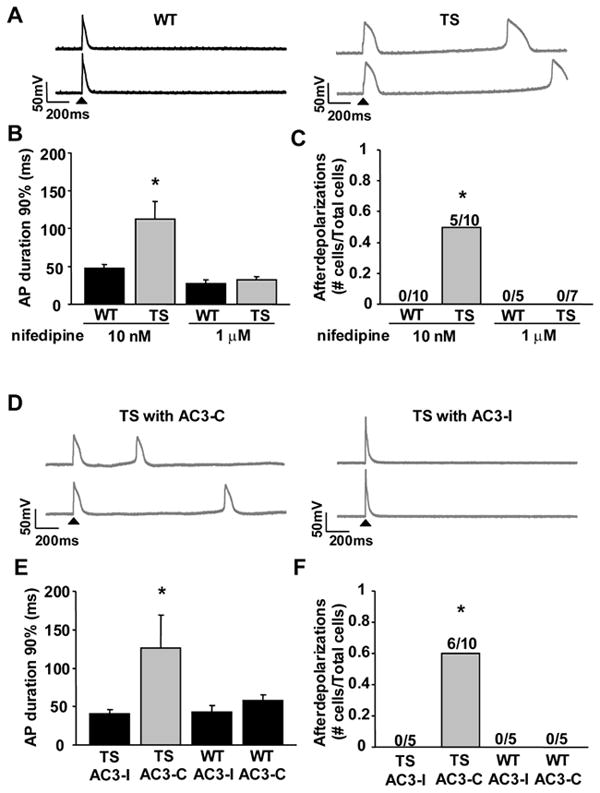
CaMKII inhibition reverses TS ventricular myocyte action potential (AP) prolongation and afterdepolarizations. (A) Action potential recordings from WT and TS ventricular myocytes. The first action potential for each sweep was initiated by injected current (arrow head), but the subsequent action potentials in TS arose from spontaneous afterdepolarizations. (B) CaV1.2 TS results in an increased action potential duration (N=5-10 cells/group, *P=0.018) and (C) afterdepolarizations (N=5-10 cells/group, *P=0.033). Numerals indicate the fraction of cells studied with afterdepolarizations. 1μm Nifedipine inhibited a majority of ICa and so prevented the CaV1.2 TS increase in (B) action potential duration (N=5-7 cells/group, P-0.39) and (C) frequency of afterdepolarizations (N=5-7 cells/group, P=1.0). (D) Action potential recordings from TS ventricular myocytes with either the CaMKII inhibitory peptide, AC3-I, or a control peptide, AC3-C. (E and F) Dialyzing AC3-I restored action potential duration in TS to WT levels and prevented afterdepolarizations (N=5-10 cells/group, TS with AC3-I compared to WT: APD90% P=0.403, afterdepolarizations P=1.0). AC3-I resulted in a non-significant shortening of the WT AP duration (N=5 cells/group, P=0.25) and no significant change in afterdepolarizations (N=5 cells/group, P=1.0) compared to WT with no peptide. (D-F) Dialyzing the control peptide, AC3-C, did not alter the TS mutation affects on action potential duration or afterdepolarizations (N=5-10 cells/group, TS with AC3-C compared to TS with AC3-I: APD90% *P=0.017, afterdepolarizations *P=0.044). AC3-C did not alter the AP duration (N=5 cells/group, P=0.28) or afterdepolarizations (N=5 cells/group, P=1.0) of WT with no peptide.
Action potential durations from WT and TS ventricular myocytes in 1μM nifedipine were reduced to equivalent times and neither WT nor TS ventricular myocytes exhibited afterdepolarizations under these conditions (Figure 3B,C). The 1μM nifedipine bath solution overcomes the dihydropyridine resistance mutation and inhibited the total peak ICa by >50% (Figure 1F, double arrows). These findings indicate that the observed TS phenotypes were initiated by increased ICa.
We tested the role of CaMKII activity in the observed proarrhythmic cellular phenotypes observed from TS ventricular myocytes by dialysis of AC3-I, a selective CaMKII inhibitory peptide 4, 19. AC3-I normalized the action potential duration in TS to WT levels (P=0.40, Figure 3D,E). The inactive control peptide, AC3-C4, 19, had no effect, suggesting that CaMKII-dependent increases in ICa contributed to action potential prolongation in TS. The CaMKII inhibitory peptide also eliminated afterdepolarizations in TS ventricular myocytes (P=1.0, Figure 3D,F), whereas AC3-C did not (P=0.04, Figure 3D,F). These data support the concept that CaMKII activity is required for the proarrhythmic electrophysiological phenotypes in TS ventricular myocytes.
