

Abstract
Coordinate regulation of ribosomal protein (RP) genes is key for controlling cell growth. In yeast, it is unclear how this regulation achieves the required equimolar amounts of the different RP components, given that some RP genes exist in duplicate copies, while others have only one copy. Here, we tested whether the solution to this challenge is partly encoded within the DNA sequence of the RP promoters, by fusing 110 different RP promoters to a fluorescent gene reporter, allowing us to robustly detect differences in their promoter activities that are as small as ∼10%. We found that single-copy RP promoters have significantly higher activities, suggesting that proper RP stoichiometry is indeed partly encoded within the RP promoters. Notably, we also partially uncovered how this regulation is encoded by finding that RP promoters with higher activity have more nucleosome-disfavoring sequences and characteristic spatial organizations of these sequences and of binding sites for key RP regulators. Mutations in these elements result in a significant decrease of RP promoter activity. Thus, our results suggest that intrinsic (DNA-dependent) nucleosome organization may be a key mechanism by which genomes encode biologically meaningful promoter activities. Our approach can readily be applied to uncover how transcriptional programs of other promoters are encoded.
The yeast genome contains 137 genes that encode for ribosomal proteins (RP), of which 19 encode a unique RP, and 118 (59 pairs) each encode a duplicated RP that exists in two copies. Together, RP transcription accounts for ∼50% of the transcripts produced by RNA polymerase II (Warner 1999). Transcription of RP genes is coordinately regulated in response to different growth stimuli and environmental conditions, and this coordinate regulation is a key mechanism by which cells adjust their protein synthesis capacity to physiological needs (Ju and Warner 1994; Gasch et al. 2000; Causton et al. 2001). The mRNA levels of RP genes are also under tight regulation, since abnormal levels of RP transcription lead to reduced fitness (Li et al. 1996; Deutschbauer et al. 2005). Several transcriptional regulators of RP genes were identified and shown to associate directly or indirectly with many RP promoters, including Rap1 (Shore 1994; Lieb et al. 2001; Lee et al. 2002), Fhl1 (Lee et al. 2002; Schawalder et al. 2004; Wade et al. 2004), Ifh1 (Schawalder et al. 2004; Wade et al. 2004), Sfp1 (Marion et al. 2004), Crf1 (Martin et al. 2004), and Hmo1 (Hall et al. 2006), and binding sites for Rap1 are required for proper RP transcription (Woudt et al. 1986; Moehle and Hinnebusch 1991; Shore 1994). However, a key unresolved question regarding RP regulation concerns the ability of yeast cells to achieve the required equimolar amounts of the different RP components of the ribosome (Spahn et al. 2001), given the copy-number differences that exist among the single-copy and duplicated RP genes (Warner 1999).
Measurements of mRNA abundance of the RPs using DNA microarrays (Holstege et al. 1998) or RNA-seq (Nagalakshmi et al. 2008) can reveal whether equimolar amounts of RP genes are achieved at the transcript level. However, since they measure the combined effect on mRNA levels of transcription, genomic context, degradation, and other post-transcriptional effects, these technologies cannot determine the relative contribution of each regulatory mechanism. In addition, since RNA-seq and microarrays are based on sequencing or hybridization of sequences specific to each measured gene, respectively, the resulting measurements may have gene-specific systematic biases (Oshlack and Wakefield 2009). This is particularly problematic in the case of RP genes, since transcripts of duplicated RPs are highly similar to each other at the sequence level, which makes determining their expression levels by sequence-based methods problematic. The same limitations apply to measurements based on quantitative real-time PCR.
Approaches based on fusion of promoters to fluorescent reporters can be used to determine the relative contribution of transcription to the resulting mRNA levels, since they provide direct real-time measurements of promoter activity with a high accuracy of ∼10%, in a way that is independent of the sequence of the measured gene (Kalir et al. 2001). Variants of this approach were successfully applied to reveal ordered activation of genes in various pathways in bacteria (Kalir et al. 2001; Zaslaver et al. 2004) and to generate libraries of synthetic promoters in bacteria (Cox et al. 2007) and yeast (Ligr et al. 2006; Murphy et al. 2007; Gertz et al. 2009), which provided much insight into the rules that underlie combinatorial cis-regulation. However, since they require genetic engineering of one strain for each tested promoter, these approaches are harder to implement. To date, there is no large-scale library of fluorescent fusions of natural promoters for any eukaryote.
Here, we fused 110 of the 137 RP promoters in yeast to a yellow fluorescent protein reporter (YFP), allowing us to measure promoter activities for most RP genes in living cells and with high accuracy and temporal resolution. Each promoter was directly fused to YFP and genomically integrated into a different strain at a fixed genomic location, thereby controlling for both post-translational and genomic context effects. Thus, since all 110 strains are isogenic except for the specific RP promoter integrated into each of them, differences between their measured YFP fluorescence are attributable only to differences in the sequences of the integrated RP promoter. This property makes our system highly suitable for studying how transcriptional control of RP genes is encoded within their promoters. We note, however, that since locations of transcription start sites are only partially known, we extended each promoter up to the translation start site of its corresponding RP gene, and thus, some of the differences in the YFP of the various RP promoters may also be due to the differential effects that their 5′ untranslated regions have on the stability and translation of the YFP mRNA. Nevertheless, our analyses and subsequent mutational experiments unravel some of the sequence rules by which RP promoters encode their measured promoter activities. Our findings suggest that DNA-encoded nucleosome organization of promoters is an important determinant of this encoding, and that the yeast genome utilizes this encoding in part to compensate for the copy-number differences that exist between its duplicated and single-copy RP genes.
