

Abstract
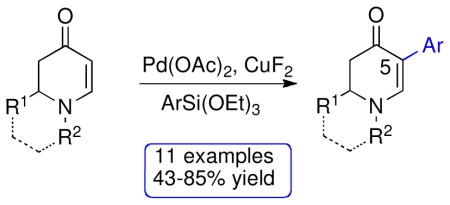
2,3-Dihydropyridin-4(1H)-ones undergo direct C–H functionalization at C5 in the palladium(II)-catalyzed Hiyama reaction, using triethoxy(aryl)silanes and dimethylphenylsilanol. The reagent CuF2 has a dual role in the reactions with triethoxy(aryl)silanes. It is a source of fluoride to activate the silane in the Hiyama reaction and also serves as the reoxidant to convert Pd(0) to Pd(II) in the catalytic cycle.
Cross-coupling reactions that directly convert C–H to C–C bonds are valuable atom efficient chemistry processes when compared with conventional cross coupling reactions.1, 2 A transition metal-catalyzed C–H functionalization obviates the need for a preactivation step to set up the substrate for the cross-coupling reaction. As a C–C bond-forming strategy, direct arylation of C–H bonds has attracted considerable attention.3 However, the majority of these reactions have been performed employing aromatic C–H bonds and often involve a directing group. 4 We have recently shown that enaminones (2,3-dihydropyridin-4(1H)-ones) can be substrates for direct C–H functionalization in Pd(II)-catalyzed Suzuki-type reactions.5 We have proposed that the innate nucleophilicity of C5 of enaminones allows for the direct reaction with electrophilic Pd(II), which is followed by deprotonation of the palladium intermediate, transmetalation, and reductive elimination (Figure 1). 6 The catalytic cycle is completed by reoxidation of Pd(0) to Pd(II) with an appropriate oxidant such as Cu(OAc)2.
Figure 1.

Proposed catalytic cycle for the direct arylation of enaminones 1.
We have recently demonstrated the utility of this chemistry for the concise enantiospecific synthesis of the phenanthropiperidine alkaloids ipalbidine and antofine,7 tylocrebrine,8 and boehmeriasin A.9
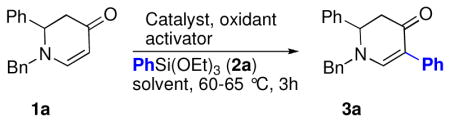
Based on our results with the Suzuki-type reactions, we hypothesized that the palladated enaminone intermediate could participate in other cross-coupling reactions, such as the Hiyama coupling. Hiyama reactions typically take place between organohalides (alkyl, aryl, and alkenyl) and organosilanes. 10 Organosilanes have a number of advantages that make them desirable organic reagents for cross-coupling reactions in comparison to organoboranes or organostannanes. They are stable, relatively non-toxic, and can be easily prepared. Nonetheless, only a few cases of direct C-H arylation with organosilane coupling partners have been reported. Yang et al.11 described the direct Hiyama ortho arylation of acetanilides and Zhou et al.4k the reaction of cyclic N-acetyl enamides with arylsilanes. In both cases, the palladation is believed to be dependent upon neighboring group assistance. In order to explore whether enaminones could participate directly in the Hiyama reaction, we selected 1-benzyl-2-phenyl-2,3-dihydropyridin-4(1H)-one (1a) as the substrate, Pd(OAc)2 as the catalyst, Cu(OAc)2 as the reoxidant, and TBAF to activate the triethoxy(phenyl)silane (2a) (Table 1, entry 1). The initial result was quite discouraging because only a trace amount of the desired reaction product 3a could be identified, accompanied by the biphenyl homocoupled product. Similar results were obtained when potassium fluoride was employed (Table 1, entry 2). Silver(I) fluoride appeared to be a better activator, compared to the more commonly used TBAF and KF, possibly due to the oxidative effect of silver(I) (Table 1, entry 3).
Table 1.