In WT ventricular myocytes the CaMKII inhibitory peptide, AC3-I, resulted in a non-significant (P=0.28) shortening of the action potential duration (Figure 3E) compared to WT ventricular myocytes dialyzed with the control peptide, AC3-C. WT ventricular myocytes did not exhibit afterdepolarizations after dialysis with AC3-I or AC3-C (Figure 3F). We assessed additional action potential parameters, including resting cell membrane potential and peak cell membrane depolarization amplitude. Both TS and WT ventricular myocytes exhibited equivalent resting membrane potentials and peak action potential amplitudes (Supplemental Table 1). Action potential parameters from WT ventricular myocytes, in the presence of 10nM nifedipine, were similar to uninfected ventricular myocytes, cultured for the same time period (24-36 hours) and recorded without nifedipine (Supplemental Table 1). These controls suggest that viral expression of CaV1.2 does not alter the action potential when peak ICa is adjusted to normal levels (by 10nM nifedipine) and that the proarrhythmic phenotype observed in TS ventricular myocytes was due to the TS mutation.
Taken together, these findings are the first to demonstrate experimentally that the action potential phenotypes observed in TS ventricular myocytes were dependent upon increased Ca2+ entry through CaV1.2. Our findings suggest that the TS VDI defect is insufficient, in the absence of increased CaMKII activity, to cause significant action potential prolongation in ventricular myocytes.
TS reduces VDI in ventricular myocytes independent of CaMKII activity
Expression of TS CaV1.2 in Xenopus oocytes1, 2 and heterologous cells1, 2, 12 (Figure 1D) showed a loss of CaV1.2 VDI. The Xenopus ooctye experiments1, 2 included Ca2+ independent conditions that would not favor CaMKII activation because Ba2+ substituted Ca2+ as the charge carrier. To test the effect of the TS mutation on VDI in ventricular myocytes under conditions not permissive to CaMKII activation, we recorded ICa from TS and WT ventricular myocytes (10nM nifedipine) using Ba2+ (1.8mM) as the charge carrier and under high intracellular Ca2+ buffering (20mM BAPTA). TS ventricular myocytes exhibited a loss of VDI as a significant (p = 0.008, Figure 4A) rightward shift compared to WT. The TS V1/2 (-30.75 mV) shifted to more positive potentials compared to WT V1/2 (-35.89 mV). In contrast, the peak ICa elicited by the conditioning pulses showed no difference between WT and TS (Figure 4B), confirming equivalent expression of exogenous WT and TS CaV1.2. No differences were observed in peak ICa or VDI recorded from adult ventricular myocytes expressing WT dihydropyridine-resistant CaV1.2, with 10nM nifedipine, compared to uninfected adult ventricular myocytes, without nifedipine (Supplemental Table 2). These findings show that TS causes a loss of CaV1.2 VDI in ventricular myocytes, establishing the initial requirement for increased cellular Ca2+ entry necessary to recruit CaMKII.
Figure 4.
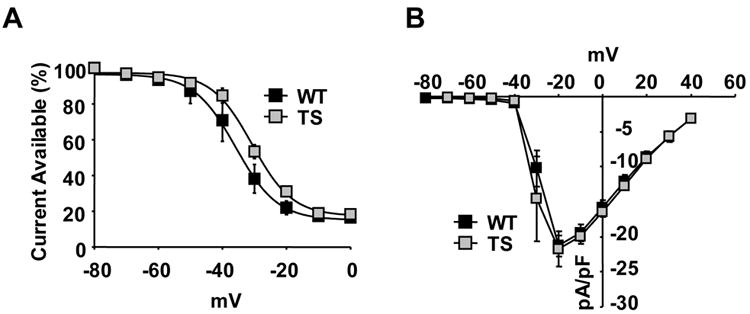
TS mutation shifts the VDI independent of Ca2+ signaling. (A) The TS mutation shifts the CaV1.2 IBa VDI (N=5 cells/point, *P=0.008), (B) without changing the current-voltage (IV) relationship (N=5 cells/group, P=0.88).
CaMKII is required for TS effects on ICa
To test the importance of CaMKII for additional ICa changes other than VDI in our TS model, we measured CaMKII dependent ICa facilitation 18, 20. ICa facilitation consists of dynamic increases in peak ICa and slowing of inactivation with repetitive depolarizations21, 22. TS ventricular myocytes exhibited maximal peak ICa during the first depolarization, whereas WT attained peak ICa after the initial depolarization (Figure 5A, Supplemental Figure 3). Subsequent depolarizations showed no difference in peak ICa between TS and WT (Figure 5A, Supplemental Figure 3). To measure the effects of ICa facilitation on cellular Ca2+ entry, we integrated total ICa during the voltage clamp command step. Integrated ICa was significantly greater in TS compared to WT during all depolarization steps (First step P=0.029, Remaining steps P<0.001, Figure 5B). We found the fast component of ICa inactivation (τfast) was slower in TS compared to WT (First step P=0.006, Remaining steps P<0.001, Figure 5C), consistent with increased ICa facilitation and augmented cellular Ca2+ entry in TS ventricular myocytes.
Figure 5.