Results
Single-copy RP genes have higher mRNA levels than duplicated RPs
We first tested whether already at the transcript level, yeast cells compensate for the gene copy differences that exist between its duplicated and single-copy RP genes. Since RPs are needed in equimolar within the ribosome (Spahn et al. 2001), and since pairs of duplicated RP genes encode nearly identical proteins and are thus functionally redundant within the ribosome (Lucioli et al. 1988; Rotenberg et al. 1988), we may expect their transcripts to have lower expression levels than transcripts of RP genes that have only one copy in the yeast genome. To test this hypothesis, we obtained mRNA abundance measurements from three different DNA microarray technologies (Holstege et al. 1998; David et al. 2006; Lipson et al. 2009) and three different RNA-seq techniques (Nagalakshmi et al. 2008; Lipson et al. 2009; Yassour et al. 2009). Indeed, in all six data sets, transcripts of single-copy RP genes are expressed at significantly higher levels than transcripts of duplicated RP genes (Fig. 1, P-values range from P < 10−9 to P < 0.02). Although all of these data sets are publicly available, we are not aware of studies that reported such a finding. This result suggests that yeast compensates for the copy-number differences that exist among its RP genes at the transcript level in order to achieve the required stoichiometry of the RPs.
Figure 1.

Single-copy RP genes have higher mRNA levels than duplicated RPs. For several data sets, a ranking of their measured RP activities is shown, with single-copy RPs marked as orange bars and duplicated RPs marked as blue bars. RPs for which a measurement was missing in a given data set appear as gray bars and are sorted to the bottom of the list. The ranking is shown for our library of RP promoter fusions (leftmost column), for transcription rate measurements using DNA microarrays (Holstege et al. 1998) (second column), and for mRNA abundance measurements using DNA microarrays (David et al. 2006; Lipson et al. 2009) or RNA-seq (Nagalakshmi et al. 2008; Lipson et al. 2009; Yassour et al. 2009). For each data set, the rank-sum test P-value that tests whether the ranking of single-copy RPs is higher than that of duplicated RPs is shown.
Obtaining accurate measurements of RP promoter activities
Next, we sought to test whether these higher expression levels of the single-copy RP genes are encoded within their promoter regions, and if so, how is it achieved at the sequence level. To this end, we devised an experimental system for accurately measuring RP promoter activities based on fusing promoters to fluorescent reporters. We designed a single master strain and separately integrated into it different RP promoters, generating one strain for every RP promoter (Fig. 2A). To eliminate variability due to genomic context, all of the promoters were integrated into the same genomic location. At this same location, we also integrated into the master strain the promoter for TEF2, a translation elongation factor, followed by a red fluorescent protein (mCherry). Thus, the activity of mCherry should be the same across all RP promoter strains, allowing us to use the mCherry measurements to estimate the experimental error of our system, and to identify mutant strains with general deficiencies in their transcriptional activity.
Figure 2.

Overview of our experimental system. (A) Illustration of the master strain into which we integrated all RP promoters. At a fixed chromosomal location, the master strain contains a gene that encodes for a red fluorescent protein (mCherry), followed by the promoter for TEF2, and a gene that encodes for a yellow fluorescent protein (YFP). Every RP promoter is integrated into this strain, together with a selection marker, between the TEF2 promoter and the YFP gene. (B) Strains with different RP promoters have highly similar growth rates. Shown is the growth of 71 different RP promoter strains, measured as optical density (OD). Measurements were obtained from a single 96-well plate, with glucose-rich media and a small number of cells from each strain inserted into each well at time zero. The exponential growth phase is indicated (vertical dashed gray lines). (C) Same as in B, but where the measurements correspond to mCherry intensity. Note the small variability in the intensity of mCherry, which is driven by the same control promoter across the different strains. (D) Same as in C, but where the measurements correspond to YFP intensity. Note the large variability in the intensity of YFP, which is driven by a different RP promoter in each strain. (E) Same as in C, but where the mCherry production rate of each strain is shown, measured as the difference between the mCherry levels of different timepoints, divided by the integral of the OD during the same time period (see Methods). (F) Same as in E, but where the YFP production rate of each strain is shown.
We integrated each promoter directly upstream of a yellow fluorescent protein (YFP). We chose YFP over GFP, since yeast cells autofluoresce much less at this wavelength, thereby increasing our measurement sensitivity (Supplemental Fig. 1). The direct fusion of promoters to YFP results in the exact same protein being produced from every promoter, thereby eliminating post-translational regulation as a source of variability in YFP levels. In addition, the YFP is stable and long-lived, such that the difference in YFP levels across time provides a direct measure of the amount of YFP produced (Fig. 2). As the promoter sequence of each RP gene, we took the genomic region located between its translation start site (TrSS) and the end of its upstream neighboring gene (for all promoter sequences see Supplemental Data1). We used translation start sites, because transcription start sites are not accurately defined in yeast. Thus, aside from a short 5′ untranslated region that may affect the stability and translation of the YFP mRNA, all promoters should produce the same transcript. This design removes post-transcriptional regulation that stems from the coding region and 3′ untranslated region of the transcripts as a source of variability between strains.