Reaction Optimization for the Hiyama Coupling Reaction of Enaminone 1a
 | ||||
|---|---|---|---|---|
| entrya | catalyst | oxidant/activator | solvent | yield (%)b |
| 1 | Pd(OAc)2 | Cu(OAc)2/TBAF | THF | 0c trace |
| 2 | Pd(OAc)2 | Cu(OAc)2/KF | dioxane | trace |
| 3 | Pd(OAc)2 | Cu(OAc)2/AgF | dioxane | 30% |
| 4 | Pd(OAc)2 | Cu(OTf)2/AgF | dioxane | 0d |
| 5e | Pd(OAc)2 | AgF2 | dioxane | 50 62f |
| 6 | White catalystg | AgF2 | dioxane | 40 |
| 7 | Pd(TFA)2 | AgF2 | dioxane | 48 |
| 8 | PdCl2 | AgF2 | dioxane | trace |
| 9 | PdI2 | AgF2 | dioxane | 0 |
| 10 | NiBr2 | AgF2 | dioxane | 0 |
| 11 | NiF2 | AgF2 | dioxane | trace |
| 12 | Pd(OAc)2 | CuF2 | dioxaneh tBuOH/AcOH (4:1)i |
0 82 |
| 13j | Pd(OAc)2 | Cu(OAc)2/TBAF | DMF | 80 |
| 14j | Pd(OAc)2 | Cu(OAc)2/KOTMS | DME | 77 |
Reaction conditions unless otherwise specified: enaminone 1a (0.1 M), siloxane 2a (2 equiv), catalyst (0.3 equiv), oxidant (2 equiv), activator (2 equiv), 3h.
Isolated yield.
PhSiMeCl2 was used.
Enaminone 1a decomposed.
A typical reaction was completed within 1 h when AgF2 was used.
Siloxane 2a (3.5 equiv).
1,2-Bis(phenylsulfinyl)-ethane palladium(II) acetate.
Enaminone 1a was recovered.
Pd(OAc)2 (0.25 equiv), CuF2 (2.5 equiv), siloxane 2a (2.5 equiv), 65 °C, 3h.
PhSiMe2OH (2b) was used instead of 2a.
Since copper and silver are both in group 11 in the periodic table, we next explored Ag(II) in this reaction because it is expected to possess chemical properties similar to Cu(II). We selected AgF2, which contains two equivalents of fluoride and is also an oxidant to test the proposition that AgF2 could not only provide the necessary fluoride atoms for the activation of the silane, but also function as the reoxidant in the palladium catalytic cycle. Gratifyingly, the Hiyama coupling reaction proceeded smoothly and with a satisfactory yield (Table 1, entry 5). When different catalysts were examined in this reaction, Pd(TFA)2 exhibited comparable activity while other Pd(II) or Ni(II) salts showed inferior catalytic capabilities (Table 1, entries 6–11). The reaction with AgF2, however, required the use of a large excess of organosilane. Therefore, we decided to replace AgF2 with CuF2. An initial attempt with CuF2, using dioxane as the solvent, did not provide the reaction product (Table 1, entry 12). However, when the solvent was changed to a 4:1 mixture of tert-butyl alcohol and acetic acid, the yield of the Hiyama coupling product increased to 82% (Table 1, entry 12). We also examined dimethylphenylsilanol (2b), an organosilanol introduced by Denmark, as a potential coupling partner. 12 Both fluoride-containing (Table 1, entry 13) and fluoride-free (Table 1, entry 14) conditions gave excellent results with this reactant. The clean, complete conversion clearly demonstrated the utility of these reagents as alternative coupling partners. To the best of our knowledge, this is the first case where an organosilanol has been used in a direct C–H cross-coupling reaction.
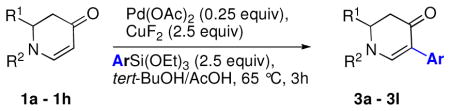
The optimized reaction conditions from Table 1, entry 12 were next used to examine the scope of the reaction with different enaminones and substituted triethoxy(aryl)silanes (Table 2). The Hiyama coupling reactions of enaminone 1a worked well with substituted triethoxy(phenyl)silanes that carry a 4-methyl group (Table 2, entry 1), the electron-withdrawing 4-trifluoromethyl group (Table 2, entry 2), and the electron-donating 4-methoxy group (Table 2, entry 3). The sterically hindered α-naphthyltrimethoxysilane was also a suitable coupling partner (Table 2, entry 6).
Table 2.