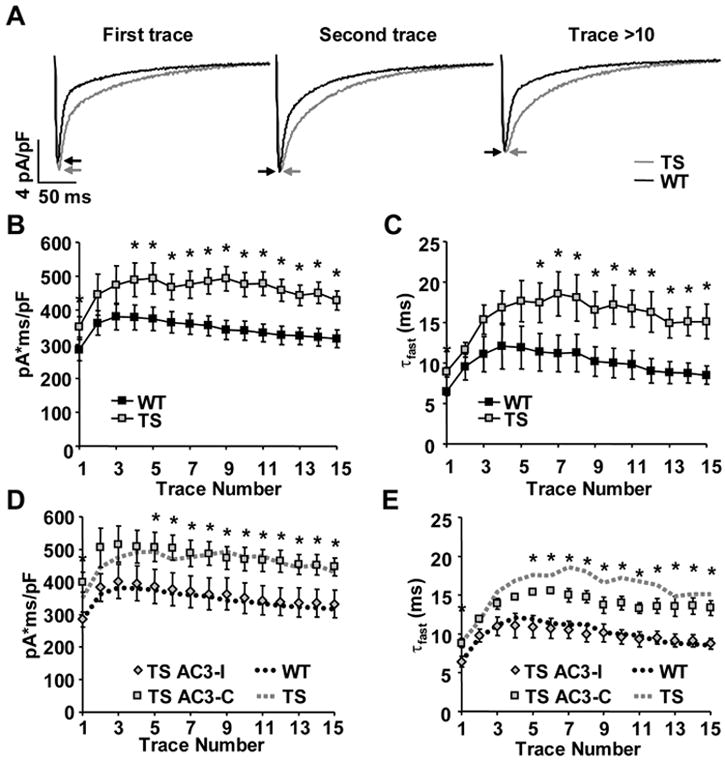
TS mutation enhances ICa facilitation. (A) TS ventricular myocytes exhibit increased peak ICa (arrows) during the first depolarizing voltage clamp command step (-80mV to 0mV, 300ms, 0.5Hz) and slowing of inactivation during all depolarizing steps. (B) Integrated ICa evoked by repetitive depolarizing voltage command steps (as in A above) is greater in TS mutation than WT (N=6-7 cells/point, ANOVA P<0.001, * P<0.05). (C) The time constant of the fast component of ICa inactivation (τfast) is significantly slower in TS ventricular myocytes than WT (N=6-7 cells/point, ANOVA P<0.001, * P<0.05). (D and E) Integrated ICa and τfast were restored to WT levels in TS ventricular myocytes dialyzed with the CaMKII inhibitory peptide, AC3-I (N=5-6 cells/point, TS with AC3-I compared to WT: integrated ICa ANOVA P=0.522, τfast ANOVA P=0.294). Dialyzing the control peptide, AC3-C, did not alter the TS mutation affects on ICa facilitation (N=5 cells/group, TS with AC3-C compared to TS with AC3-I: integrated ICa ANOVA P<0.001, * P<0.05; τfast ANOVA P<0.001, * P<0.05).
AC3-I restored the dynamic response characteristics of integrated ICa and τfast in TS to levels recorded from WT cells (integrated ICa P=0.522, τfast P=0.294, Figure 5E,F). In contrast, dialysis of AC3-C had no effect of τfast or integrated ICa. Dialysis of the CaMKII inhibitory peptide prevented ICa facilitation in WT ventricular myocytes (Supplemental Figure 3), whereas the control peptide had no effect on WT ventricular myocyte ICa facilitation. These measurements show that CaMKII is a significant determinant of ICa from TS mutant channels, and that CaMKII actions are distinct from the previously reported shift in VDI.
TS augments intracellular Ca2+
Mathematical modeling studies predicted alterations in intracellular Ca2+ handling in TS, including increased Ca2+ transient amplitude and increased SR Ca2+ content1. We recorded Ca2+ transients (Figure 6A) from WT and TS ventricular myocytes loaded with fluo-3 AM and field stimulated at 1Hz23. TS caused a significant increase in the peak Ca2+ transient compared to WT (P=0.04, Figure 6B), which is consistent with computer models1, 23, 24. Interestingly, the 50% decay time for Ca2+ transients in TS was significantly shortened over WT (P=0.02, Figure 6C). A faster decay time implicates increased SERCA activity25, which was not predicted by modeling studies, but is associated with CaMKII signaling26-29. These experimental data reveal that TS alters intracellular Ca2+ handling by increasing the peak Ca2+ transient amplitude and enhancing the decay of the intracellular Ca2+ transient.
Figure 6.