To efficiently generate large-scale libraries of native promoters, we devised an automated, bacteria-free robotic procedure for cloning promoters into our master strain, which is capable of constructing a library of 96 different native promoters within 2 wk, with two additional weeks needed for clone validation (Supplemental Fig. 2). We applied this procedure to all 137 RP promoters, and successfully generated promoter strains for 110 of them, which we subsequently validated by sequencing the inserted promoter.
We measured the activity of these different RP promoter strains in multiwell plates by transferring a small number of cells from each strain into fresh medium of the tested growth condition and continuously tracking the growth (measured as optical density [OD]), mCherry, and YFP fluorescence of the growing cell population at high temporal resolution, using a robotically automated plate fluorometer (Fig. 2B,C,D). To extract promoter activities from the resulting measurements, we developed a data processing pipeline that detects and removes outliers, subtracts the measured autofluorescence of both the yeast cells and the growth medium, averages replicate measurements, and identifies the different growth phases of the cell population (Supplemental Fig. 3). As a single measure of promoter activity, we then take the amount of YFP fluorescence produced during the exponential growth phase, divided by the integral of the OD during the same time period. This results in a measure, termed “promoter activity,” which represents the average rate of YFP production from each promoter, per cell per second, during the exponential phase (Fig. 2E,F). (For all promoter activities see Supplemental Data1).
We performed several tests to gauge the accuracy and sensitivity of our system. First, as expected, we found that the growth curves of all strains are nearly identical (Fig. 2B). Second, we found that the YFP levels of independent clones of the same promoter sequence are indistinguishable from those of replicate measurements of the same clone, indicating that our library construction procedure does not introduce mutations that have global effects on transcription or translation (Supplemental Fig. 4). Third, we validated that signals measured in the YFP wavelength are not affected by the presence of the mCherry protein by showing that the YFP level of a strain that has mCherry is the same as that of a strain that does not have mCherry (Supplemental Fig. 5). Fourth, since the mCherry and YFP promoters are separated by 1317 bp of the selection marker, we asked whether higher promoter activities from the YFP promoter result in increased mCherry transcription through long-range interactions, but found essentially no correlation between the YFP and mCherry promoter activities across the different RP promoter strains (R = 0.05, Supplemental Fig. 6). Fifth, we confirmed that protein levels of the YFP are an accurate proxy for the corresponding mRNA levels by comparing the measured fluorescence of YFP to quantitative real-time PCR (qPCR) measurements of its mRNA, and finding good agreement between the two measurements (R = 0.96, Supplemental Fig. 7). This high correspondence between the protein and mRNA levels of YFP further suggests that at least within our promoter set, the different 5′ untranslated regions of each promoter have minor effects on YFP translation rates.
Finally, we used two measures to assess the experimental error of our system. First, we examined the promoter activity of mCherry, which should be the same across all promoter strains, and found that the average difference between any two strains was ∼5% (Fig. 2E; Supplemental Fig. 6). Second, using replicate measurements, we found that on average, the relative error in the estimated YFP promoter activity of an RP promoter is ∼2%, indicating that at a 95% confidence interval around the mean promoter activity we can distinguish between any two promoters whose activities differ by as little as ∼8% (Supplemental Fig. 8). Indeed, most RP promoters have significantly different promoter activities (Supplemental Fig. 9).
Taken together, our system provides measurements of promoter activity for most RP genes in living cells and with high accuracy, sensitivity, and temporal resolution, while controlling for genomic context, post-transcriptional, and post-translational effects, such that differences in the measured YFP activity of different RP promoters likely reflect true differences in their promoter activities.
Promoters of single-copy RP genes encode higher promoter activities
To compare the activities of promoters of single-copy RPs with those of duplicated RPs, we grew the different RP promoter strains in rich media with glucose, as well as in seven other growth conditions that span various carbon sources and environmental stresses. The correlations between the RP promoter activities across all growth conditions were high (R = 0.94–0.99, Supplemental Fig. 10), and we thus used the activities measured in glucose in all of our subsequent analyses. Notably, we found that promoters of single-copy RPs have significantly higher activities than promoters of duplicated RPs (2.22 ± 0.59 vs. 1.48 ± 0.5, P < 10−6) (Fig. 3A), and 13 of the 16 measured single-copy RP promoters are among the 24 (of 110) RP promoters with the highest activities.
Figure 3.

Promoters of duplicated RP genes encode lower promoter activities. (A) Shown is a histogram of promoter activities of the promoters whose corresponding RP gene exists in two duplicate copies in the yeast genome (blue) and another histogram for the activities of promoters whose corresponding gene exists in one copy (orange). Also shown is the T-test P-value of the difference between the two histograms (top left). (B) The promoter activity variability of RP promoters is significantly reduced when summing pairs of duplicated RP promoters. For each of the 41 pairs of duplicate RP genes for which we have measurements for the two corresponding promoters, we added the promoter activities of the two promoters and computed the coefficient of variation (CV) of promoter activities of the promoter set that consists of these combined promoters and of RP promoters that have a single copy in the yeast genome (red arrow, CV = 0.27). For comparison, we computed the same coefficient of variation of activities when the pair of promoters whose activities are added were chosen at random. A histogram of 107 such permutations is shown (blue), and the P-value of the real RP promoter pairing is indicated (top left).
