

Abstract
We recently observed an autoimmune profile in 24-week-old C57BL/6 mice that received a 2.5 or 5.0 μg/kg mid-gestation dose of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) (Mustafa et al., 2008). The clinical signs were consistent with a lupus-like syndrome and included: increased autoantibody levels, renal IgG and C3 immune complex deposition with associated inflammation, and increased peripheral Vβ+ T cells. No studies currently exist following the progression of such disease into middle or advanced ages, when human autoimmune diseases may manifest. Therefore in the present study, littermates of mice from the previous 24 week prenatal TCDD study were allowed to age to 48 weeks, considered early geriatric in mice. Similarities and differences in the disease profile based on age and sex were observed. Peripheral autoreactive Vβ+ T cells were increased in both sexes at 48 weeks, in contrast to males only at 24 weeks. Activated T cells from 48-week-old prenatal TCDD females over-produced the proinflammatory cytokine IFN-γ while males over-produced IL-10, effects again not seen at 24 weeks. Splenic transitional-2 B cells (CD21intCD24hi) were increased in males while transitional-1 B cells (CD23neg CD1neg) were increased in females at 48 weeks. Autoantibodies to cardiolipin and CD138+ spleen plasma cells were significantly increased in the aged males but not females. Anti-IgG and anti-C3 immune complex renal deposition were also significantly increased in the prenatal TCDD males but not females. These selective changes in the aged male mice may be noteworthy, in that the prevalence of SLE in humans shifts dramatically toward males with aging. The collective findings in aged mice suggest that prenatal TCDD permanently biases the postnatal immune response in C57BL/6 mice toward autoimmunity, and support a significant B cell component to the induced renal autoimmune disease.
Keywords: TCDD, C57BL/6 mice, autoimmunity, T cell, B cell, thymus, bone marrow, spleen
Introduction
Prenatal TCDD exposure affects thymus biology and T cell maturation, manifested perinatally by severe but transient thymic atrophy and changes in proportions of thymocyte subpopulations (Camacho et al. 2005; Fine et al. 1990a; Kamath et al. 1997). TCDD may in part elicit such thymic toxicity by altering transcription of genes controlling proliferation and trafficking in the most immature thymocyte phenotypes, specifically TN2 (CD44+, CD25+) and TN3 (CD44-, CD25+) (Frericks et al. 2006; McMillan et al. 2007). The majority of available prenatal TCDD studies have focused on the suppressive effects of this agent on cell-mediated immunity, which may be permanent (Gehrs and Smialowicz 1997; Holladay and Smialowicz 2000).
In the thymus, generating a repertoire of T cells capable of recognizing a diverse and broad array of MHC-restricted antigens is critical for survival. Yet, to avoid autoreactivity, the diversity of MHC-restricted antigen recognition is limited through the process of negative selection (Palmer 2003; Sebzda et al. 1999). TCDD appears to alter the dynamics of thymic selection in part through dysregulation of coreceptors, MHC expression and costimulatory molecules that are believed to play an important role in thymic selection (Fisher et al. 2004). Beginning in the developing thymus, MHC molecules act as thymic self-antigen-presenting molecules in a process whereby thymocytes expressing T-cell receptors (TCRs) with high affinity to self-antigen are eliminated. Prenatal exposure of mice to TCDD produced long-term alterations in thymic MHC antigen expression (Holladay et al. 2011), an effect that predictably may contribute to an altered postnatal T cell repertoire.
In addition to potentially facilitating the escape of autoreactive thymocytes during negative selection, TCDD enhanced extrathymic production of T cells in compartments where negative selection is absent or less stringent compared to thymus. Silverstone et al. (1994) showed that TCDD induced T cell differentiation in the liver of young adult mice. These extrathymic-derived T cells expressed elevated levels of CD4+Vβ17a and Vβ3+TCR. Such TCR variable β (Vβ) chains are usually deleted in the thymus by reaction with self-MHC and minor lymphocyte stimulatory antigens (Okuyama et al., 1992; Hanawa et al., 1993) and have been associated with autoimmunity in experimental mouse models (Rocha et al. 1992). Thus, multiple effects of TCDD on T-lineage cells suggest the potential for increased autoreactivity.
There is a deficit of information about effects of early TCDD exposure on postnatal B cell development and function, particularly involving enhanced immune reactivity (Holsapple et al. 1991; Luster et al. 2003). Recent adult mouse studies suggest the disruption of normal B cell function may precipitate autoimmune responses by events other than production of autoantibodies, including altered antigen presentation and shifts in cytokine production that cause modulation of other immune cell types. For these reasons, B cells may play a more diverse and important role in autoimmunity than previously proposed (Fujimoto and Sato 2007; Martin and Chan 2006).
We recently detected altered B lymphopoiesis and B cell subpopulations in the spleens of C57BL/6 mice at 24 weeks following a single prenatal dose of TCDD (Mustafa et al. 2008). These studies suggested that prenatal TCDD exposure caused at least a highly persistent change in differentiation and activation of B cells, possibly shifting the maturation of naïve B cells toward the marginal zone in the spleen. Similar expansion of these B cell clones into the marginal zone has been associated with autoimmune disease (i.e. murine lupus) (Carnrot et al. 2011; Grimaldi et al. 2001). The prenatal TCDD-exposed 24-week-old mice also exhibited increased autoantibody titers to dsDNA, ssDNA, cardiolipin and glomerular substrate in kidneys, suggesting a collection of altered humoral responses that may contribute to the lupus-like syndrome.
Aging of the immune system is associated with gradual deterioration of its function, or immunosenecence, and exerts significant effects on both innate and adaptive immune cells as well as the precursor cells. Aging decreases the responsiveness of B cells to antigens (Klinman and Kline 1997; Zharhary and Klinman 1983), diminishes immature splenic B cell numbers (Kline et al. 1999), reduces costimulatory molecules (LeMaoult et al. 1997; Weksler and Szabo 2000; Zheng et al. 1997), dysregulates B-cell receptor signaling (Souvannavong et al. 1998; Van der Put et al. 2003), but at the same time enhances T-cell-independent immunoglobulin and low-affinity auto-antibody production (Cancro and Smith 2003; Dunn-Walters and Ademokun 2010; Johnson et al. 2002). Furthermore, age-modulated dysregulation of T-cell/B-cell interactions in mice and humans has been related to defective humoral immunity with enhanced autoantibody levels and chronic inflammatory status (Krabbe et al. 2004; Lazuardi et al. 2005; Yang et al. 1996). Aging is also associated with expression of certain autoimmune diseases, which may significantly impact the quality of life in the elderly (Klinman and Kline 1997). It was therefore important to follow prenatal TCDD mice that showed an autoimmune signature at 24 weeks-of-age (Mustafa et al., 2008) into more advanced ages.
The present study was conducted to determine if a lupus-like syndrome induced by prenatal TCDD in C57BL/6 mice, would continue to progress as the animal approached the geriatric stage. We also evaluated how aging influenced the prenatal TCDD--mediated disease progression across sex. A more detailed analysis of bone marrow B lymphopoiesis and renal pathology was included based on changes in these endpoints that were detected at 24 weeks. Selected parameters of cell-mediated and antibody-mediated immunity were also examined in both primary and secondary lymphoid organs. We hypothesized that immune-mediated renal disease would continue to progress with aging and that the immune modulation in these late-adult mice would continue to be differentially expressed based on sex.
Methods and Materials
Mice and TCDD exposure
C57BL/6 mice were obtained from Charles River Laboratories (Portage, MA) at 4–5 weeks-of-age. Mice were acclimated to the animal care facility for at least 2 weeks prior to breeding. Briefly, 80 female C57BL/6 mice were bred overnight using one C57BL/6 male per two females, and plug positive mice the next morning were designated gestation day (gd) 0. Pregnant C57BL/6 mice were orally gavaged on gd 12 with 0, 2.5 or 5.0 μg/kg TCDD dissolved in corn oil (n=12 pregnant mice/treatment). The F1 offspring remained with dams until weaning, and then were separated by treatment and sex, and a randomly selected group of mice from each treatment group was allowed to mature to 48 weeks-of-age. All animals were fed a commercial pelleted diet and provided water ad libitum, and were housed under controlled conditions of temperature (22°C), humidity (40–60%), and lighting (12:12 light:dark cycle). Animal maintenance, care and use were at all times in accordance with Institutional Animal Care and Use Committee (IACUC) guidelines at Virginia Tech, and approved prior to initiation of experiments.
Body weights and tissue collection
Mice were euthanized by cervical dislocation and weighed. The thymus, spleen, bone marrow and axillary and inguinal lymph nodes (LN) were immediately collected post-euthanasia under aseptic conditions, using dissection scissors and curved forceps. Axillary and inguinal LN were pooled for flow cytometry. Non-LN organs were weighed and then placed individually into pre-labeled sterile Petri dishes (Corning, Corning, NY), containing 8 ml of RPMI-1640 culture medium (Mediatech, Herndon, VA). Dishes were placed on ice until tissue dissociation.
Cell dissociation and enrichment
Each organ was gently dissociated over a stainless steel sieve screen (Sigma, St. Louis, MO) using curved forceps. Cells were then pipetted through the sieve screen following dissociation to remove debris. Cells were washed in RPMI-1640 for 10 min, 240×g, and 23°C. The supernatant was discarded and, with the exception of spleen, the cell pellet was resuspended in 8 ml of RPMI-1640. Spleen cells were resuspended in 1 ml incomplete RPMI-1640. To each tube, 2 ml of 0.83% ammonium chloride lysis buffer (ACK, pH 7.29) were added, to lyse red blood cells, and tubes were incubated for 5 min at 23°C. After the lysis incubation, the cells were resuspended in 5 ml of incomplete RPMI-1640 and washed twice (7 min, 290×g and 7°C). The splenic leukocyte-rich cells were then resuspended in 5 ml complete RPMI-1640 media containing 10% heat-inactivated FBS (Atlanta Biologicals, Atlanta, GA), 2 mM L-glutamine (ICN, Costa Mesa, CA), 50 IU/ml penicillin (ICN), and 50 mg/ml of streptomycin (ICN), and maintained at 7-10°C. For bone marrow isolation, femurs were removed and the bone marrow cavities flushed with 2% FBS-PBS, washed once (7 min, 290×g and 7°C), resuspended in 1 ml incomplete RPMI-1640 media and stored at 4°C.
Cell enumeration
Cells were enumerated and size-analyzed using a Beckman Multisizer 3® cell counter (Beckman Coulter, Fullerton, CA) according to the manufacturer's protocol. Briefly, a 10 μL aliquot of enriched cell suspension was transferred to a plastic counting-chamber containing 10 ml of PBS (Mediatech). Plastic chamber was capped, mixed by repeated gentle inversion, and counted. Cells were adjusted to 5.0×106 cells/ml in complete media.