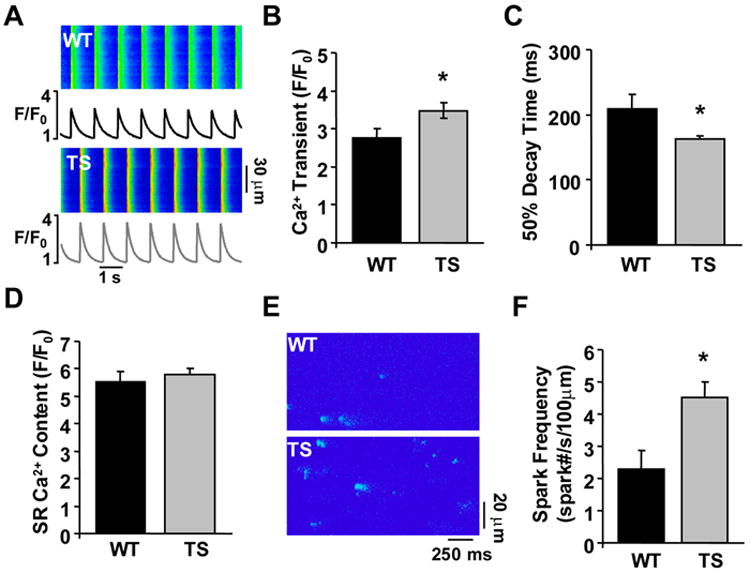
The TS mutation augments intracellular Ca2+ handling. (A) Confocal Ca2+ transient recordings from WT and TS ventricular myocytes. (B) Summary data showing TS mutation causes an increase in the peak Ca2+ transient during 1Hz stimulations (N=14-28 cells/group, *P=0.042). (C) Summary data showing the 50% decay time of the whole cell Ca2+ transients were faster in TS ventricular myocytes (N=14-28 cells/group, *P=0.047). (D) No difference was observed in SR Ca2+ content between TS and WT ventricular myocytes (N=14-28 cells/group, P=0.524). (E) Ca2+ sparks recorded from WT and TS ventricular myocytes. (F) Summary data showing TS infected ventricular myocytes exhibited an increased frequency of Ca2+ sparks during diastole (N=22-37 cells/group, *P=0.001).
Mathematical modeling also predicted increased SR Ca2+ content with TS, due to enhanced ICa from TS CaV1.2. Surprisingly, we found that TS SR Ca2+ content was not different than WT (P=0.55, Figure 6D). We considered that increased SR Ca2+ leak in TS balances faster SR Ca2+ uptake25, thereby preventing a net increase in SR Ca2+ content compared to WT. Increased SR Ca2+ leak is implicated in CaMKII signaling6, 30, 31 and in triggering DADs16, 17, a prominent feature of the TS ventricular myocytes (Figure 3A,C). We assessed diastolic SR Ca2+ leak by measuring spontaneous Ca2+ sparks from TS and WT ventricular myocytes (Figure 6E)32. The SR Ca2+ sparks were significantly increased in TS compared to WT (P=0.001, Figure 6E,F), indicating increased SR Ca2+ leak in TS. The spark amplitude for TS was significantly greater than WT (P=0.002, Supplemental Table 3), consistent with the increase in peak Ca2+ transient observed with TS. The effects of TS on intracellular Ca2+ handling had two unexpected results, first the faster decay time and second the increase in spark frequency. Taken together, these data suggest that SR Ca2+ cycling is enhanced in TS ventricular myocytes, resulting in significantly increased SR Ca2+ uptake and diastolic Ca2+ leak, but without a change in SR Ca2+ content.
In contrast to TS, WT exhibited no difference in the Ca2+ transient peak amplitude or decay time as compared to uninfected ventricular myocytes (Supplemental Table 3). No significant changes were observed in the width or duration of the Ca2+ sparks (Supplemental Table 3) between WT, TS and uninfected cells. The spark frequency and profile of individual sparks showed no difference between WT and uninfected ventricular myocytes (Supplemental Table 3)33.
Revised TS mathematical modeling
Several studies have modeled the impact of TS on myocardial electrophysiology by using a shift in CaV1.2 VDI estimated from measurements in non-myocytes1, 2, 24. Using data from our TS ventricular myocyte model, we developed a new mathematical model of TS incorporating CaMKII signaling (Figure 7A). As the basis for our new model of TS, we used the Luo-Rudy dynamic model (LRd), 9, 10 because of its established utility in studying cardiac arrhythmia mechanisms.
Figure 7.
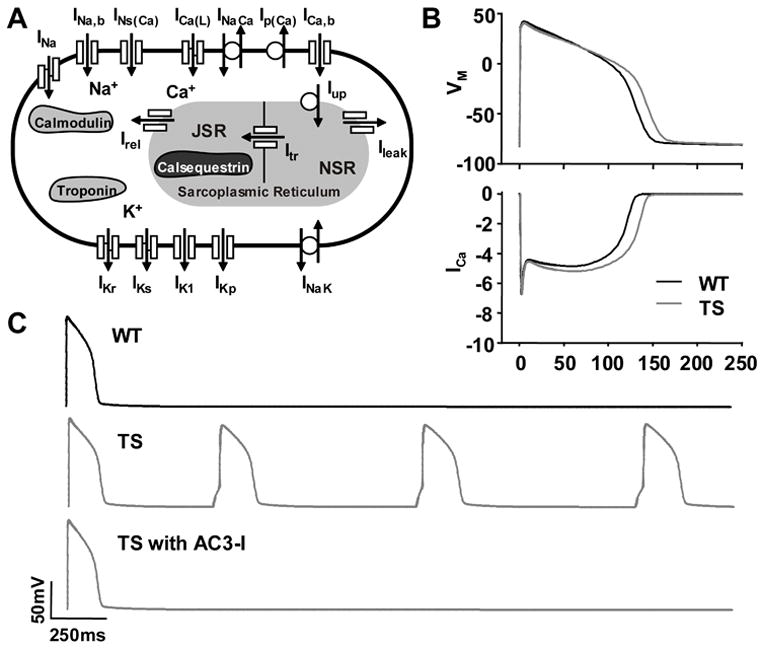
LRd modeling of WT, TS and TS with AC3-I based upon experimental data from ventricular myocytes. (A) Schematic of LRd model. (B) LRd model indicates CaMKII activation in TS causes increased ICa and action potential prolongation (CL = 700ms) and (C) afterdepolarizations.