To ask whether the lower promoter activities of the duplicated RPs may help to achieve equimolar amounts of RPs, we compared the promoter activity variability of RP promoters with the variability achieved when treating every pair of duplicated RPs as a single promoter, whose promoter activity is the sum of the activity of its two corresponding promoters. Notably, summing the promoter activities of duplicated RPs reduces the promoter activity variability of RP promoters by ∼30% (coefficient of variation 0.27 vs. 0.38, P < 10−5) (Fig. 3B), suggesting that RP promoter sequences have evolved in part to achieve the proper stoichiometry of RPs required within the ribosome. Note, however, that even after summing duplicate RPs, there still remains considerable variability (0.27). This remaining variability may be small enough to be tolerated by yeast cells. Alternatively, this variability may be further reduced at the post-transcriptional or post-translational level, although we find no such evidence, at least at the post-transcriptional level, since applying the above computation to RNA-seq data (Yassour et al. 2009) results in an mRNA level variability similar to the promoter activity variability obtained with our data (0.24 vs. 0.27).
If the duplicated RP promoters indeed have lower promoter activities, in part to achieve proper RP stoichiometry, then we may expect the combined promoter activity of a pair of duplicated RP promoters to be more important than the way in which the two activities are distributed between the two promoters. In this view, we may further expect that duplicated promoter pairs can diverge in their promoter activities and promoter sequences, as long as their combined activity is maintained. Indeed, we find that the promoter activities and, separately, the promoter sequences, of pairs of duplicated RP promoters are as similar to each other as are random pairings of RP promoters, and there is no correlation between the sequence similarity and promoter activity of duplicated RP promoters (Supplemental Fig. 11).
Together, these results suggest that the compensation that we observed at the transcript levels between duplicated and single-copy RPs is achieved in part by the DNA sequence of RP promoters through their encoding of higher activities for promoters of single-copy RPs.
Strong RP promoters have distinct organizations of transcription-factor binding sites
Since differences in RP promoter activity measured in our system are attributable only to differences in the sequence of the integrated RP promoters, we next sought to identify the sequence features that account for the measured activity differences. Thus, our goal was to explain, as best as possible and from DNA sequence alone, the measured RP promoter activities.
As a first step, we examined one possible partitioning of the RP promoters into three groups of promoters with either high (topmost 25%), intermediate (middle 50%), or low (bottommost 25%) promoter activities, with the boundary between the groups adjusted by one to two promoters based on the ability to experimentally distinguish promoter activities (Supplemental Fig. 9). Notably, although this partitioning was based solely on promoter activity, the resulting promoter groups differed from each other in the number and/or spatial organization of TATA boxes (Basehoar et al. 2004) and of binding sites for Rap1, Fhl1, and Sfp1, the key known transcriptional regulators of RP genes (Figs. 4, 5; Badis et al. 2008; Zhu et al. 2009). This suggests that the contribution of a factor to the overall promoter activity depends on the specific organization of its sites within the promoter.
Figure 4.

Detailed view of RP promoters and their associated sequence features. RP promoters are sorted according to their measured promoter activities (right bar graph), and for every promoter, shown are the locations of TATA boxes (pink circles), and of binding sites for Rap1 (red), Fhl1 (green), and Sfp1 (blue). Sites for TATA are taken as defined in Basehoar et al. (2004). Sites for the three other factors were computed using their experimentally derived binding specificities (Badis et al. 2008; Zhu et al. 2009), and are shown above the binding site threshold determined by our computational model (see Methods, thresholds are Rap1 = 4.4, Fhl1 = 7.6, Sfp1 = 7.1). For Rap1 and Fhl1, sites are only shown in one of the two possible site orientations. In addition, shown is the per-basepair nucleosome occupancy of every promoter (occupancy is shown in a white to black scale, with white corresponding to no occupancy and black to full occupancy), predicted using a computational model of nucleosome sequence preferences (Kaplan et al. 2009). Also shown is a matrix (left) summary of the number of factor sites that appear in every RP promoter (counts for Rap1 are only shown for the 400 bp upstream of the TrSS; for Fhl1 and Sfp1, 300 bp; and for TATA, 200 bp), along with a column representing whether the corresponding RP gene exists in a single-copy in the yeast genome (first column, black), and whether it is an essential gene (second column, gray). Two horizontal dashed red lines indicate a partitioning of RP promoters into three groups of promoters with either high, intermediate, or low promoter activities. The length of each native promoter is indicated (cyan vertical line) if it is shorter than 600 bp. For locations of transcription start sites, see Supplemental Figure 18.
Figure 5.

RP promoters with high promoter activities have distinct organizations of transcription-factor binding sites. (A) For each group of RP promoters with high (red), intermediate (green), and low (blue) promoter activities, defined as in Figure 4, the average number of Fhl1 binding sites per promoter as a function of the distance from the translation start site is shown. The plots show a moving average using a window of 50 bp. Fhl1 sites are shown in only one of the two possible orientations and using the binding strength threshold from Figure 4 (see Supplemental Fig. 12 for plots using many possible thresholds). (B) Same as in A, for Sfp1 binding sites, but where sites are counted in both orientations (see Supplemental Fig. 13 for many site thresholds). (C) Same as in A, for Rap1 sites (see Supplemental Fig. 14 for many thresholds). (D) Same as in A, for TATA boxes (see Supplemental Fig. 15 for many thresholds).