Substrate Scope of Hiyama Coupling Reactions
 | |||
|---|---|---|---|
| entrya | enaminone | product | yield (%)b |
| 1 |
 1a |
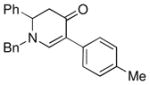 3b |
85 |
| 2 |
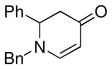 1a |
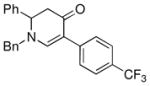 3c |
73 |
| 3 |
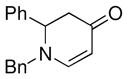 1a |
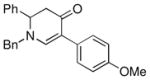 3d |
50 |
| 4 |
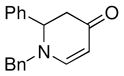 1a |
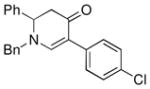 3e |
65 |
| 5 |
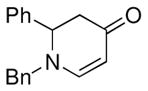 1a |
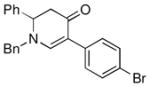 3f |
61 |
| 6c |
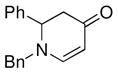 1a |
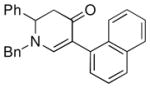 3g |
43 |
| 7 |
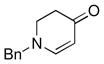 1b |
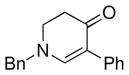 3h |
62 |
| 8 |
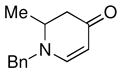 1c |
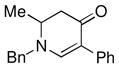 3i |
81 |
| 9 |
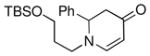 1d |
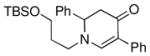 3j |
75 |
| 10 |
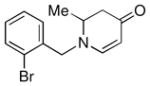 1e |
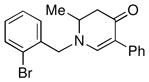 3k |
68 |
| 11 |
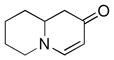 1f |
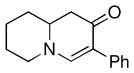 3l |
72 |
| 12d |
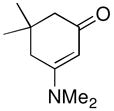 1g |
not observed | 0 |
| 13d |
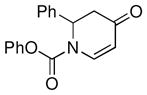 1h |
not observed | 0 |
Reaction conditions: enaminone (0.1 M), Pd(OAc)2 (0.25 equiv), CuF2 (2.5 equiv), ArSi(OEt)3 (2.5 equiv) in tBuOH/AcOH (4:1), 65 °C, 3h.
Isolated yield.
α-Naphthyltrimethoxysilane was used.
Starting material recovered.
Both monocyclic and bicyclic enaminones were sufficiently active in the direct coupling reactions (Table 2, entries 7, 8 and 11). TBAF, often used in Hiyama coupling reactions to activate the organosilicon reagent, also deprotects silyl protecting groups and thus presents a limitation of this method. It is of note that the TBS group was stable in the presence of CuF2 (Table 2, entry 9). We observed that halides on both the enaminone and organosiloxane remained intact, presenting positions for further derivatization (Table 2, entries 4, 5 and 10). Steric (Table 2, entry 12) or electronic factors (Table 2, entry 13) are presumably responsible for the lack of reactivity of enaminones 1g and 1h.
In summary, effective protocols for the Hiyama coupling reaction with enaminones have been developed. In the case of triethoxy(aryl)silanes as the coupling partner, AgF2 and CuF2 were employed for the first time as bifunctional activator/re-oxidants. Dimethylpheny-lsilanol was successfully used as well, without fluoride activation. All reactions took place in open air. To the best of our knowledge, this is the first direct Hiyama coupling reaction to take place without neighboring group assistance and with an organosilanol. The reaction conditions are compatible with the TBS protecting groups, and thus, could find utility in multi-step organic syntheses.
Supplementary Material
Acknowledgments
This work was supported by NIH grant GM081267 and by the University of Minnesota through the Vince and McKnight Endowed Chairs. The authors wish to thank Dr. Haibo Ge for some initial efforts with this chemistry and Yiyun Yu for the resynthesis of two compounds.