Our model of TS incorporated three modifications to match our experimental observations. First, we shifted the CaV1.2 steady-state VDI in the LRd model to simulate the measured TS defect on channel gating. Second, we simulated the downstream CaMKII effect on CaV1.2 ICa facilitation associated with TS by slowing ICa inactivation to increase integrated ICa as measured experimentally (Supplemental Figure 4). Third, we simulated the CaMKII actions on intracellular Ca2+ handling associated with TS by increasing the mean open time of the ryanodine receptor SR Ca2+ release channels, decreasing the threshold for spontaneous SR Ca2+ release and increasing SR Ca2+ release. Consistent with our experimental measurements, the new model of TS predicted an increase in the intracellular Ca2+ transient amplitude without any change in SR Ca2+ load compared to WT (Supplemental Figure 4). The model also predicted an increase in action potential duration (Figure 7B) and afterdepolarizations (Figure 7C) during a pause after pacing. We also simulated CaMKII inhibition using the TS LRd model by reversing the simulated downstream CaMKII effects, but leaving in place the shift in CaV1.2 VDI we measured under conditions not permissive for CaMKII activity (Figure 4A). The resulting TS LRd model with ‘CaMKII inhibition’ prevented action potential prolongation and afterdepolarizations (Figure 7C). Our mathematical models of TS, with and without CaMKII inhibition, are consistent with our experimental data from our TS ventricular myocyte model.
Discussion
TS is the first arrhythmia syndrome (LQT8) due to a genetic mutation in the CaV1.2 pore-forming α subunit.1, 2 In comparison to cardiac Na+ and K+ channels, CaV1.2 has proven to be remarkably resistant to genetic disease. One key difference between Ca2+, Na+ and K+ is the prominent role Ca2+ plays as a second messenger. TS patients not only have extremely profound QT interval prolongation, but also structural cardiac abnormalities, which are not typical of Na+ or K+ channel gene-related long QT syndrome patients. QT interval prolongation reflects increased duration of the ventricular action potential. The action potential duration prolongation in TS was attributed entirely to the defect in VDI1, but this defect in TS VDI was ascertained in heterologous (non-myocardial) cells, where action potentials could not be directly measured. Furthermore, heterologous cells lack the highly ordered ultrastructure that is present in ventricular myocytes, for Ca2+ homeostasis and excitation-contraction coupling. The ventricular myocyte TS model allowed us to measure electrophysiological, intracellular Ca2+ handling and Ca2+ mediated signaling changes that occur downstream to the loss of VDI.
Despite the relatively modest reduction in CaV1.2 VDI measured in our TS model, we found action potential prolongation and spontaneous afterdepolarizations that were due to secondary activation of CaMKII. We conclude that the shift in VDI provides the initial stimulus to trigger intracellular Ca2+ signaling that includes CaMKII activation. Increased CaMKII activity appears to be necessary for the cellular phenotype of prolonged action potentials and afterdepolarizations, in so far as CaMKII inhibition prevents these phenotypes. CaMKII inhibition may be a viable alternative therapeutic approach for TS patients treated with the ICa antagonist verapamil34. Our results showed that CaMKII amplifies Ca2+ entry through CaV1.2 in TS, by slowing τfast, and shifting the V1/2 of ICa inactivation. Our studies do not exclude the possibility that CaMKII inhibition could also affect other depolarizing or repolarizing currents, such as Na+ current35 or K+ current36. Our findings that SR Ca2+ leak is increased in TS is consistent with other reports that show proarrhythmic actions of CaMKII are due to increasing SR Ca2+ leak37, thereby enabling a transient inward current16 (INCX) that triggers DADs. Thus, our data support the concept that the ryanodine receptor is a secondary proarrhythmic target for excessive CaMKII activity in TS. Our data highlight how small changes in cellular Ca2+ entry through CaV1.2 can lead to unanticipated, maladaptive and far-reaching changes in Ca2+ activated signaling.