Flow cytometric evaluation of cell-surface markers
Cell suspensions (5 × 105/100 μl) from the thymus, spleen, lymph node, and bone marrow were dispensed into individual wells of a 96-well round-bottom tissue culture plate (Corning). Monoclonal antibodies (mAbs) with phycoerythrin (PE) fluorescent labels were used according to manufacturer's (BDPharmingen; San Diego, CA) recommendation at a concentration of 0.2 μg/μl; mAbs with fluorescein isothiocyanate (FITC) fluorescent labels were similarly used at the recommended concentration of 0.5 μg/μl. Cells were stained as previously described for 24-week-old littermates of the present mice (Gogal et al. 2001; Klein et al. 2006). Briefly, lymphocyte aliquots (5×105 cells/ 100 μl) from thymus, spleen, lymph node and bone marrow were incubated with the following primary mAbs: PE-anti CD4, FITC-anti CD8, FITC-anti CD25, FITC-anti CD19, PE-anti CD5, PE-anti CD3, PE-anti CD45/B220, PE-Cy5 anti CD45/B220, PE-Cy5 anti CD93 (AA4.1) (eBioscience, San Diego, CA); FITC-anti Vβ3 TcR (KJ25), FITC-anti Vβ17a TcR (KJ23), FITC-anti CD45/B220, PE-anti CD24 (HSA), FITC-anti CD1, PE-anti CD23, PE-anti CD138 (BDPharmingen). For double or triple staining protocols, mAbs with different fluorescent labels were simultaneously added to the sample. For bone marrow analysis, aliquots of 5×105 cells were pre-blocked with anti-FcγIII/IIR (clone 2.4G2, Rat IgG2b). Following staining, cells were washed and evaluated on a Coulter Epics XL flow cytometer (Beckman Coulter). From each sample, 10,000 events were collected and analyzed using the FlowJo software program (Tree Star, San Carlos, CA). Dead cells, clumps, and debris were excluded electronically by gating on forward scatter (FSC) versus side scatter (SSC). The distribution of B cell subsets gating was as previously described (Grimaldi et al. 2001).
ELISA for autoantibodies to double-stranded DNA, single-stranded DNA and cardiolipin
To detect the presence of dsDNA antibody, 96-well medium-binding microtiter plates (Corning) were coated with heat-denatured calf thymus DNA (100 μg/ml; Sigma). For ssDNA antibody titers, DNA from calf thymus was used (10 μg/ml; Sigma), and for autoantibodies to cardiolipin a solution from bovine heart was used (10 μg/ml; Sigma). All pre-coated plates were incubated at 4°C overnight. Plates were washed three times with 300 μl PBS/0.05% Tween20 (Mediatech), blocked with 1% BSA (Sigma) for 2 h at 23°C, washed and then incubated with diluted serum samples collected via retro-orbital bleeding prior to euthanasia. For all three ELISA assays, serial dilutions of each mouse serum were performed to optimize optical density readings. Serum diluted 1/100 was determined to be the optimal dilution for reading the optical density among the samples. After 3 h at 23°C, the serum-coated plates were washed three times with 300 μl of PBS-0.05% Tween 20. To each well, 0.2 ml of diluted alkaline phosphatase-conjugated anti-IgG antibody (1/3000) (Sigma) were added and the plates were incubated for 60 min at 23°C. Plates were then washed three times with 300 μl of PBS-0.05% Tween20. To each well, 0.2 ml prepared substrate for alkaline phosphatase conjugated secondary antibody (SIGMA FAST™ p-Nitrophenyl phosphate tablets) was added and allowed to develop for 45 min at 23°C before adding 50 μL of 3 M NaOH stop solution (Sigma). The absorbance (A405) of the initial dilution was measured. Optical density (OD) readings represent the average from sera from each mouse performed in duplicate.
Histology of the kidneys
Kidneys were collected at the time of euthanasia and immediately fixed in 10% formalin (Fisher Scientific, Pittsburgh, PA). After 48 h in formalin, the tissues were removed, routinely processed and embedded in a paraffin block. Following embedding, 5 μm sections were cut from each tissue block, and stained with hematoxylin and eosin (H&E, Richard-Allen Scientific, Kalamazoo, MI) using standard histologic methods. The prepared slides were then evaluated, with a light microscope, in a blinded-manner by a veterinary pathologist (co-author PS). For each kidney, 100 consecutive renal cortex glomeruli were evaluated. Each glomerulus was scored for the presence of fibrinoid necrosis or crescents and extent of lymphocytic infiltration. The perivascular scores were evaluated at 40×. Number of inflammatory foci and their relative size were evaluated and scored together in the following manner; a score of “0” indicated no inflammation associated with vessels (these include both arteries and veins, though usually the veins appear to be the main structure associated with these), a score of “1” indicated small foci and only one or two in the section, a score of “2” indicated more than two, and of a size that was obvious and a score of “3” indicated widespread, large foci around several vascular structures.
Immunohistochemistry of the kidneys: C3 and IgG deposition
Frozen kidneys were cut into 5 μm sections and stained with FITC conjugated antibodies (Mustafa et al. 2008). Briefly, tissue sections were thawed at room temperature and dried for 30 min. Slides were fixed in acetone for 10 min and then washed with PBS thrice for 3 min/wash. Goat anti-mouse IgG diluted 1:100 (MPBiomedicals, Santa Ana, CA) or goat anti-mouse C3 diluted 1:100 (MP Biomedicals) were incubated with tissues sections in a humid chamber for 60 min at 23°C. The sections were then rinsed thrice for 5 min/wash with PBS. The slides were mounted using Vectashield™ mounting media (Vector Labs, Burlingame, CA) and then examined using an Olympus BX-60 fluorescence microscope (Center Valley, PA). The severity of glomerulonephritis and immune complex deposition was scored using a range from 0 to 3+, where 0 corresponded to a non-autoimmune healthy mouse and 3+ to the maximal alteration observed in the study. All slides were scored in a blinded manner independently by two investigators with previous experience (co-authors CR and RG) and averaged for the final score.
Lymphocyte proliferation assay
Splenocytes were plated into each well (5 × 105 cells/ 100 μL per well) of a 96-well round-bottom tissue-culture plate (Corning Cell Wells™, Corning). Cells were exposed to mitogens as follows: 100 μl of: ConcanavalinA (ConA, 10 μg/ml, Sigma), lipopolysaccharide (LPS, 50 μg/ml, Sigma), or phorbol myristate acetate (PMA, 10 ng/ml, Sigma) plus ionomycin (0.5 μg/ml, Sigma) in complete media. Non-stimulated control cultures contained 100 μl of complete media alone. Triplicate wells were used for each stimulant. Following 48 h incubation, 20 μl of Alamar Blue™ dye (Serotec, Raleigh, NC) (10% of incubation volume) were added to each well of the culture plates. At 24 and 48 h post addition, degree of absorbance was determined under dual wavelength (570 and 600 nm) using a Molecular Devices plate reader (Menlo Park, CA) (Klein et al. 2006; Mustafa et al. 2008).
Cytokine ELISA Assays
Splenocytes were plated into each well (1 ml in complete media; 5×106 cells per well) of a 24-well tissue-culture plate (Corning). Cells were co-cultured with 1 ml ConA (10 μg/ml) and incubated at 37°C, 5% CO2 for 48 h. The plates were centrifuged (7°C, 250×g, 7 min), and the supernatants were transferred to sterile 12×75 mm cultured tubes (Fisher). Supernatants were stored at -80 °C until use. Levels of interleukin-2 (IL-2), IL-4, IL-10, IL-12, and interferon-gamma (IFN-γ) were determined using ELISA kits (Ready-to-use; eBioscience) according to the manufacturer's instructions.
Statistical analysis
Data were expressed as arithmetical mean ± SEM. Analysis of variance (ANOVA) was used with Dunnett's test to establish significant differences between treatment groups and control. The pregnant dam was maintained as the statistical unit in all cases such that each offspring analyzed represented a separate dam. Group size was six C57BL/6 offspring per sex for all experiments (n=6). Results described as different in this report indicate significantly different at p <0.05.
Results
Body and organ weights, and organ cellularity
Body weight was reduced relative to controls in 48-week-old female, but not male offspring of TCDD-treated dams, at the 5.0 μg/kg dose. Thymus weights and thymus cellularity were significantly decreased in the 5.0 μg/kg TCDD females but not males. Thymic weight/body weight ratios showed a numerical trend toward increase in males and decrease in females, at 5.0 μg/kg TCDD. Spleen weight/body weight ratios were near a significant increased (p=0.054) at 48 weeks, in the 5.0 μg/kg TCDD females. Additionally, spleen weight and cellularity also showed numerical trends toward increase in the 5.0 μg/kg TCDD females (Table 1).
Table 1. Body and organ weight and organ cellularity at 48 weeks of age.
| sex | 0 μg/kg mean ± SEM |
2.5 μg/kg mean ± SEM |
5.0 μg/kg mean ± SEM |
|
|---|---|---|---|---|
| Body weight (g) | ♀ | 30.2 ± 0.6 | 30.7 ± 1.8 | 27.3 ± 0.7* |
| ♂ | 44.5 ± 2.8 | 41.6 ± 3.3 | 44.5 ± 0.5 | |
|
| ||||
| Spleen weight (g) | ♀ | 0.14 ± 0.02 | 0.13 ± 0.03 | 0.18 ± 0.02 |
| ♂ | 0.11 ± 0.01 | 0.09 ± 0.01 | 0.10 ± 0.01 | |
|
| ||||
| Spleen/body weight ratio | ♀ | 0.45 ± 0.07 | 0.50 ± 0.04 | 0.64 ± 0.05 |
| ♂ | 0.25 ± 0.01 | 0.23 ± 0.03 | 0.22 ± 0.01 | |
|
| ||||
| Spleen cellularity (× 106) | ♀ | 57.7 ± 4.8 | 54.4 ± 9.2 | 75.2 ± 3.4 |
| ♂ | 89.9 ± 5.4 | 82.1 ± 5.5 | 91.0 ± 4.7 | |
|
| ||||
| Thymus weight (g) | ♀ | 0.06 ± 0.01 | 0.06 ± 0.01 | 0.05 ± 0.01* |
| ♂ | 0.06 ± 0.01 | 0.07 ± 0.01 | 0.08 ± 0.01 | |
|
| ||||
| Thymus/body weight ratio | ♀ | 0.19 ± 0.01 | 0.19 ± 0.01 | 0.17 ± 0.01 |
| ♂ | 0.14 ± 0.01 | 0.16 ± 0.01 | 0.19 ± 0.03 | |
|
| ||||
| Thymus cellularity (× 106) | ♀ | 76.2 ± 9.7 | 67.2 ± 7.0 | 49.2 ± 6.1* |
| ♂ | 72.0 ± 6.5 | 63.5 ± 7.2 | 71.3 ± 2.6 | |
|
| ||||
| Bone marrow cellularity (× 106) | ♀ | 12.7 ± 1.3 | 15.2 ± 2.6 | 14.4 ± 1.0 |
| ♂ | 9.3 ± 1.7 | 11.5 ± 1.0 | 7.8 ± 1.1 | |
|
| ||||
| Lymph node cellularity (× 106) | ♀ | 5.0 ± 0.5 | 3.3 ± 0.6* | 4.9 ± 1.4 |
| ♂ | 7.8 ± 1.8 | 7.1 ± 0.6 | 7.1 ± 1.1 | |
N≥6 mice/treatment/sex;
p<0.05, Dunnett's test.