Footnotes
Supporting Information Available: Representative experimental procedures, characterization data and spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Godula K, Sames D. Science. 2006;312:67. doi: 10.1126/science.1114731. [DOI] [PubMed] [Google Scholar]; (b) Ellman JA. Science. 2007;316:1131. doi: 10.1126/science.1143373. [DOI] [PubMed] [Google Scholar]
- 2.For recent reviews, see: Dyker G, Handb C-H. Transform. 2005;2:465.Alberico D, Scott ME, Lautens M. Chem Rev. 2007;107:465. doi: 10.1021/cr0509760.Daugulis O, Do HQ, Shabashov D. Acc Chem Res. 2009;42:1074. doi: 10.1021/ar9000058.Chen X, Engle Keary M, Wang D-H, Yu J-Q. Angew Chem, Int Ed. 2009;48:5094. doi: 10.1002/anie.200806273.Sun CL, Li BJ, Shi ZJ. Chem Commun. 2010;46:677. doi: 10.1039/b908581e.
- 3.For a recent review, see: Lyons TW, Sanford MS. Chem Rev. 2010;110:1147. doi: 10.1021/cr900184e.
- 4.For related reactions see: Liu Y, Li D, Park CM. Angew Chem, Int Ed. 2011;50:7333–7335. doi: 10.1002/anie.201101550.Bai YG, Zeng J, Cai ST, Liu XW. Org Lett. 2011;13:4394–4397. doi: 10.1021/ol201734w.Cheng D, Gallagher T. Org Lett. 2009;11:2639–2641. doi: 10.1021/ol900627q.Colbon P, Ruan J, Purdie M, Xiao J. Org Lett. 2010;12:3670–3673. doi: 10.1021/ol101466g.Pelletier G, Larivee A, Charette AB. Org Lett. 2008;10:4791–4794. doi: 10.1021/ol8018709.Ramtohul YK, Chartrand A. Org Lett. 2007;9:1029–1032. doi: 10.1021/ol063057k.Sorensen US, Pombo-Villar E. Helv Chim Acta. 2004;87:82–89.Wuertz S, Rakshit S, Neumann JJ, Droege T, Glorius F. Angew Chem, Int Ed. 2008;47:7230–7233. doi: 10.1002/anie.200802482.Xu YH, Lu J, Loh TP. J Am Chem Soc. 2009;131:1372–1373. doi: 10.1021/ja8084548.Zhou H, Chung WJ, Xu YH, Loh TP. Chem Commun. 2009:3472–3474. doi: 10.1039/b903151k.Zhou H, Xu YH, Chung WJ, Loh TP. Angew Chem, Int Ed. 2009;48:5355–5357. doi: 10.1002/anie.200901884.Heck RF. J Am Chem Soc. 1968;90:5535–5538.
- 5.Ge H, Niphakis MJ, Georg GI. J Am Chem Soc. 2008;130:3708. doi: 10.1021/ja710221c. [DOI] [PubMed] [Google Scholar]
- 6.Niphakis MJ, Turunen BJ, Georg GI. J Org Chem. 2010;75 doi: 10.1021/jo100907u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niphakis MJ, Georg GI. J Org Chem. 2010;75:6019. doi: 10.1021/jo101051w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niphakis MJ, Georg GI. Org Lett. 2011;13:196. doi: 10.1021/ol1023954. [DOI] [PubMed] [Google Scholar]
- 9.Leighty MW, Georg GI. ACS Med Chem Lett. 2011;2:313. doi: 10.1021/ml1003074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For recent reviews, see: Hiyama T, Shirakawa E. Handb Organopalladium Chem Org Synth. 2002;1:285.Denmark SE, Ober MH. Aldrichim Acta. 2003;36:75.Denmark SE, Sweis RF. In: Metal-Catalyzed Cross-Coupling Reactions. 2. de Meijere A, Diederich F, editors. Wiley-VCH; Weinheim, Germany: 2004. p. 163.Hiyama T. Chem Rec. 2008;8:337. doi: 10.1002/tcr.20162.
- 11.Yang S, Li B, Wan X, Shi Z. J Am Chem Soc. 2007;129:6066. doi: 10.1021/ja070767s. [DOI] [PubMed] [Google Scholar]
- 12.For recent reviews, see: Denmark SE, Sweis RF. Acc Chem Res. 2002;35:835. doi: 10.1021/ar020001r.Denmark SE, Baird JD. Chem-Eur J. 2006;12:4954. doi: 10.1002/chem.200600034.Denmark SE, Regens CS. Acc Chem Res. 2008;41:1486. doi: 10.1021/ar800037p.Denmark SE. J Org Chem. 2009;74:2915. doi: 10.1021/jo900032x.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.