Interestingly a connection between CaMKII and a TS mutation was suggested based upon single channel recordings from heterologous expression of TS CaV1.2 in baby hamster kidney 6 cells38. These experiments found that TS CaV1.2 were more likely than WT to exhibit frequent, long openings, so called mode 2 gating that are the single channel mechanism underlying CaMKII-mediated ICa facilitation20. Our new studies add to evidence supporting a connection between TS and CaMKII by showing that CaMKII is critical for increased ICa facilitation action potential prolongation and afterdepolarizations in our TS ventricular myocyte model. Enhanced CaMKII activity increases ICa facilitation,17 which may cause generation of EADs5.
Although major Ca2+ homeostatic proteins are conserved in ventricular myocytes across mammalian species, differences exist between species regarding the quantitative contribution of these components to the action potential 39. Thus, one goal of future studies should be to determine if CaMKII, or other Ca2+-activated signaling molecules, contribute to TS phenotypes in ventricular myocytes from other species. However, the use of our TS adult ventricular myocyte model has contributed new insights about arrhythmia mechanisms in TS, by illustrating how a concise defect in CaV1.2 gating can initiate downstream recruitment of CaMKII that ultimately enables the electrophysiological cellular disease phenotype in TS. The CNS defects of TS patients may also be due to secondary recruitment of Ca2+ activated signaling molecules, including CaMKII. Over-expression of CaMKII is known to interfere with neuronal growth and differentiation40 and a constitutively active CaMKII within the mouse brain causes significantly impaired spatial memory41. CaMKII recruitment in TS ventricular myocytes also suggests the possibility that other disease phenotypes in TS patients (e.g. structural heart disease or mental retardation), may be initiated by defects in VDI but carried forward, indirectly, by recruitment of Ca2+-dependent signaling molecules.
Supplementary Material
Clinical Perspective.
Timothy Syndrome (TS) is a genetic disorder causing excessive cellular Ca2+ entry due to defective voltage dependent inactivation (VDI) of the predominant myocardial L-type Ca2+ channel (CaV1.2) current (ICa). Timothy Syndrome patients die on average at 2.5 years, due to malignant cardiac arrhythmias. TS is a ‘model’ disease whereby a concise biophysical defect in ICa activates a cellular signaling cascade that is required for the cardiac disease phenotypes. Our studies showed that loss of VDI leads to cellular arrhythmias by recruiting activity of the calmodulin dependent protein kinase II (CaMKII). The role of CaMKII was not anticipated by previous computer models that relied on data obtained from non-excitable, heterologous cells. Our studies show that the TS VDI defect activated CaMKII and CaMKII was the feed forward signal required for the proarrhythmic cellular phenotypes in TS. These findings have potentially broad implications for other pathological phenotypes in Timothy Syndrome, such as autism, and for other genetic diseases affecting Ca2+ channels, such as migraine headache, myasthenia, ataxia and malignant hyperthermia. Diseases associated with excitable cells, where ion channels frequently constitute a final common pathway, require careful consideration of the connections between ion channel gating and signaling cascades in order to more comprehensively understand underlying mechanisms.
Acknowledgments
We thank the University of Iowa Gene Transfer Vector Core (an NIH-funded resource) for their help in preparing the lenti virus, in particular Maria Scheel and Dalyz Ochoa for their assistance and expertise.
Funding Sources Work funded by NIH R01 HL 079031, R01 HL 62494, and R01 HL 70250 (MEA); NIH R01 HL084583, R01 HL083422, and Pew Scholars Trust (PJM); the University of Iowa Cardiovascular Center Interdisciplinary Research Fellowship (TH) and the University of Iowa Research Foundation. William Thiel was supported in part by an American Heart Association pre-doctoral fellowship award.
Footnotes
Conflict of Interest Disclosures none
Contributor Information
William H. Thiel, Vanderbilt University, University of Iowa.
Biyi Chen, University of Iowa.
Thomas J. Hund, University of Iowa.
Olha M. Koval, University of Iowa.
Anil Purohit, University of Iowa.
Long-Sheng Song, University of Iowa.
Peter J. Mohler, University of Iowa.
Mark E. Anderson, University of Iowa.