Regarding Fhl1, ChIP-chip experiments revealed that it associates almost exclusively with RP promoters, suggesting that it is a key regulator of RP genes (Harbison et al. 2004; Schawalder et al. 2004; Wade et al. 2004). However, since the association of Fhl1 to some RP promoters depends on Rap1 (Wade et al. 2004), and since a computational analysis of Fhl1-associated promoters identified the Rap1 binding site motif instead of the motif for Fhl1 (Harbison et al. 2004), it is unclear how many RP promoters are directly bound by Fhl1. Two independent studies have recently identified novel binding specificities for Fhl1 using an in vitro approach that measures binding to all possible 8-mers (Badis et al. 2008; Zhu et al. 2009). Using these new binding specificities, we found relatively few Fhl1 binding sites across the RP promoters, but these sites were strongly enriched in RP promoters with high promoter activities, in one of the two possible orientations of the site, and in a specific location within the promoters (Figs. 4, 5A). For example, at some threshold of Fhl1 binding strength, 27% of the RP promoters with high promoter activity have Fhl1 sites at specific locations (175 ± 47 bp upstream of the TrSS) compared with only 3.5% occurrences in the promoters with intermediate or low activities, where the sites are also more dispersed (224 ± 190 bp, P < 10−4; results are not sensitive to threshold selection) (Supplemental Fig. 12). These results suggest that Fhl1 binds relatively few RP promoters directly, but that direct binding of Fhl1 strongly increases transcription, in a manner that likely depends on the orientation of the Fhl1 site and on its distance from the transcription start site.
Sfp1 has also been shown to regulate RP transcription (Marion et al. 2004), but like Fhl1, a computational analysis of Sfp1-associated promoters identified the Rap1 motif instead of the Sfp1 motif (Harbison et al. 2004; MacIsaac et al. 2006), leaving open the question of which RP promoters are bound directly by Sfp1. Notably, using the Sfp1 motif determined in vitro (Badis et al. 2008; Zhu et al. 2009), we found that similar to Fhl1, RP promoters with high promoter activities are strongly enriched with Sfp1 sites at specific locations within the promoters, though in this case Sfp1 appears to directly bind more promoters than Fhl1 and the enrichment does not depend on the orientation of the site (Figs. 4, 5B). For example, at some Sfp1 binding threshold, we find localized sites (180 ± 120 bp upstream of the TrSS) for Sfp1 at 73% of the RP promoters with high promoter activities, compared with fewer (35% at 200 ± 200 bp) at RP promoters with intermediate and low-transcription activities, respectively (results are not sensitive to threshold selection; Supplemental Fig. 13). These results suggest that Sfp1 may be a direct and relatively prevalent transcriptional activator of many RP genes.
Unlike Fhl1 and Sfp1, which are strongly enriched in promoters with high promoter activities, we found that most RP promoters, including those with intermediate and low activities, have binding sites for Rap1, and these are strongly enriched in one orientation, consistent with previous studies (Lieb et al. 2001; Harbison et al. 2004). However, although the number of promoters with Rap1 sites does not differ between RP promoters with different promoter activities, the spatial organization of Rap1 sites does differ, such that Rap1 sites in promoters with high activities are significantly closer to the TrSS compared with promoters from the other groups, especially the one with intermediate promoter activities (P < 10−4) (Figs. 4, 5C). For example, at some threshold of the Rap1 motif, Rap1 sites in promoters with high promoter activities are located, on average, 220 ± 80 bp from the TrSS, compared with a spatial distribution of 310 ± 73 bp in promoters with intermediate activities (P < 10−7; results are not sensitive to threshold selection; Supplemental Fig. 14). The association of different promoter activities with distinct architectures of Rap1 sites suggests that the contribution of Rap1 to transcription depends on the precise location and organization of its sites. However, unlike Fhl1 and Sfp1, whose enrichment in the strong promoters suggests a simple mechanism by which their contribution increases with their number of sites, no analogous simple mechanism can explain the contribution of Rap1 to the overall promoter activity.
Finally, since TATA boxes are important for assembling the transcription machinery at promoters, we examined their distribution within RP promoters. Notably, although ∼20% of the promoters in yeast have TATA boxes in the 200 bp upstream of the TrSS (Basehoar et al. 2004), only nine (8%) RP promoters have TATA boxes, but these are strongly enriched in the high-activity promoters, with TATA boxes appearing in five of the eight (63%) strongest RP promoters (Figs. 4, 5D) (P < 10−4; results are not sensitive to threshold selection; Supplemental Fig. 15). These results suggest that the appearance of a TATA box within an RP promoter increases its promoter activity, presumably through enhanced recruitment of TBP to the promoter.
Taken together, the distinct organizations of transcription-factor binding sites that we find, solely from sequence analysis, between promoters with high and low promoter activities may partly explain how the measured activities of RP promoters are encoded in their DNA sequence and provide insights into the way by which each of these factors contributes to transcription. The mechanism by which Fhl1, Sfp1, and TATA boxes contribute appears to be relatively simple, such that, in general, more sites for these factors in proximity to the TrSS likely result in higher promoter activities. In contrast, although we find significant differences in the distribution of Rap1 sites between promoters with high and low promoter activities, the mechanism by which these differences contribute differentially to transcription is unclear.