B lymphoid progenitors in bone marrow
At 48 weeks-of-age, the 5.0 μg/kg TCDD females showed a near significant decrease (p=0.053) and the 5.0 μg/kg TCDD males showed a significant decrease in total B220 cells (B220+). The percentage of these B220+ cells that were B220hi, representing small pre-B cells and immature B lymphocytes, was significantly decreased while the percentage that were B220lo, representing B lineage committed progenitors, pro-B cells and large pre-B cells, was significantly increased in 5.0 μg/kg TCDD females. The percentage of B220loCD24- cells that expressed AA4.1 (B220loCD24-AA4.1+), representing B lineage committed progenitors, was near significantly (p=0.054) increased in the 5.0 μg/kg TCDD females and significantly increased in the 2.5 and 5.0 μg/kg TCDD males. The percentage of B220 IgM+, representing immature B cells, was significantly decreased in 5.0 μg/kg TCDD males (Table 2). Figure 1a shows a schematic summary of the effect of prenatal TCDD on adult bone marrow B cell maturation.
Table 2. B lymphoid progenitors in the bone marrow at 48 weeks of age.
| Bone Marrow | sex | 0 μg/kg mean ± SEM |
2.5 μg/kg mean ± SEM |
5.0 μg/kg mean ± SEM |
|---|---|---|---|---|
| Total B220+ (%) | ♀ | 11.8 ± 1.5 | 9.1 ± 1.0 | 8.4 ± 0.6 |
| ♂ | 10.2 ± 0.6 | 8.9 ± 0.8 | 6.7 ± 0.4* | |
|
| ||||
| B220lo (% of B220+) | ♀ | 47.5 ± 1.5 | 53.7 ± 1.9* | 58.2 ± 2.4* |
| ♂ | 46.6 ± 0.7 | 48.5 ± 2.3 | 47.3 ± 0.6 | |
|
| ||||
| B220loCD24+ (% of B220lo) | ♀ | 67.8 ± 4.4 | 66.4 ± 3.1 | 69.7 ± 2.1 |
| ♂ | 71.9 ± 2.8 | 61.7 ± 3.3 | 55.5 ± 3.6* | |
|
| ||||
| B220loCD24hi (% of B220lo) | ♀ | 26.7 ± 2.5 | 27.6 ± 3.2 | 33.0 ± 2.4 |
| ♂ | 32.8 ± 3.7 | 24.4 ± 1.7* | 29.2 ± 2.0 | |
|
| ||||
| B220loCD24lo (% of B220lo) | ♀ | 73.3 ± 2.5 | 72.4 ± 3.2 | 67.0 ± 2.4 |
| ♂ | 67.2 ± 3.7 | 75.6 ± 1.7* | 70.8 ± 2.0 | |
|
| ||||
| B220loCD24neg (% of B220lo) | ♀ | 32.2 ± 4.4 | 33.6 ± 3.1 | 30.3 ± 2.1 |
| ♂ | 28.1 ± 2.8 | 38.3 ± 3.3 | 44.6 ± 3.6* | |
|
| ||||
| B220loCD24-AA41+ (%B220loCD24-) | ♀ | 68.2 ± 2.1 | 71.3 ± 5.0 | 80.4 ± 3.0 |
| ♂ | 73.3 ± 6.4 | 90.8 ± 1.6* | 91.7 ± 1.6* | |
|
| ||||
| B220hi (% of B220+) | ♀ | 52.5 ± 1.5 | 46.4 ± 1.9* | 41.8 ± 2.4* |
| ♂ | 53.4 ± 0.7 | 51.5 ± 2.3 | 52.7 ± 0.6 | |
|
| ||||
| B220hiCD24+ (% of B220hi) | ♀ | 90.9 ± 1.4 | 92.6 ± 0.8 | 93.5 ± 0.5 |
| ♂ | 70.7 ± 1.6 | 61.2 ± 4.4* | 57.5 ± 1.6 | |
|
| ||||
| B220hiCD24hi (% of B220hi) | ♀ | 30.6 ± 3.7 | 35.3 ± 2.5 | 35.9 ± 2.4 |
| ♂ | 28.0 ± 3.1 | 24.6 ± 2.4 | 19.2 ± 1.6* | |
|
| ||||
| B220hiIgM+ (% of B220hi) | ♀ | 86.5 ± 1.6 | 87.4 ± 1.4 | 86.9 ± 1.0 |
| ♂ | 91.6 ± 0.5 | 92.5 ± 0.6 | 91.8 ± 0.5 | |
|
| ||||
| B220loIgM+ (% of B220lo) | ♀ | 32.4 ± 5.0 | 35.0 ± 4.9 | 33.7 ± 2.9 |
| ♂ | 40.4 ± 1.0 | 36.7 ± 2.5 | 34.9 ± 0.7* | |
N≥6 mice/treatment/sex;
p<0.05, Dunnett's test.
Figure 1.
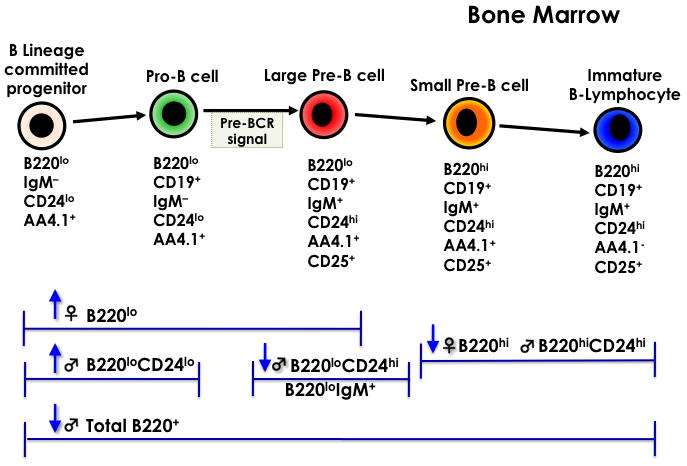
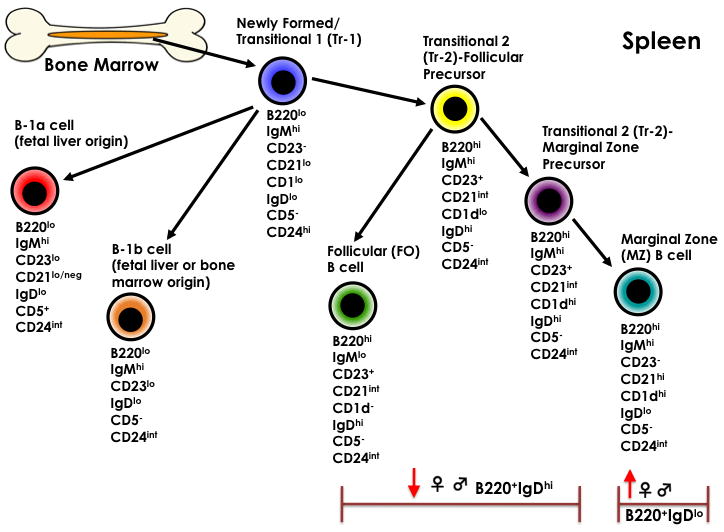
A. Bone Marrow B-cell Lymphopoiesis. B. B cell maturation in the spleen. The cell-surface markers below each cell type were evaluated. Arrows pointed up indicate a significant increase and arrows pointed down indicate a significant decrease from controls (n=5 mice/treatment/sex, p ≤ 0.05, Dunnett's test).
Thymic T cell differentiation
Both female and male offspring of dams dosed with 5.0 μg/kg TCDD exhibited marginal but significant thymic phenotypic changes at 48 weeks-of-age. The percentage of CD4-CD8- double negative thymocytes was increased by gd 12 TCDD in 5.0 μg/kg females. In 5.0 μg/kg males, the percentage of CD4+CD8+ double positive thymocytes was significantly decreased and the percentage of CD4+CD8- was near significantly increased (p=0.54) (Table 3).
Table 3. Thymic T cell differentiation at 48 weeks of age.
| Thymus | sex | 0 μg/kg mean ± SEM |
2.5 μg/kg mean ± SEM |
5.0 μg/kg mean ± SEM |
|---|---|---|---|---|
| CD4-CD8- (%) | ♀ | 17.1 ± 0.6 | 18.6 ± 1.2 | 21.8 ± 1.2* |
| ♂ | 13.2 ± 0.8 | 12.2 ± 0.8 | 15.0 ± 1.2 | |
|
| ||||
| CD4+CD8+ (%) | ♀ | 67.8 ± 1.6 | 63.3 ± 1.4 | 62.3 ± 2.0 |
| ♂ | 65.0 ± 1.1 | 64.7 ± 1.3 | 58.8 ± 1.4* | |
|
| ||||
| CD4-CD8+ (%) | ♀ | 5.1 ± 1.0 | 3.9 ± 0.3 | 3.6 ± 0.2 |
| ♂ | 7.7 ± 0.3 | 8.4 ± 0.6 | 9.4 ± 0.7 | |
|
| ||||
| CD4+CD8- (%) | ♀ | 10.0 ± 1.0 | 14.2 ± 1.7 | 12.4 ± 1.3 |
| ♂ | 14.2 ± 0.4 | 14.7 ± 0.5 | 16.8 ± 1.2 | |
N≥6 mice/treatment/sex;
p<0.05, Dunnett's test.
Phenotype of spleen B and T cells, and migration of mature B cells in the spleen
The relative percentage of spleen leukocytes that expressed B220 did not differ across treatment or sex compared to controls (Table 4). However, there was a significant difference within B cell subsets expressing CD21/CD24 and CD1/CD23 phenotypes. The 5.0 μg/kg TCDD males showed a significantly increased relative percentage of B transitional-2 cells (CD21hiCD24hi). In females, the B transitional-1 cells (CD23neg CD1neg) were significantly increased while other B cell subsets expressing CD1/CD23 did not differ from control groups. The percentages of splenic immature B220+CD24hi transitional B cells and B220+CD24lo-int mature B were unchanged; however, there was a significant decrease in mature IgDhi splenic B cells and a significant increase in IgDneg-lo splenic B cells at 5.0 μg/kg TCDD regardless the gender. Additionally, CD138+ plasma cells were significantly increased in 5.0 μg/kg TCDD males. The relative percentage of CD19+CD5+ B cells (representing B-1a cells, a B cell subset of fetal origin only, rather than bone marrow) was unchanged. Figure 1b shows a schematic summary of these effects of prenatal TCDD on adult spleen B cell populations. The relative percentage of spleen T cells expressing CD4/CD8 markers did not change in any treatment group or by sex (Table 5).