Reference List
- 1.Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, Sanguinetti MC, Keating MT. Inaugural Article: Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. PNAS. 2005;102:8089–96. doi: 10.1073/pnas.0502506102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. CaV1.2 Calcium Channel Dysfunction Causes a Multisystem Disorder Including Arrhythmia and Autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 3.Wu Y, Kimbrough JT, Colbran RJ, Anderson ME. Calmodulin kinase is functionally targeted to the action potential plateau for regulation of L-type Ca2+ current in rabbit cardiomyocytes. J Physiol. 2004;554:145–55. doi: 10.1113/jphysiol.2003.053314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409–17. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 5.Wu Y, Temple J, Zhang R, Dzhura I, Zhang W, Trimble R, Roden DM, Passier R, Olson EN, Colbran RJ, Anderson ME. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 2002;106:1288–93. doi: 10.1161/01.cir.0000027583.73268.e7. [DOI] [PubMed] [Google Scholar]
- 6.Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH, Bers DM. Transgenic CaMKII{delta}C Overexpression Uniquely Alters Cardiac Myocyte Ca2+ Handling: Reduced SR Ca2+ Load and Activated SR Ca2+ Release. Circ Res. 2003;92:904–11. doi: 10.1161/01.RES.0000069685.20258.F1. [DOI] [PubMed] [Google Scholar]
- 7.Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294:333–9. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- 8.Grueter CE, Abiria SA, Dzhura I, Wu Y, Ham AJ, Mohler PJ, Anderson ME, Colbran RJ. L-type Ca2+ channel facilitation mediated by phosphorylation of the beta subunit by CaMKII. Mol Cell. 2006;23:641–50. doi: 10.1016/j.molcel.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 9.Faber GM, Rudy Y. Action Potential and Contractility Changes in [Na+]i Overloaded Cardiac Myocytes: A Simulation Study. Biophys J. 2000;78:2392–404. doi: 10.1016/S0006-3495(00)76783-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ Res. 1994;74:1071–96. doi: 10.1161/01.res.74.6.1071. [DOI] [PubMed] [Google Scholar]
- 11.Altier C, Dubel SJ, Barrere C, Jarvis SE, Stotz SC, Spaetgens RL, Scott JD, Cornet V, De Waard M, Zamponi GW, Nargeot J, Bourinet E. Trafficking of L-type calcium channels mediated by the postsynaptic scaffolding protein AKAP79. J Biol Chem. 2002;277:33598–603. doi: 10.1074/jbc.M202476200. [DOI] [PubMed] [Google Scholar]
- 12.Barrett CF, Tsien RW. The Timothy syndrome mutation differentially affects voltage- and calcium-dependent inactivation of CaV1.2 L-type calcium channels. PNAS. 2008;105:2157–62. doi: 10.1073/pnas.0710501105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 14.Lou LL, Lloyd SJ, Schulman H. Activation of the multifunctional Ca2+/calmodulin-dependent protein kinase by autophosphorylation: ATP modulates production of an autonomous enzyme. Proc Natl Acad Sci U S A. 1986;83:9497–501. doi: 10.1073/pnas.83.24.9497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roden DM, Lazzara R, Rosen M, Schwartz PJ, Towbin J, Vincent GM. Multiple Mechanisms in the Long-QT Syndrome: Current Knowledge, Gaps, and Future Directions. Circulation. 1996;94:1996–2012. doi: 10.1161/01.cir.94.8.1996. [DOI] [PubMed] [Google Scholar]
- 16.Wu Y, Roden DM, Anderson ME. Calmodulin kinase inhibition prevents development of the arrhythmogenic transient inward current. Circ Res. 1999;84:906–12. doi: 10.1161/01.res.84.8.906. [DOI] [PubMed] [Google Scholar]
- 17.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/Calmodulin-Dependent Protein Kinase Modulates Cardiac Ryanodine Receptor Phosphorylation and Sarcoplasmic Reticulum Ca2+ Leak in Heart Failure. Circ Res. 2005;97:1314–22. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 18.Wu Y, MacMillan LB, McNeill RB, Colbran RJ, Anderson ME. CaM kinase augments cardiac L-type Ca2+ current: a cellular mechanism for long Q-T arrhythmias. Am J Physiol. 1999;276:H2168–H2178. doi: 10.1152/ajpheart.1999.276.6.H2168. [DOI] [PubMed] [Google Scholar]
- 19.Wu Y, Colbran RJ, Anderson ME. Calmodulin kinase is a molecular switch for cardiac excitation-contraction coupling. Proc Natl Acad Sci U S A. 2001;98:2877–81. doi: 10.1073/pnas.051449198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dzhura I, Wu Y, Colbran RJ, Balser JR, Anderson ME. Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat Cell Biol. 2000;2:173–7. doi: 10.1038/35004052. [DOI] [PubMed] [Google Scholar]
- 21.Yuan W, Bers DM. Ca-dependent facilitation of cardiac Ca current is due to Ca-calmodulin-dependent protein kinase. Am J Physiol. 1994;267:H982–H993. doi: 10.1152/ajpheart.1994.267.3.H982. [DOI] [PubMed] [Google Scholar]
- 22.Anderson ME, Braun AP, Schulman H, Premack BA. Multifunctional Ca2+/calmodulin-dependent protein kinase mediates Ca(2+)-induced enhancement of the L-type Ca2+ current in rabbit ventricular myocytes. Circ Res. 1994;75:854–61. doi: 10.1161/01.res.75.5.854. [DOI] [PubMed] [Google Scholar]