Stronger RP promoters are less favorable for nucleosome formation
As another type of sequence element that may influence promoter activity, we examined the intrinsic (DNA-encoded) nucleosome organization of RP promoters. Since nucleosomes occlude their wrapped DNA from access to most transcription factors (Kornberg and Lorch 1999), we asked whether RP promoters that are more energetically favorable for nucleosome wrapping may be less accessible to binding by transcription factors and by the transcription initiation machinery, and thus have lower promoter activity. In vitro (Polach and Widom 1995), and for two yeast genes in vivo (Svaren et al. 1994; Iyer and Struhl 1995; Lam et al. 2008), it was indeed shown that transcription factors have reduced access to sites that are wrapped within a nucleosome prior to production of that factor. However, we are not aware of a large set of genes for which part of the differences between their promoter activities within the same species were suggested to be linked to the intrinsic nucleosome affinity of their promoter sequences.
To test whether there is a correspondence between promoter activity and intrinsic nucleosome organization across RP promoters, we used a computational model of nucleosome sequence preferences (Kaplan et al. 2009) to predict the DNA-encoded nucleosome organization of every RP promoter. Indeed, we found a relatively high anticorrelation between the activity of the promoter and the average nucleosome occupancy predicted within the 200 bp upstream of the TrSS (R = −0.46, P < 10−5) (Figs. 4, 6A). In addition, we found distinct nucleosome organizations across the three different promoter groups defined above, such that promoters in the low and especially the intermediate promoter activity groups are predicted to have a strongly positioned nucleosome centered ∼200 bp upstream of the TrSS, whereas this nucleosome does not appear in the predictions of the high-activity promoters (Figs. 4, 6B). Examining the predicted nucleosome occupancy over binding sites for the transcriptional regulators of RP genes, we also found stronger nucleosome depletion over binding sites for Fhl1 and Rap1, and to a lesser extent, Sfp1 in the high-activity promoters, compared with the two other groups (Supplemental Fig. 16). Notably, we found experimental validation for all of these nucleosome model predictions, as they are recapitulated when examining the nucleosome occupancy of RP promoters both in a genome-wide reconstitution of nucleosomes in vitro (Kaplan et al. 2009), where nucleosome occupancy is governed only by the intrinsic sequence preferences of nucleosomes, as well as in a genome-wide map of nucleosome occupancy in vivo (Fig. 6C,D; Kaplan et al. 2009; Supplemental Fig. 16).
Figure 6.

RP promoters with high promoter activities have a lower intrinsic affinity to nucleosomes. (A) For each RP promoter, shown is the lowest average nucleosome occupancy, predicted by a computational model of nucleosome sequence preferences (Kaplan et al. 2009), across any 10-bp region within 200 bp upstream of the translation start site (y-axis). RP promoters are plotted by their measured promoter activity (x-axis). Also shown is a moving average of consecutive RP promoters using a window of 11 promoters (red line). (B) For each group of RP promoters with high (red), intermediate (green), and low (blue) promoter activities, defined as in Figure 4, the average model-predicted (Kaplan et al. 2009) nucleosome occupancy across the group promoters is shown as a function of the distance from the translation start site. (C) Same as in B, but using measurements of nucleosome occupancy in vitro (Kaplan et al. 2009), in which nucleosome positions are dominated by nucleosome sequence preferences. (D) Same as in B, but using measurements of nucleosome occupancy in vivo (Kaplan et al. 2009) during growth in glucose-rich media.
The finding that transcription-factor binding sites are located in regions of low nucleosome occupancy in vivo leaves open the question of whether the low occupancy is a consequence of factor binding or whether the sequences in these regions have low intrinsic affinity for nucleosomes. However, the low occupancy that these regions exhibit in vitro, in the absence of the transcription factors, suggests that the low occupancy is largely due to sequences in these regions being less favorable for nucleosome formation. To further examine this point, we asked whether homopolymeric stretches of A nucleotides, referred to as poly(dA:dT) tracts, may underlie the enhanced nucleosome depletion of the high-activity promoters, since these tracts are a key component of the above nucleosome model and are known to strongly disfavor nucleosome formation (Segal and Widom 2009). Indeed, we found significantly more poly(dA:dT) elements in the high-activity promoters (P < 10−3, Supplemental Figs. 17, 18). For example, 38% of the high-activity promoters contain perfect poly(dA:dT) tracts of a length of 10 bp or more within the 300 bp upstream of their TrSS, compared with only 11% and 13% of the intermediate or low-activity promoters, respectively (P < 0.006 and P < 0.03). Consistent with the suggestion that these elements, through the enhanced accessibility created by their nucleosome exclusion effect, increase transcriptional activity of the promoters in which they appear, two earlier studies (Rotenberg and Woolford 1986; Goncalves et al. 1995) showed that for two RP genes, deletion of a T-rich element from their promoter resulted in a significant reduction in transcription.
Together, these results suggest that the DNA-encoded nucleosome organization of RP promoters may be important for their transcription, and a key genetic mechanism by which their differential promoter activities may be achieved.