Table 4. Spleen B cell populations at 48 weeks of age.
| Spleen | sex | 0 μg/kg mean ± SEM |
2.5 μg/kg mean ± SEM |
5.0 μg/kg mean ± SEM |
|---|---|---|---|---|
| B220+ (%) | ♀ | 35.4 ± 1.5 | 36.5 ± 3.1 | 33.2 ± 2.1 |
| ♂ | 27.3 ± 0.8 | 25.0 ± 1.4 | 25.9 ± 1.5 | |
|
| ||||
| B220+CD24hi (%) | ♀ | 78.9 ± 2.3 | 76.8 ± 2.8 | 79.9 ± 1.8 |
| ♂ | 74.9 ± 2.0 | 70.4 ± 2.4 | 79.5 ± 1.3 | |
|
| ||||
| B220+CD24lo (%) | ♀ | 21.1 ± 2.3 | 23.2 ± 2.8 | 20.1 ± 1.8 |
| ♂ | 25.1 ± 2.0 | 29.6 ± 2.4 | 20.5 ± 1.3 | |
|
| ||||
| CD19+CD5+ (%) | ♀ | 5.7 ± 2.4 | 9.0 ± 1.9 | 4.7 ± 1.5 |
| ♂ | 6.6 ± 0.6 | 7.6 ± 0.7 | 7.4 ± 0.8 | |
|
| ||||
| CD23negCD1neg (%) | ♀ | 23.5 ± 2.3 | 22.1 ± 2.6 | 33.6 ± 3.1* |
| ♂ | 25.8 ± 1.8 | 27.2 ± 1.0 | 25.6 ± 1.2 | |
|
| ||||
| CD21intCD24lo-int (%) | ♀ | 60.7 ± 2.9 | 64.7 ± 0.5 | 66.7 ± 3.6 |
| ♂ | 60.8 ± 1.5 | 58.0 ± 3.0 | 58.6 ± 1.3 | |
|
| ||||
| CD21hiCD24int (%) | ♀ | 13.6 ± 0.8 | 11.9 ± 0.9 | 11.3 ± 1.3 |
| ♂ | 11.8 ± 0.8 | 11.6 ± 0.6 | 10.9 ± 0.5 | |
|
| ||||
| CD21-CD24hi (%) | ♀ | 12.4 ± 1.0 | 13.1 ± 1.1 | 11.7 ± 1.6 |
| ♂ | 16.2 ± 1.0 | 17.0 ± 1.3 | 14.4 ± 0.4 | |
|
| ||||
| CD21hiCD24hi (%) | ♀ | 13.0 ± 1.5 | 9.9 ± 0.4 | 9.9 ± 1.9 |
| ♂ | 10.7 ± 1.4 | 13.0 ± 1.5 | 15.3 ± 1.0* | |
|
| ||||
| B220+IgDhi (%) | ♀ | 69.3 ± 2.4 | 62.8 ± 3.2 | 60.2 ± 1.4* |
| ♂ | 58.5 ± 0.5 | 58.6 ± 1.5 | 53.2 ± 1.3* | |
|
| ||||
| B220+IgDneg-lo (%) | ♀ | 30.8 ± 2.4 | 37.2 ± 3.2 | 39.9 ± 1.4* |
| ♂ | 41.5 ± 0.9 | 41.4 ± 1.5 | 46.8 ± 1.3* | |
|
| ||||
| CD138pos | ♀ | 9.0 ± 1.1 | 11.3 ± 1.8 | 12.7 ± 1.1 |
| ♂ | 3.8 ± 1.8 | 5.7 ± 2.0 | 13.0 ± 3.5* | |
N≥6 mice/treatment/sex;
p<0.05, Dunnett's test.
Table 5. Spleen T cell populations at 48 weeks of age.
| Spleen | sex | 0 μg/kg mean ± SEM |
2.5 μg/kg mean ± SEM |
5.0 μg/kg mean ± SEM |
|---|---|---|---|---|
| CD4+CD8- (%) | ♀ | 16.9 ± 1.5 | 17.7 ± 1.5 | 16.9 ± 2.0 |
| ♂ | 29.1 ± 1.0 | 31.5 ± 1.5 | 31.3 ± 1.2 | |
|
| ||||
| CD4-CD8+ (%) | ♀ | 7.7 ± 0.7 | 6.8 ± 1.0 | 6.1 ± 0.8 |
| ♂ | 12.4 ± 0.8 | 12.9 ± 0.5 | 11.0 ± 0.6 | |
|
| ||||
| CD4-CD8- (%) | ♀ | 75.1 ± 1.9 | 75.1 ± 2.2 | 76.7 ± 2.6 |
| ♂ | 61.2 ± 1.5 | 57.9 ± 1.7 | 59.2 ± 1.4 | |
N≥6 mice/treatment/sex;
p<0.05, Dunnett's test.
T cell subset expression in lymph nodes
Prenatal TCDD at 5.0 μg/kg caused a significant decrease in the percentage of CD4-CD8+ cells in the lymph nodes of both male and female 48-week-old mice. The percentage of CD3+ cells expressing Vβ3+TcR showed numeric (non-significant) and dose-related increasing trends in female mice, and were significantly increased in 5.0 μg/kg males. The percentage of CD4+ T cells expressing Vβ17a+TcR was significantly increased in both sexes. CD4+CD8- cells in lymph nodes of male, but not female, 5.0 μg/kg mice were significantly increased. The CD4+ T cells expressing CD25, which includes T regulatory and activated T cells, showed a numeric (non-significant) trend toward decrease in both females and males by 5.0 μg/kg prenatal TCDD (Table 6).
Table 6. Lymph node T cell populations at 48 weeks of age.
| Lymph node | sex | 0 μg/kg mean ± SEM |
2.5 μg/kg mean ± SEM |
5.0 μg/kg mean ± SEM |
|---|---|---|---|---|
| CD4+CD8- (%) | ♀ | 27.7 ± 3.1 | 27.3 ± 1.2 | 24.4 ± 1.9 |
| ♂ | 28.4 ± 1.1 | 32.0 ± 2.3 | 37.7 ± 1.1* | |
|
| ||||
| CD4-CD8+ (%) | ♀ | 20.1 ± 3.4 | 13.7 ± 1.5 | 11.1 ± 0.9* |
| ♂ | 24.4 ± 2.0 | 24.6 ± 1.5 | 19.3 ± 1.0* | |
|
| ||||
| CD4-CD8- (%) | ♀ | 51.4 ± 6.4 | 58.3 ± 2.5 | 63.9 ± 2.2 |
| ♂ | 43.2 ± 2.6 | 40.0 ± 2.9 | 39.7 ± 1.3 | |
|
| ||||
| CD4+Vβ17TcR+ (%) | ♀ | 3.6 ± 0.3 | 4.9 ± 1.0 | 5.4 ± 0.5 |
| ♂ | 6.1 ± 0.3 | 7.3 ± 0.9 | 8.7 ± 0.8* | |
|
| ||||
| CD3+Vβ3TcR+ (%) | ♀ | 5.2 ± 0.4 | 6.4 ± 1.0 | 8.1 ± 1.0* |
| ♂ | 5.1 ± 0.4 | 6.4 ± 1.0 | 8.4 ± 1.2* | |
|
| ||||
| CD4+CD25+ (%) | ♀ | 8.7 ± 1.0 | 6.8 ± 1.0 | 6.1 ± 0.4 |
| ♂ | 10.2 ± 1.2 | 13.0 ± 2.0 | 7.2 ± 1.8 | |
N≥6 mice/treatment/sex;
p<0.05, Dunnett's test.
Mitogen-stimulated splenic lymphocyte proliferation
Mitogen stimulation of enriched, cultured splenic lymphocytes was employed to assess the influence of prenatal TCDD on lymphocyte functionality in the adult mouse. There was a trend toward decreased spontaneous proliferation in 5.0 μg/kg TCDD males at 48 hr that became significant at 72 hr, where a minimal (14%) decrease in spontaneous proliferation was observed (data not shown).
Antibody titers to ssDNA, dsDNA and cardiolipin
The detection of dsDNA, sDNA and cardiolipin autoantigens was performed with ELISA assays to assess the autoreactive functionality of B cells in mice prenatally exposed to TCDD. Anti-ssDNA and anti-dsDNA anti-cardiolipin titers showed numeric but non-significant increasing trends in both the male and female offspring. Anti-cardiolipin antibodies were significantly increased in male offspring by 5.0 μg/kg TCDD (Figure 2).
Figure 2.
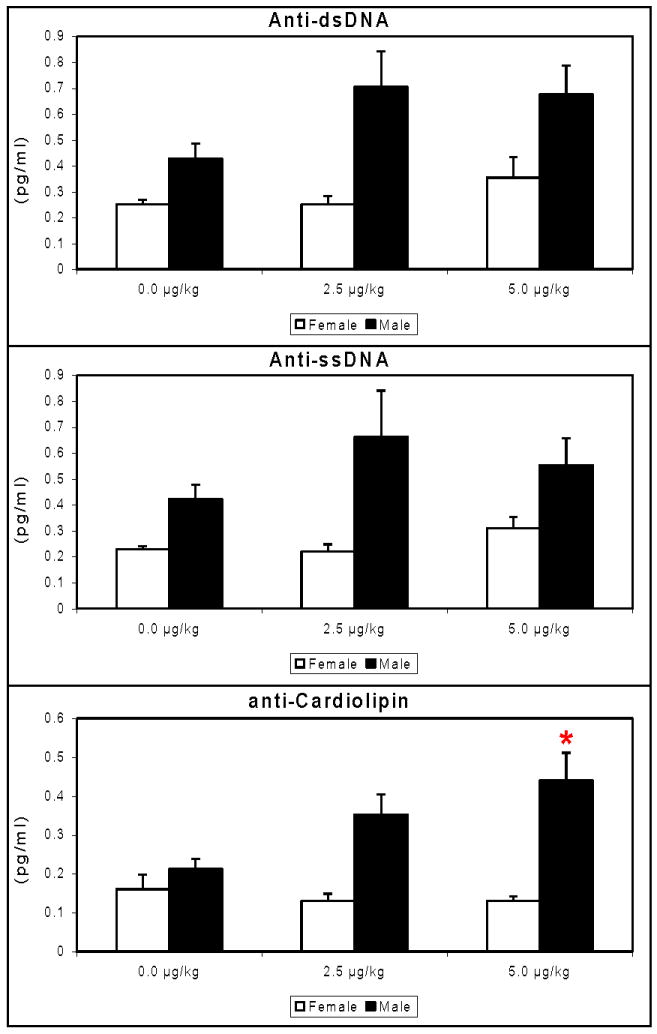
Sera from 48 week-old C57BL/6 mice that were prenatally exposed to 0.0, 2.5 and 5.0 μg/kg TCDD were analyzed for the presence of autoantibodies to dsDNA (A), ssDNA (B) and cardiolipin (C), (n=5/gender; *=p ≤ 0.05, Dunnett's t-test).