- 23.Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H. Orphaned ryanodine receptors in the failing heart. Proceedings of the National Academy of Sciences. 2006;103:4305–10. doi: 10.1073/pnas.0509324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faber GM, Silva J, Livshitz L, Rudy Y. Kinetic Properties of the Cardiac L-Type Ca2+ Channel and Its Role in Myocyte Electrophysiology: A Theoretical Investigation. Biophys J. 2007;92:1522–43. doi: 10.1529/biophysj.106.088807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. 2. Dordrecht, the Netherlands: Kluwer Academic Publishers; 2001. [Google Scholar]
- 26.Bassani RA, Mattiazzi A, Bers DM. CaMKII is responsible for activity-dependent acceleration of relaxation in rat ventricular myocytes. Am J Physiol. 1995;268:H703–H712. doi: 10.1152/ajpheart.1995.268.2.H703. [DOI] [PubMed] [Google Scholar]
- 27.DeSantiago J, Maier LS, Bers DM. Frequency-dependent acceleration of relaxation in the heart depends on CaMKII, but not phospholamban. J Mol Cell Cardiol. 2002;34:975–84. doi: 10.1006/jmcc.2002.2034. [DOI] [PubMed] [Google Scholar]
- 28.Picht E, DeSantiago J, Huke S, Kaetzel MA, Dedman JR, Bers DM. CaMKII inhibition targeted to the sarcoplasmic reticulum inhibits frequency-dependent acceleration of relaxation and Ca2+ current facilitation. J Mol Cell Cardiol. 2007;42:196–205. doi: 10.1016/j.yjmcc.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hagemann D, Kuschel M, Kuramochi T, Zhu W, Cheng H, Xiao RP. Frequency-encoding Thr17 phospholamban phosphorylation is independent of Ser16 phosphorylation in cardiac myocytes. J Biol Chem. 2000;275:22532–6. doi: 10.1074/jbc.C000253200. [DOI] [PubMed] [Google Scholar]
- 30.Guo T, Zhang T, Mestril R, Bers DM. Ca2+/Calmodulin-Dependent Protein Kinase II Phosphorylation of Ryanodine Receptor Does Affect Calcium Sparks in Mouse Ventricular Myocytes. Circ Res. 2006;99:398–406. doi: 10.1161/01.RES.0000236756.06252.13. [DOI] [PubMed] [Google Scholar]
- 31.Wehrens XHT, Lehnart SE, Reiken SR, Marks AR. Ca2+/Calmodulin-Dependent Protein Kinase II Phosphorylation Regulates the Cardiac Ryanodine Receptor. Circ Res. 2004;94:e61–e70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- 32.Wang SQ, Wei C, Zhao G, Brochet DXP, Shen J, Song LS, Wang W, Yang D, Cheng H. Imaging Microdomain Ca2+ in Muscle Cells. Circ Res. 2004;94:1011–22. doi: 10.1161/01.RES.0000125883.68447.A1. [DOI] [PubMed] [Google Scholar]
- 33.Song LS, Guia A, Muth JN, Rubio M, Wang SQ, Xiao RP, Josephson IR, Lakatta EG, Schwartz A, Cheng H. Ca(2+) signaling in cardiac myocytes overexpressing the alpha(1) subunit of L-type Ca(2+) channel. Circ Res. 2002;90:174–81. doi: 10.1161/hh0202.103230. [DOI] [PubMed] [Google Scholar]
- 34.Jacobs A, Knight BP, McDonald KT, Burke MC. Verapamil decreases ventricular tachyarrhythmias in a patient with Timothy syndrome (LQT8) Heart Rhythm. 2006:967–70. doi: 10.1016/j.hrthm.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 35.Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006;116:3127–38. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li J, Marionneau C, Zhang R, Shah V, Hell JW, Nerbonne JM, Anderson ME. Calmodulin Kinase II Inhibition Shortens Action Potential Duration by Upregulation of K+ Currents. Circ Res. 2006;99:1092–9. doi: 10.1161/01.RES.0000249369.71709.5c. [DOI] [PubMed] [Google Scholar]
- 37.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/Calmodulin-Dependent Protein Kinase Modulates Cardiac Ryanodine Receptor Phosphorylation and Sarcoplasmic Reticulum Ca2+ Leak in Heart Failure. Circ Res. 2005;97:1314–22. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 38.Erxleben C, Liao Y, Gentile S, Chin D, Gomez-Alegria C, Mori Y, Birnbaumer L, Armstrong DL. Cyclosporin and Timothy syndrome increase mode 2 gating of CaV1.2 calcium channels through aberrant phosphorylation of S6 helices. PNAS. 2006;103:3932–7. doi: 10.1073/pnas.0511322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bassani JW, Bassani RA, Bers DM. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J Physiol. 1994;476:279–93. doi: 10.1113/jphysiol.1994.sp020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Masse T, Kelly PT. Overexpression of Ca2+/Calmodulin-Dependent Protein Kinase II in PC12 Cells Alters Cell Growth, Morphology, and Nerve Growth Factor-Induced Differentiation. J Neurosci. 1997;17:924–31. doi: 10.1523/JNEUROSCI.17-03-00924.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bach ME, Hawkins RD, Osman M, Kandel ER, Mayford M. Impairment of spatial but not contextual memory in CaMKII mutant mice with a selective loss of hippocampal LTP in the range of the theta frequency. Cell. 1995;81:905–15. doi: 10.1016/0092-8674(95)90010-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.