A computational model of cis-regulation of RP promoters
Finally, we asked what fraction of the variability in RP promoter activities can be explained by the above binding sites and nucleosome positioning signals. To this end, we first plotted the distribution of these sequence features across the different RP promoter groups (Fig. 7). These combined plots revealed different architectures in each promoter group. Promoters with high activities have, on average, Rap1 sites, followed by sites for Fhl1 and Sfp1, and in some promoters, the Fhl1 and Sfp1 sites are followed by TATA boxes (Fig. 7A). All of these sites tend to reside in the ∼300 bp upstream of the TrSS, and this region also has low nucleosome affinity. RP promoters with intermediate activities are characterized by Rap1 sites, followed by a region with a strong affinity for nucleosomes (Fig. 7B). These promoters have very few TATA boxes and sites for Fhl1 and Sfp1, and the Rap1 sites are located further upstream compared with their average location in the high-activity promoters. Finally, RP promoters with low activities exhibit, on average, a less coherent organization of the above sequence features, with a more diffuse distribution of Rap1 sites, and with very few TATA boxes, Fhl1, and Sfp1 sites (Fig. 7C). We note, however, that despite these differences in promoter architecture, any one of these groups contains some individual promoters whose organization is more similar to that of another group, suggesting that these differences alone are not sufficient to explain the promoter activity differences between these groups.
Figure 7.

RP promoters with high and low promoter activities have different promoter architectures. (A) For the group of RP promoters with high promoter activities, defined as in Figure 4, shown are the average (per promoter) number of binding sites for Fhl1 (green), Sfp1 (blue), and Rap1 (red), TATA boxes (light blue), and model-predicted nucleosome occupancy (Kaplan et al. 2009) (purple), as a function of the distance from the translation start site. Sites for Fhl1, Sfp1, and Rap1, and TATA boxes are shown using the same binding strength threshold used in Figure 4. Sites for Fhl1 and Rap1 and for TATA boxes are only counted in one of the two possible site orientations. To allow a comparison of all data types to each other, every data type was normalized to its maximum value across A–C. The plots are shown as moving averages using a window of 50 bp. (B) Same as in A, but for the group of RP promoters with intermediate promoter activities. (C) Same as in A, but for RP promoters with low promoter activities.
To obtain a quantitative measure of the degree to which these distinct promoter architectures can explain the measured promoter activities, we devised a simple and mechanistically motivated computational model that predicts promoter activities from the combination of sequence features for Fhl1, Sfp1, Rap1, TATA, and nucleosomes. We made a simplifying assumption, whereby the predicted activity of a promoter is equal to the sum of the contribution to transcription of each TATA box and each site for Fhl1, Sfp1, and Rap1 that appears in the promoter, using a factor-specific threshold to determine binding sites. Motivated by the spatial localization of sites for these factors (Fig. 5), we consider potential binding sites for these factors only within specific regions (for Rap1, 400 bp upstream of the TrSS; for Fhl1 and Sfp1, 300 bp upstream; and for TATA, 200 bp upstream). To integrate the effect of nucleosomes into our model, the contribution of each binding site was proportional to its binding probability, which we computed using a thermodynamic approach that models the competition between transcription factors and nucleosomes (Raveh-Sadka et al. 2009). In addition, we also modeled the effect of the DNA-encoded nucleosome organization on binding of the general transcription initiation machinery, independent of any binding site. Overall, our model has three parameters for each factor and one parameter for the factor-independent effect of nucleosomes, for a total of 13 parameters (see Methods section).
To assess the predictive power of our model, we compared its predicted promoter activities with those measured, using a cross-validation scheme in which we randomly partitioned the RP promoters into five equally sized sets, and the activity of every promoter was predicted using a model whose parameters were learned using the RP promoters of the four sets (i.e., using 80% of the data) that do not include that promoter and its paralog, if it exists. Notably, the promoter activities predicted by the model on held-out promoters were highly correlated to those measured (R = 0.64) and explained a large fraction (40%, compared with a maximum explained fraction of 3.5% in 1000 promoter activity permutations, P < 0.001) of the variability in the measured activities (Fig. 8A,B,C). We obtained equivalent results using a leave-one-out cross-validation scheme.
Figure 8.

A large fraction of the promoter activity variability of RP promoters can be predicted from their DNA sequence. (A) Fraction of the variance of RP promoter activities that is explained by model-predicted promoter activities, for six different models. The full model (leftmost column) combines sites for Rap1, Fhl1, and Sfp1, as well as TATA boxes and computational predictions of nucleosome occupancy (Kaplan et al. 2009). The models in columns two to six represent models that used only Rap1 sites (column two), Fhl1 sites (column three), Sfp1 sites (column four), TATA boxes (column five), or nucleosome occupancy predictions (column six). The predictions of each model were computed in a fivefold cross-validation scheme, whereby the RP promoters were randomly partitioned into five equally sized sets, and the activities of RP promoters in each set were predicted using a model whose parameters were learned using the RP promoters of the other four sets (i.e., using 80% of the data). When randomly partitioning the promoters into five sets, promoter pairs of duplicated RP genes were always assigned to the same set. (B) Histogram of the fraction of variance explained by 1000 models in which the RP promoter activities were permuted. The fraction of variance explained by the full model from A is indicated (red arrow). (C) Detailed view of the predictions of the full model from A, showing the measured (x-axis) and model-predicted (y-axis) promoter activity of every RP promoter. The fraction of the variance of RP promoter activities explained by the model is indicated in the top left corner. (D) Same as in C, for a model that only used Fhl1 binding sites. (E) Same as in C, for a model that only used TATA boxes. (F) Same as in C, for a model that only used Sfp1 binding sites. (G) Same as in C, for a model that only used predictions of intrinsic nucleosome occupancy. (H) Same as in C, for a model that only used Rap1 binding sites.