Kidney pathology
Since previous C57BL/6 mice treated with TCDD in a manner identical to the present mice showed immune complex deposition in the kidney at 24 weeks-of-age (Mustafa et al. 2008), suggestive of an early stage autoimmune response, histopathologic examination of the kidney was performed at 48 weeks-of-age. Such immune complex deposition in kidneys is also a common clinical signalment in human lupus patients, thus clinically relevant. Glomeruli evaluated for evidence of fibrinoid necrosis (N) or crescents (C) showed an increasing non-significant trend by treatment and sex. The mean number of parenchyma mononuclear inflammatory foci (I) also numerically increased, non-significantly, by treatment and sex turning significant by 5.0 μg/kg TCDD males (Figure 3).
Figure 3.
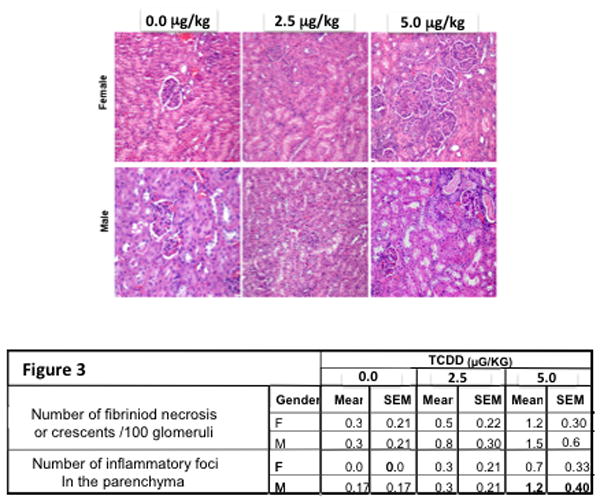
The kidneys from 24-week-old SNF1 mice that were prenatally exposed to 0, 2.5 or 5.0 μg/kg TCDD were collected, fixed, sectioned and stained with H&E. Images are representative of renal sections by treatment and sex. All sections were scored for number of fibrinoid necrosis cells or crescents/100 glomeruli and number of inflammatory foci in the parenchyma. The data are based on 5 mice/treatment/sex (Bold=p ≤ 0.05, Dunnett's test).
Deposition of anti-IgG and anti-C3 immune complexes in the kidney
Immunofluorescent staining was performed to elucidate IgG and C3 involvement in immune complex deposition. Kidney sections from the 48-week-old 5.0 μg/kg TCDD-treated males were significantly increased for both anti-IgG and anti-C3. A TCDD dose-dependent increasing trend in deposition of immune complexes, for both anti-IgG and anti-C3 probes, was present in the other treatment groups (Figure 4).
Figure 4.
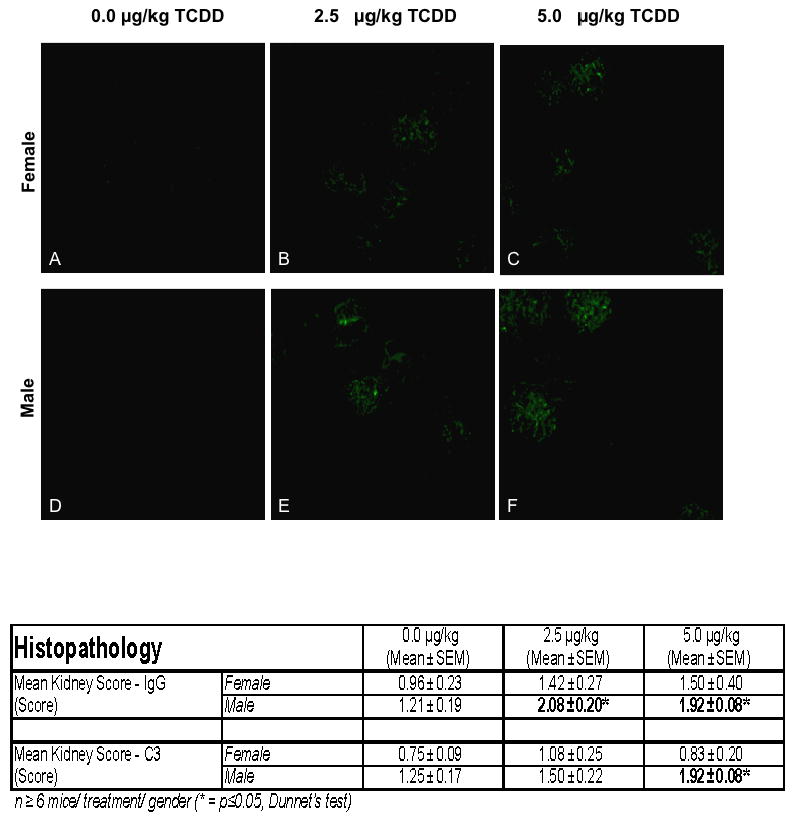
The kidneys from 24 week-old C57BL/6 mice that were prenatally exposed to 0.0, 2.5 and 5.0 μg/kg TCDD were collected, fixed, section and stained with FITC-labeled anti-IgG. The above figures are representative of kidneys stained with FITC-anti-IgG from control female (A) and male (D) or 2.5 μg/kg TCDD-exposed female (B) and male (E) mice or 5.0 μg/kg TCDD-exposed female (C) and male (F) mice. The table data show the mean± SEM of the IgG and C3 disposition scores of 5 mice/treatment/gender.
Th1/Th2 cytokine balance
Female mice showed increased IFN-γ production relative to controls as a consequence of prenatal 5.0 μg/kg TCDD. The 5.0 μg/kg TCDD mice also showed increased IL-10 production (Figure 5).
Figure 5.
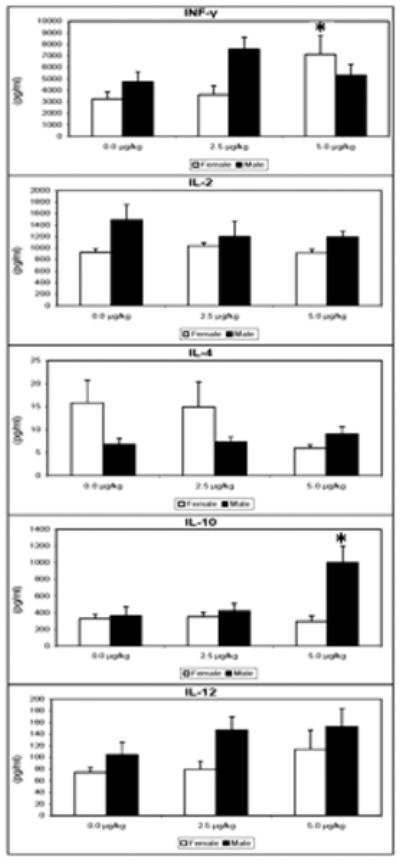
Supernatants were collected from splenocytes, of 24 week-old mice that were prenatally exposed to 0.0, 2.5 and 5.0 μg/kg TCDD, cultured for 48 hr with Con A (10 μg/mL). The levels of IL-2, IL-4, IL-10, IL-12 and INF-γ were determined using murine cytokine ELISA kits. The data are presented in a scatter-plot analysis with mean bars (n = 5 mice/treatment/ gender, *= p ≤ 0.05, Dunnett's t-test).
Discussion
Comparing the present mice to high-exposure human cohorts is difficult beyond total exposure estimates during early life. The low-dose mice received a bolus dose of 2.5 μg TCDD/kg/day on gd 12. Approximately 0.5% of gavaged TCDD crosses the rodent placenta (Weber and Birnbaum 1985; Weber et al. 1985), indicating TCDD exposure to the fetal mice was approximately 2.5 μg/kg × 0.005 = 0.0125 μg/kg or 12.5 ng/kg. Gehrs and Smialowicz (1999) used a similar single gestational TCDD exposure as low as 0.1 μg/kg in rats, and found suppressed T cell function at 18 months of age. This exposure was 25 times lower than the present mice and would translate to about 0.5 ng/kg exposure to the fetal rats. In either rodent scenario, the transplacental exposure would be increased postnatally via undefined lactational transfer.
Human TCDD exposure has been estimated at about 0.2-1.0 pg/kg per day (predominantly via food) and for infants, up to 30 pg/kg per day via mothers' milk (Neubert et al. 1990). Human infant exposure to 30 pg/kg TCDD via lactation for the first 6 months of life of the child would translate to a total dose of 30 pg/kg/day × 182.5 days = 5475 pg/kg TCDD, or about 5.5 ng/kg. Neubert et al. (1990) further indicated that in certain geographic and occupational human populations, the actual TCDD exposure to the infant could be 200-300 fold higher. This would translate to 5.5 ng/kg × 200-300= 1.10-1.65 μg/kg TCDD exposure. Such postnatal (lactational) exposure would be additive to the transplacental TCDD exposure that would have occurred in these infants, but is yet to be quantitated. Based on these data, it appears that human infant cohorts may exist where early-life total exposure to TCDD is in the range of or greater than rodent studies that demonstrated persistent postnatal immunotoxicity. As such, concerns over the effects of TCDD exposure during critical stages of human immune development appear to be justified.
Thymic development in rodents is highly sensitive to TCDD. Prenatal TCDD induces severe but typically transient perinatal thymic atrophy and reduced thymocyte numbers, however causes less dramatic but highly persistent postnatal alterations in thymocyte subpopulations (Blaylock et al. 1992; Esser 1994; Holladay et al. 1991; Lundberg et al. 1992; Oughton et al. 1995). For instance thymocyte subpopulations in offspring from female F344 rats exposed to TCDD (0.1-3.0 μg/kg) on gestational day 14 (gd14) showed increased γδTCR+ cells, and decreased γδTCR+/CD4-CD8- and MHCI- MHCII- cells at 4-5 months of age (Gehrs and Smialowicz 1997). These rats also showed depressed delayed-type hypersensitivity (DTH) responses that did not correlate with altered thymic subpopulations, lasted until 4 months of age in the females and until 19 months of age in males, and were more inhibited in both sexes at 14 months than at 4 months. These DTH results suggest possible worsening of immune functional lesions caused by prenatal TCDD, with aging, and emphasize the importance of immunologically following such rodents to later life stages. Similar to the aged rats, the present C57BL/6 mice continued to show altered thymocyte populations at 48 weeks-of-age, in the form of increased CD4-CD8- cells in females and decreased CD4+CD8+ cells in males. Peripheral Vβ+T cells were increased in both sexes at 48 weeks-of-age, in contrast to only in the males at 24 weeks-of-age. Very limited similar data are available for humans, however perinatal exposure of Japanese infants to dioxin-like contaminants through lactation markedly enhanced peripheral CD8+ and CD3+ T lymphocytes, as well as CD4+/CD8+ T cell ratios (Nagayama et al. 2007). These results suggest possible similar alteration of T cell production in humans who receive early TCDD exposure.
Cytokine production and signaling play key roles in immune system regulation. The extent and postnatal duration to which cytokine pathways may be impaired by developmental exposure to TCDD remain largely undefined, however a few available reports suggest postnatal cytokine effects. In BALB/c mice, perinatal TCDD inhibited IL-4 production in OVA-sensitized young post-pubertal (6-week old) mice (Tarkowski et al. 2010). Using an influenza virus infection model, prenatal TCDD enhanced IFN-γ levels in the lungs of adult (6-10 weeks old) male and female C57BL/6 mice, and depressed IL-12 levels in the female mice (Vorderstrasse et al. 2006). Our previous 24-week-old C57BL/6 mice showed decreased IL-12 production by con-A stimulated T cells collected from females, and decreased IL-10 production in males after prenatal TCDD exposure (Mustafa et al. 2008). This profile changed in the 48-week old early geriatric prenatal TCDD mice. Activated T cells from female mice that were exposed to 5 μg/kg TCDD during gestation produced 2.3 times more IFN-γ than controls, while males produced 2.8 times more IL-10 than controls. Increased IFN-γ in the aged females suggests a proinflammatory cytokine profile and is noteworthy, in that this cytokine has been associated with pathogenesis of various autoimmune diseases including systemic lupus erythematosus (SLE) (Haas et al. 1997), autoimmune insulitis (Campbell et al. 1991), Sjogren's syndrome (Hayashi et al. 1996), and autoimmune arthritis (Billiau 1996).
It was recently noted that elderly human females may display increased IL-4, IL-10 and IFNγ production by T cells (Pietschmann et al. 2003), in a chronic subclinical inflammatory profile defined as inflamm-aging (Finch and Crimmins 2004). Franceschi et al. (2000) suggested that inflamm-aging could be caused by early- or late-life exposure to various inflammatory factors including environmental chemicals, viral, or bacterial antigens. Reports of increased IFN-γ in adult mice after prenatal TCDD may support an environmental contribution to inflamm-aging in elderly individuals, due to altered cytokine/immune homeostasis. The increased IL-10 production by T cells from male mice that received prenatal TCDD may also be noteworthy. This cytokine has been considered the hallmark human SLE disease marker. Serum levels of IL-10 are elevated in SLE patients and increased IL-10 correlates with SLE disease activity (Hagiwara et al. 1996; Horwitz et al. 1998; Park et al. 1998). A possible correlation between increased IL-10 levels in the 48-week prenatal TCDD males and the observed phenotypic and histopathologic shifts toward enhanced lupus-like autoimmune disease has not yet been examined, and represents an important direction for future experiments.
Bone marrow cells, the progenitors for lymphocytes, are sensitive targets of prenatal TCDD (Fine et al. 1990b; Fine et al. 1990a; Mustafa et al. 2009; Mustafa et al. 2008). Luster et al. (1980) showed that 5 μg/kg prenatal TCDD diminished bone marrow cellularity/femur by 4% in male and 3% in female B6C3F1 mice on postnatal day 14 (Luster et al. 1980). The terminal deoxynucleotidyl transferase (TdT)-synthesizing fetal liver and bone marrow population, representing lymphocyte stem cells, was also found to be reduced by TCDD at concentrations as low as 200 fg/mg tissue (Fine et al. 1990b). The present mice showed multiple TCDD-related differences in B cells at 48 weeks-of-age including an increase in the earliest bone marrow B progenitor (B220loCD24-AA4.1+) and a decrease in total bone marrow-derived lymphoid cells expressing B220. The increase in the earliest B progenitor cells may agree with an increase in pre-pro-B cells detected in a S17 stromal line exposed to TCDD (Wyman et al. 2002). Functional or other changes in bone marrow B-lineage cells that may accompany these alterations in phenotypic distribution have not been studied but may be important. The molecular pathways controlling B cell lymphopoiesis, B-cell receptor (BCR) positive and negative selection processes, and B cell trafficking from bone marrow are mediated by several different bone marrow-level signal-transduction pathways (Monroe et al. 2003) and related proteins (e.g., NFκB, CD22, and CD19), which are affected by inappropriate activation of the aryl hydrocarbon receptor (AhR) (Masten and Shiverick 1995; Tian et al. 1999). It is not yet known if these B cell pathways may be altered by prenatal TCDD and contributes to the observed autoimmune phenotype.
In addition to altered B lymphopoiesis in bone marrow, prenatal TCDD caused changes in spleen transitional B cell numbers and shifted B cell differentiation toward the marginal zone (MZ) in the 48-week-old mice, effects not detected at 24 weeks (Mustafa et al. 2008). In particular, the 48-week mice showed increased CD23-CD1- transitional 1 (T1) B cells in females and increased CD21hiCD24hi T2 B cells in males. During the transitional stage, autoreactive B cells that have not encountered self-antigen in the bone marrow are likely to be targets for negative selection in the spleen (Monroe et al. 2003; Sandel and Monroe 1999). Transitional 1 B cells represent recent immigrants from the bone marrow and are believed to be direct precursors of T2 B cells (Loder et al. 1999). Signaling through the BCR initiates signal transduction events that direct the differentiation of transitional B cells into the mature marginal zone or follicular B subset or B1 cell subset (Allman and Pillai 2008; Martin and Kearney 2000). Over-expression of B cell-activating factor of the TNF family (BAFF) in transgenic (Tg) mice correlates with a lupus phenotype as well as decreased T1 B cells and expansion of T2 and marginal zone B cell compartments, similar to the phenotypes of the present prenatal TCDD mice (Batten et al. 2000). This appears to be the result of impaired negative selection of the autoreactive B cells leading to an increased number of marginal zone B cells (Batten et al. 2000). Prenatal TCDD exposure also enhanced the presence of CD138+ plasma cells in the spleens of 48-week-old male mice but not female mice, which correlated well with the increased autoantibody production detected in the males but not females. Finally, histopathologic scoring of kidneys from the male but not female mice showed increased inflammatory foci and immune complex deposition. These collective observations suggest a B cell component to the increased kidney autoimmune response detected in the 48-week-old mice, and driven by prenatal TCDD exposure.
The SLE prevalence in premenopausal females is about 10:1 compared to men of the same age, and characterized by increased autoantibody levels (dsDNA, anti-ribosomal P and anti-chromatin) (Boddaert et al. 2004; Cervera et al. 1993; Cooper et al. 2002; de Carvalho et al. 2010; Formiga et al. 1999; Hashimoto et al. 1987; Voulgari et al. 2002). The ratio of female to male lupus patients then decreases to 1.1:1 by 65 years-of-age (Pu et al. 2000), after which immune-mediated renal pathology is present in most male SLE human patients (Cervera et al. 1993; Chang et al. 1998; Hashimoto et al. 1987; Kaufman et al. 1989; Maddison 1987) as well as in lupus mouse models (Bagavant et al. 2004). Plasma estradiol and testosterone levels are altered with age, and likely explain this shifting in human disease prevalence by sex. Elderly men (82±12 of age) have higher mean estradiol levels compared to elderly women (85±7 of age) and about half of the testosterone levels observed in young males (31±5 of age) (Pietschmann et al. 2003). With increasing age, the decrease in naïve T cell populations and the shift towards a Th2 profile upon activation could enhance the B cell mediated autoreactive antibody production seen in elderly lupus patients (i.e. RF, anti-Ro, and anti-La, anti-dsDNA antibodies) (Belostocki and Paget 2002; Boren and Gershwin 2004; Catoggio et al. 1984; Ho et al. 1998; Maddison 1987; Padovan et al. 2007; Stacy et al. 2002). This dysregulation in immune homeostasis during aging could lead to central alterations in immune function, thus favoring the pathogenesis of autoimmune diseases in elderly males. It will be important to determine if prenatal TCDD leads to increased autoimmune renal disease in 48-week-old male but not female mice due to endocrine changes that have occurred in the males by this age.
To summarize, developmental exposure to TCDD appears to result in a fundamental disruption in the establishment and function of both T and B cells, including possible effects in both cell types on central tolerance. The disruption of the immune system is long-lasting in high AhR affinity C57BL/6 mice, reaching into the early geriatric stage. Such immune dysregulation could potentially contribute to increased incidence of autoimmune disease and infections observed in older individuals (Miller 1996; Prelog 2006). Factors contributing to the risk for and severity of such environmental exposure-driven autoimmune disease may then include: the individual's AhR affinity for TCDD and related ligands, genetic predisposition to autoimmune disease, and sex.
Footnotes
Prenatal TCDD altered the postnatal immune system, and differently so based on sex.
Immune dysregulation affected both T and B cells, phenotypically and functionally.
Prenatal TCDD caused a lupus-like autoimmune phenotype.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Camacho IA, Singh N, Hegde VL, Nagarkatti M, Nagarkatti PS. Treatment of mice with 2,3,7,8-tetrachlorodibenzo-p-dioxin leads to aryl hydrocarbon receptor-dependent nuclear translocation of NF-kappaB and expression of Fas ligand in thymic stromal cells and consequent apoptosis in T cells. J Immunol. 2005;175:90–103. doi: 10.4049/jimmunol.175.1.90. [DOI] [PubMed] [Google Scholar]
- Fine JS, Silverstone AE, Gasiewicz TA. Impairment of prothymocyte activity by 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Immunol. 1990a;144:1169–1176. [PubMed] [Google Scholar]
- Kamath AB, Xu H, Nagarkatti PS, Nagarkatti M. Evidence for the induction of apoptosis in thymocytes by 2,3,7,8-tetrachlorodibenzo-p-dioxin in vivo. Toxicol Appl Pharmacol. 1997;142:367–377. doi: 10.1006/taap.1996.8049. [DOI] [PubMed] [Google Scholar]
- Frericks M, Temchura VV, Majora M, Stutte S, Esser C. Transcriptional signatures of immune cells in aryl hydrocarbon receptor (AHR)-proficient and AHR-deficient mice. Biol Chem. 2006;387:1219–1226. doi: 10.1515/BC.2006.151. [DOI] [PubMed] [Google Scholar]
- McMillan BJ, McMillan SN, Glover E, Bradfield CA. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces premature activation of the KLF2 regulon during thymocyte development. J Biol Chem. 2007;282:12590–12597. doi: 10.1074/jbc.M611446200. [DOI] [PubMed] [Google Scholar]
- Gehrs BC, Smialowicz RJ. Alterations in the developing immune system of the F344 rat after perinatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin I. [correction of II]. Effects on the fetus and the neonate. Toxicology. 1997;122:219–228. doi: 10.1016/s0300-483x(97)00098-x. [DOI] [PubMed] [Google Scholar]
- Holladay SD, Smialowicz RJ. Development of the murine and human immune system: differential effects of immunotoxicants depend on time of exposure. Environ Health Perspect. 2000;108 3:463–473. doi: 10.1289/ehp.00108s3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer E. Negative selection--clearing out the bad apples from the T-cell repertoire. Nat Rev Immunol. 2003;3:383–391. doi: 10.1038/nri1085. [DOI] [PubMed] [Google Scholar]
- Sebzda E, Mariathasan S, Ohteki T, Jones R, Bachmann MF, Ohashi PS. Selection of the T cell repertoire. Annu Rev Immunol. 1999;17:829–874. doi: 10.1146/annurev.immunol.17.1.829. [DOI] [PubMed] [Google Scholar]
- Fisher MT, Nagarkatti M, Nagarkatti PS. Combined screening of thymocytes using apoptosis-specific cDNA array and promoter analysis yields novel gene targets mediating TCDD-induced toxicity. Toxicol Sci. 2004;78:116–124. doi: 10.1093/toxsci/kfh058. [DOI] [PubMed] [Google Scholar]
- Holladay SD, Mustafa A, Gogal RM., Jr Prenatal TCDD in mice increases adult autoimmunity. Reprod Toxicol. 2011 doi: 10.1016/j.reprotox.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha B, Vassalli P, Guy-Grand D. The extrathymic T-cell development pathway. Immunol Today. 1992;13:449–454. doi: 10.1016/0167-5699(92)90074-H. [DOI] [PubMed] [Google Scholar]
- Holsapple MP, Snyder NK, Wood SC, Morris DL. A review of 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced changes in immunocompetence: 1991 update. Toxicology. 1991;69:219–255. doi: 10.1016/0300-483x(91)90184-3. [DOI] [PubMed] [Google Scholar]
- Luster MI, Dean JH, Germolec DR. Consensus workshop on methods to evaluate developmental immunotoxicity. Environ Health Perspect. 2003;111:579–583. doi: 10.1289/ehp.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M, Sato S. B cell signaling and autoimmune diseases: CD19/CD22 loop as a B cell signaling device to regulate the balance of autoimmunity. J Dermatol Sci. 2007;46:1–9. doi: 10.1016/j.jdermsci.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Martin F, Chan AC. B cell immunobiology in disease: evolving concepts from the clinic. Annu Rev Immunol. 2006;24:467–496. doi: 10.1146/annurev.immunol.24.021605.090517. [DOI] [PubMed] [Google Scholar]
- Mustafa A, Holladay SD, Goff M, Witonsky SG, Kerr R, Reilly CM, Sponenberg DP, Gogal RM., Jr An enhanced postnatal autoimmune profile in 24 week-old C57BL/6 mice developmentally exposed to TCDD. Toxicol Appl Pharmacol. 2008;232:51–59. doi: 10.1016/j.taap.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnrot C, Prokopec KE, Rasbo K, Karlsson MC, Kleinau S. Marginal zone B cells are naturally reactive to collagen type II and are involved in the initiation of the immune response in collagen-induced arthritis. Cell Mol Immunol. 2011;8:296–304. doi: 10.1038/cmi.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi CM, Michael DJ, Diamond B. Cutting edge: expansion and activation of a population of autoreactive marginal zone B cells in a model of estrogen-induced lupus. J Immunol. 2001;167:1886–1890. doi: 10.4049/jimmunol.167.4.1886. [DOI] [PubMed] [Google Scholar]
- Klinman NR, Kline GH. The B-cell biology of aging. Immunol Rev. 1997;160:103–114. doi: 10.1111/j.1600-065x.1997.tb01031.x. [DOI] [PubMed] [Google Scholar]
- Zharhary D, Klinman NR. Antigen responsiveness of the mature and generative B cell populations of aged mice. J Exp Med. 1983;157:1300–1308. doi: 10.1084/jem.157.4.1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline GH, Hayden TA, Klinman NR. B cell maintenance in aged mice reflects both increased B cell longevity and decreased B cell generation. J Immunol. 1999;162:3342–3349. [PubMed] [Google Scholar]
- LeMaoult J, Szabo P, Weksler ME. Effect of age on humoral immunity, selection of the B-cell repertoire and B-cell development. Immunol Rev. 1997;160:115–126. doi: 10.1111/j.1600-065x.1997.tb01032.x. [DOI] [PubMed] [Google Scholar]
- Weksler ME, Szabo P. The effect of age on the B-cell repertoire. J Clin Immunol. 2000;20:240–249. doi: 10.1023/a:1006659401385. [DOI] [PubMed] [Google Scholar]
- Zheng B, Han S, Takahashi Y, Kelsoe G. Immunosenescence and germinal center reaction. Immunol Rev. 1997;160:63–77. doi: 10.1111/j.1600-065x.1997.tb01028.x. [DOI] [PubMed] [Google Scholar]
- Souvannavong V, Lemaire C, Andreau K, Brown S, Adam A. Age-associated modulation of apoptosis and activation in murine B lymphocytes. Mech Ageing Dev. 1998;103:285–299. doi: 10.1016/s0047-6374(98)00051-7. [DOI] [PubMed] [Google Scholar]
- Van der Put E, Sherwood EM, Blomberg BB, Riley RL. Aged mice exhibit distinct B cell precursor phenotypes differing in activation, proliferation and apoptosis. Exp Gerontol. 2003;38:1137–1147. doi: 10.1016/j.exger.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Cancro MP, Smith SH. Peripheral B cell selection and homeostasis. Immunol Res. 2003;27:141–148. doi: 10.1385/IR:27:2-3:141. [DOI] [PubMed] [Google Scholar]
- Dunn-Walters DK, Ademokun AA. B cell repertoire and ageing. Curr Opin Immunol. 2010;22:514–520. doi: 10.1016/j.coi.2010.04.009. [DOI] [PubMed] [Google Scholar]
- Johnson SA, Rozzo SJ, Cambier JC. Aging-dependent exclusion of antigen-inexperienced cells from the peripheral B cell repertoire. J Immunol. 2002;168:5014–5023. doi: 10.4049/jimmunol.168.10.5014. [DOI] [PubMed] [Google Scholar]
- Krabbe KS, Pedersen M, Bruunsgaard H. Inflammatory mediators in the elderly. Exp Gerontol. 2004;39:687–699. doi: 10.1016/j.exger.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Lazuardi L, Jenewein B, Wolf AM, Pfister G, Tzankov A, Grubeck-Loebenstein B. Age-related loss of naive T cells and dysregulation of T-cell/B-cell interactions in human lymph nodes. Immunology. 2005;114:37–43. doi: 10.1111/j.1365-2567.2004.02006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Stedra J, Cerny J. Relative contribution of T and B cells to hypermutation and selection of the antibody repertoire in germinal centers of aged mice. J Exp Med. 1996;183:959–970. doi: 10.1084/jem.183.3.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogal RM, Jr, Prater MR, Smith BJ, Johnson MS, Holladay SD. Bilateral dissected spleens and thymuses in rodents exhibit homogeneity in leukocyte markers. Toxicology. 2001;157:217–223. doi: 10.1016/s0300-483x(00)00351-6. [DOI] [PubMed] [Google Scholar]
- Klein AB, Witonsky SG, Ahmed SA, Holladay SD, Gogal RM, Jr, Link L, Reilly CM. Impact of different cell isolation techniques on lymphocyte viability and function. J Immunoassay Immunochem. 2006;27:61–76. doi: 10.1080/15321810500403755. [DOI] [PubMed] [Google Scholar]
- Weber H, Birnbaum LS. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) and 2,3,7,8-tetrachlorodibenzofuran (TCDF) in pregnant C57BL/6N mice: distribution to the embryo and excretion. Arch Toxicol. 1985;57:159–162. doi: 10.1007/BF00290880. [DOI] [PubMed] [Google Scholar]
- Weber H, Harris MW, Haseman JK, Birnbaum LS. Teratogenic potency of TCDD, TCDF and TCDD-TCDF combinations in C57BL/6N mice. Toxicol Lett. 1985;26:159–167. doi: 10.1016/0378-4274(85)90161-4. [DOI] [PubMed] [Google Scholar]
- Neubert R, Jacob-Muller U, Stahlmann R, Helge H, Neubert D. Polyhalogenated dibenzo-p-dioxins and dibenzofurans and the immune system. 1. Effects on peripheral lymphocyte subpopulations of a non-human primate (Callithrix jacchus) after treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) Arch Toxicol. 1990;64:345–359. doi: 10.1007/BF01973455. [DOI] [PubMed] [Google Scholar]
- Blaylock BL, Holladay SD, Comment CE, Heindel JJ, Luster MI. Exposure to tetrachlorodibenzo-p-dioxin (TCDD) alters fetal thymocyte maturation. Toxicol Appl Pharmacol. 1992;112:207–213. doi: 10.1016/0041-008x(92)90189-y. [DOI] [PubMed] [Google Scholar]
- Esser C. Dioxins and the immune system: mechanisms of interference. A meeting report. Int Arch Allergy Immunol. 1994;104:126–130. doi: 10.1159/000236719. [DOI] [PubMed] [Google Scholar]
- Holladay SD, Lindstrom P, Blaylock BL, Comment CE, Germolec DR, Heindell JJ, Luster MI. Perinatal thymocyte antigen expression and postnatal immune development altered by gestational exposure to tetrachlorodibenzo-p-dioxin (TCDD) Teratology. 1991;44:385–393. doi: 10.1002/tera.1420440405. [DOI] [PubMed] [Google Scholar]
- Lundberg K, Dencker L, Gronvik KO. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) inhibits the activation of antigen-specific T-cells in mice. Int J Immunopharmacol. 1992;14:699–705. doi: 10.1016/0192-0561(92)90133-6. [DOI] [PubMed] [Google Scholar]
- Oughton JA, Pereira CB, DeKrey GK, Collier JM, Frank AA, Kerkvliet NI. Phenotypic analysis of spleen, thymus, and peripheral blood cells in aged C57B1/6 mice following long-term exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Fundam Appl Toxicol. 1995;25:60–69. doi: 10.1006/faat.1995.1040. [DOI] [PubMed] [Google Scholar]
- Nagayama J, Tsuji H, Iida T, Nakagawa R, Matsueda T, Hirakawa H, Yanagawa T, Fukushige J, Watanabe T. Immunologic effects of perinatal exposure to dioxins, PCBs and organochlorine pesticides in Japanese infants. Chemosphere. 2007;67:S393–398. doi: 10.1016/j.chemosphere.2006.05.134. [DOI] [PubMed] [Google Scholar]
- Tarkowski M, Kur B, Nocun M, Sitarek K. Perinatal exposure of mice to TCDD decreases allergic sensitisation through inhibition of IL-4 production rather than T regulatory cell-mediated suppression. Int J Occup Med Environ Health. 2010;23:75–83. doi: 10.2478/v.10001-010-0006-7. [DOI] [PubMed] [Google Scholar]
- Vorderstrasse BA, Cundiff JA, Lawrence BP. A dose-response study of the effects of prenatal and lactational exposure to TCDD on the immune response to influenza a virus. J Toxicol Environ Health A. 2006;69:445–463. doi: 10.1080/15287390500246985. [DOI] [PubMed] [Google Scholar]
- Haas C, Ryffel B, Le Hir M. IFN-gamma is essential for the development of autoimmune glomerulonephritis in MRL/Ipr mice. J Immunol. 1997;158:5484–5491. [PubMed] [Google Scholar]
- Campbell IL, Kay TW, Oxbrow L, Harrison LC. Essential role for interferon-gamma and interleukin-6 in autoimmune insulin-dependent diabetes in NOD/Wehi mice. J Clin Invest. 1991;87:739–742. doi: 10.1172/JCI115055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Haneji N, Hamano H. Cytokine gene expression and autoantibody production in Sjogren's syndrome of MRL/lpr mice. Autoimmunity. 1996;23:269–277. doi: 10.3109/08916939608995349. [DOI] [PubMed] [Google Scholar]
- Billiau A. Interferon-gamma in autoimmunity. Cytokine Growth Factor Rev. 1996;7:25–34. doi: 10.1016/1359-6101(96)00004-4. [DOI] [PubMed] [Google Scholar]
- Pietschmann P, Gollob E, Brosch S, Hahn P, Kudlacek S, Willheim M, Woloszczuk W, Peterlik M, Tragl KH. The effect of age and gender on cytokine production by human peripheral blood mononuclear cells and markers of bone metabolism. Exp Gerontol. 2003;38:1119–1127. doi: 10.1016/s0531-5565(03)00189-x. [DOI] [PubMed] [Google Scholar]
- Finch CE, Crimmins EM. Inflammatory exposure and historical changes in human life-spans. Science. 2004;305:1736–1739. doi: 10.1126/science.1092556. [DOI] [PubMed] [Google Scholar]
- Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- Hagiwara E, Gourley MF, Lee S, Klinman DK. Disease severity in patients with systemic lupus erythematosus correlates with an increased ratio of interleukin-10:interferon-gamma-secreting cells in the peripheral blood. Arthritis Rheum. 1996;39:379–385. doi: 10.1002/art.1780390305. [DOI] [PubMed] [Google Scholar]
- Horwitz DA, Gray JD, Behrendsen SC, Kubin M, Rengaraju M, Ohtsuka K, Trinchieri G. Decreased production of interleukin-12 and other Th1-type cytokines in patients with recent-onset systemic lupus erythematosus. Arthritis Rheum. 1998;41:838–844. doi: 10.1002/1529-0131(199805)41:5<838::AID-ART10>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Park YB, Lee SK, Kim DS, Lee J, Lee CH, Song CH. Elevated interleukin-10 levels correlated with disease activity in systemic lupus erythematosus. Clin Exp Rheumatol. 1998;16:283–288. [PubMed] [Google Scholar]
- Fine JS, Gasiewicz TA, Fiore NC, Silverstone AE. Prothymocyte activity is reduced by perinatal 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure. J Pharmacol Exp Ther. 1990b;255:128–132. [PubMed] [Google Scholar]
- Mustafa A, Holladay SD, Goff M, Witonsky S, Kerr R, Weinstein DA, Karpuzoglu-Belgin E, Gogal RM., Jr Developmental exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin alters postnatal T cell phenotypes and T cell function and exacerbates autoimmune lupus in 24-week-old SNF1 mice. Birth Defects Res A Clin Mol Teratol. 2009;85:828–836. doi: 10.1002/bdra.20603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luster MI, Boorman GA, Dean JH, Harris MW, Luebke RW, Padarathsingh ML, Moore JA. Examination of bone marrow, immunologic parameters and host susceptibility following pre- and postnatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) Int J Immunopharmacol. 1980;2:301–310. doi: 10.1016/0192-0561(80)90030-2. [DOI] [PubMed] [Google Scholar]
- Wyman A, Lavin AL, Wilding GE, Gasiewicz TA. 2,3,7,8-tetrachlorodibenzo-p-dioxin does not directly alter the phenotype of maturing B cells in a murine coculture system. Toxicol Appl Pharmacol. 2002;180:164–177. doi: 10.1006/taap.2002.9396. [DOI] [PubMed] [Google Scholar]
- Monroe JG, Bannish G, Fuentes-Panana EM, King LB, Sandel PC, Chung J, Sater R. Positive and negative selection during B lymphocyte development. Immunol Res. 2003;27:427–442. doi: 10.1385/IR:27:2-3:427. [DOI] [PubMed] [Google Scholar]
- Masten SA, Shiverick KT. The Ah receptor recognizes DNA binding sites for the B cell transcription factor, BSAP: a possible mechanism for dioxin-mediated alteration of CD19 gene expression in human B lymphocytes. Biochem Biophys Res Commun. 1995;212:27–34. doi: 10.1006/bbrc.1995.1931. [DOI] [PubMed] [Google Scholar]
- Tian Y, Ke S, Denison MS, Rabson AB, Gallo MA. Ah receptor and NF-kappaB interactions, a potential mechanism for dioxin toxicity. J Biol Chem. 1999;274:510–515. doi: 10.1074/jbc.274.1.510. [DOI] [PubMed] [Google Scholar]
- Sandel PC, Monroe JG. Negative selection of immature B cells by receptor editing or deletion is determined by site of antigen encounter. Immunity. 1999;10:289–299. doi: 10.1016/s1074-7613(00)80029-1. [DOI] [PubMed] [Google Scholar]
- Loder F, Mutschler B, Ray RJ, Paige CJ, Sideras P, Torres R, Lamers MC, Carsetti R. B cell development in the spleen takes place in discrete steps and is determined by the quality of B cell receptor-derived signals. J Exp Med. 1999;190:75–89. doi: 10.1084/jem.190.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allman D, Pillai S. Peripheral B cell subsets. Curr Opin Immunol. 2008;20:149–157. doi: 10.1016/j.coi.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin F, Kearney JF. B-cell subsets and the mature preimmune repertoire. Marginal zone and B1 B cells as part of a “natural immune memory”. Immunol Rev. 2000;175:70–79. [PubMed] [Google Scholar]
- Batten M, Groom J, Cachero TG, Qian F, Schneider P, Tschopp J, Browning JL, Mackay F. BAFF mediates survival of peripheral immature B lymphocytes. J Exp Med. 2000;192:1453–1466. doi: 10.1084/jem.192.10.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddaert J, Huong DL, Amoura Z, Wechsler B, Godeau P, Piette JC. Late-onset systemic lupus erythematosus: a personal series of 47 patients and pooled analysis of 714 cases in the literature. Medicine (Baltimore) 2004;83:348–359. doi: 10.1097/01.md.0000147737.57861.7c. [DOI] [PubMed] [Google Scholar]
- Cervera R, Khamashta MA, Font J, Sebastiani GD, Gil A, Lavilla P, Domenech I, Aydintug AO, Jedryka-Goral A, de Ramon E, et al. Systemic lupus erythematosus: clinical and immunologic patterns of disease expression in a cohort of 1,000 patients. The European Working Party on Systemic Lupus Erythematosus. Medicine (Baltimore) 1993;72:113–124. [PubMed] [Google Scholar]
- Cooper GS, Parks CG, Treadwell EL, St Clair EW, Gilkeson GS, Cohen PL, Roubey RA, Dooley MA. Differences by race, sex and age in the clinical and immunologic features of recently diagnosed systemic lupus erythematosus patients in the southeastern United States. Lupus. 2002;11:161–167. doi: 10.1191/0961203302lu161oa. [DOI] [PubMed] [Google Scholar]
- de Carvalho JF, do Nascimento AP, Testagrossa LA, Barros RT, Bonfa E. Male gender results in more severe lupus nephritis. Rheumatol Int. 2010;30:1311–1315. doi: 10.1007/s00296-009-1151-9. [DOI] [PubMed] [Google Scholar]
- Formiga F, Moga I, Pac M, Mitjavila F, Rivera A, Pujol R. Mild presentation of systemic lupus erythematosus in elderly patients assessed by SLEDAI. SLE Disease Activity Index. Lupus. 1999;8:462–465. doi: 10.1177/096120339900800609. [DOI] [PubMed] [Google Scholar]
- Hashimoto H, Tsuda H, Hirano T, Takasaki Y, Matsumoto T, Hirose S. Differences in clinical and immunological findings of systemic lupus erythematosus related to age. J Rheumatol. 1987;14:497–501. [PubMed] [Google Scholar]
- Voulgari PV, Katsimbri P, Alamanos Y, Drosos AA. Gender and age differences in systemic lupus erythematosus. A study of 489 Greek patients with a review of the literature. Lupus. 2002;11:722–729. doi: 10.1191/0961203302lu253oa. [DOI] [PubMed] [Google Scholar]
- Pu SJ, Luo SF, Wu YJ, Cheng HS, Ho HH. The clinical features and prognosis of lupus with disease onset at age 65 and older. Lupus. 2000;9:96–100. doi: 10.1191/096120300678828109. [DOI] [PubMed] [Google Scholar]
- Chang DM, Chang CC, Kuo SY, Chu SJ, Chang ML. The clinical features and prognosis of male lupus in Taiwan. Lupus. 1998;7:462–468. doi: 10.1191/096120398678920479. [DOI] [PubMed] [Google Scholar]
- Kaufman LD, Gomez-Reino JJ, Heinicke MH, Gorevic PD. Male lupus: retrospective analysis of the clinical and laboratory features of 52 patients, with a review of the literature. Semin Arthritis Rheum. 1989;18:189–197. doi: 10.1016/0049-0172(89)90061-9. [DOI] [PubMed] [Google Scholar]
- Maddison PJ. Systemic lupus erythematosus in the elderly. J Rheumatol Suppl. 1987;14 13:182–187. [PubMed] [Google Scholar]
- Bagavant H, Deshmukh US, Gaskin F, Fu SM. Lupus glomerulonephritis revisited 2004: autoimmunity and end-organ damage. Scand J Immunol. 2004;60:52–63. doi: 10.1111/j.0300-9475.2004.01463.x. [DOI] [PubMed] [Google Scholar]
- Belostocki KB, Paget SA. Inflammatory rheumatologic disorders in the elderly. Unusual presentations, altered outlooks. Postgrad Med. 2002;111:72–74. 77–78, 81–73. doi: 10.3810/pgm.2002.04.1154. [DOI] [PubMed] [Google Scholar]
- Boren E, Gershwin ME. Inflamm-aging: autoimmunity, and the immune-risk phenotype. Autoimmun Rev. 2004;3:401–406. doi: 10.1016/j.autrev.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Catoggio LJ, Skinner RP, Smith G, Maddison PJ. Systemic lupus erythematosus in the elderly: clinical and serological characteristics. J Rheumatol. 1984;11:175–181. [PubMed] [Google Scholar]
- Ho CT, Mok CC, Lau CS, Wong RW. Late onset systemic lupus erythematosus in southern Chinese. Ann Rheum Dis. 1998;57:437–440. doi: 10.1136/ard.57.7.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padovan M, Govoni M, Castellino G, Rizzo N, Fotinidi M, Trotta F. Late onset systemic lupus erythematosus: no substantial differences using different cut-off ages. Rheumatol Int. 2007;27:735–741. doi: 10.1007/s00296-006-0284-3. [DOI] [PubMed] [Google Scholar]
- Stacy S, Krolick KA, Infante AJ, Kraig E. Immunological memory and late onset autoimmunity. Mech Ageing Dev. 2002;123:975–985. doi: 10.1016/s0047-6374(02)00035-0. [DOI] [PubMed] [Google Scholar]
- Miller RA. The aging immune system: primer and prospectus. Science. 1996;273:70–74. doi: 10.1126/science.273.5271.70. [DOI] [PubMed] [Google Scholar]
- Prelog M. Aging of the immune system: a risk factor for autoimmunity? Autoimmun Rev. 2006;5:136–139. doi: 10.1016/j.autrev.2005.09.008. [DOI] [PubMed] [Google Scholar]