To assess the relative contribution of each type of sequence feature, we constructed separate models that each used only one type of feature. As expected from their enrichment in RP promoters with high promoter activities (Fig. 5), models that used only TATA boxes, Sfp1 sites, or Fhl1 sites each explained a significant fraction of the variability in the measured promoter activities (Fig. 8D,E,F). However, the total number of RP promoters explained by Fhl1-only and TATA-only models is very small (<8%), since these sites appear in very few RP promoters (Figs. 4, 8D,E). In notable contrast, a model that only uses the DNA-encoded nucleosome organization also explains a significant fraction of the overall variability, but its predictive power is not dominated by a small number of promoters, suggesting that intrinsic nucleosome affinity may be an important determinant of the activity of many RP promoters (Fig. 8G). Finally, using our simple modeling assumptions, a model that only uses Rap1 sites has no predictive power, consistent with our inability to explain how the distinct organizations of Rap1 sites that we found between promoters with high and low activities contribute differentially to transcription (Fig. 8H).
We emphasize that our model is not aimed at providing an accurate mechanistic description of RP transcriptional regulation. Rather, it is aimed at providing one estimate for the fraction of the measured variability in promoter activity that can be explained by a simple integration of the above sequence features, and at highlighting gaps in our understanding of their contribution. In this respect, our results suggest that a large fraction, but certainly not all, of the activity variability can be explained by a simple combination of the DNA-encoded nucleosome organization, and of TATA boxes and sites for Fhl1 and Sfp1, with the DNA-encoded nucleosome organization likely being important for many RP promoters. In addition, our model highlights the gap that currently exists in our understanding of how Rap1 contributes to transcription.
Experimental validation of RP promoter elements
Our above analyses provide strong evidence for the role of several types of promoter elements, including novel elements, in determining RP expression. However, as they were derived computationally, these insights are correlative. To validate their role, we carried out experiments in which we separately tested the contribution of each of the implicated elements to RP promoter activity (Fig. 9). To this end, we designed mutations to each of the implicated elements in several different promoters, constructed the corresponding promoter variants, integrated them into the same yeast strain into which the natural RP promoters were integrated, and measured the activity of the mutated promoters (Supplemental Figs. 20, 21; Supplemental Data2). In all cases, we took care to permute, but not delete base pairs, since deletions would also change the distance of the tested regulatory element from the gene start. Furthermore, aside from nucleosome disfavoring sequences, whose deletion requires more sequence changes, we always mutated elements with a minimal number (two or three) of base-pair changes and selected changes that preserved the original G/C content. As a measure of the effect expected from arbitrary mutations in RP promoters, we generated random mutations in eight RP promoters, and found detectable effects in only two of these cases, and even in these two cases, the effects were small (10% and 12%) (Fig. 9A).
Figure 9.

Experimental validation of regulatory elements in RP promoters. (A) The effect of random mutations on RP promoter activity for eight RP promoters. For each promoter, shown is the activity of the natural promoter and a promoter in which a random mutation (1–8 bp sequence changes) was generated. Error bars represent two standard errors computed from 24 replicates. The magnitude of the effect of the mutation on promoter activity is indicated above the activity bars of each promoter pair, where two stars mark promoter activity differences that are statistically significant. (B) Same as in A, for five different mutations of Fhl1 binding sites in five different RP promoters. Mutations of Fhl1 sites were done by 2-bp changes that preserved the G/C content. (C) Same as in A, for three different mutations of Sfp1 sites in three different promoters. Mutations of Sfp1 sites were done by 2-bp changes that preserved the G/C content. (D) Same as in A, for four different mutations of TATA boxes in four different RP promoters. Mutations of TATA boxes were done by 2–3 bp changes that preserved the G/C content. (E) Same as in A, for nine different mutations of nucleosome disfavoring sequences in nine different RP promoters. Mutations to these A/T-rich nucleosome disfavoring sequences were done by replacing 16 A/T base pairs with G/C base pairs in a region of 31 base pairs within the promoter that had the lowest predicted nucleosome occupancy (Kaplan et al. 2009). (F) Summary and comparison of the effect of the mutations according to the type of element mutated. For each type of mutation from A to E, the average and standard deviation of the effect on promoter activity of all of the different mutations done to these elements are shown, where the effect is taken from the numbers above the various bars in A–E, such that the wild-type promoter (WT, leftmost bar) is defined to have a promoter activity of 1.
Notably, we found significant changes in promoter activity for all types of regulatory elements that we tested. In the case of Fhl1, for which our experiments provide the first in vivo test of its newly reported binding specificities (Badis et al. 2008; Zhu et al. 2009), three of five site deletions that we tested resulted in significant reductions of over 20% in promoter activity (Fig. 9B). For Sfp1, for which we also provide the first in vivo test of new binding specificities reported for it (Badis et al. 2008; Zhu et al. 2009), all three site deletions resulted in significant and large reductions in promoter activity (36%–68%) (Fig. 9C). Similarly, all four mutations that we performed in TATA boxes resulted in significant and large reductions (39%–64%) (Fig. 9D). Finally, seven of nine mutations to nucleosome disfavoring sequences that we performed resulted in significant reductions in promoter activity, with a magnitude that spanned a large dynamic range (11%–86%) (Fig. 9E). We did not mutate Rap1 binding sites, as their role was previously established with similar mutagenesis experiments (Woudt et al. 1986; Moehle and Hinnebusch 1991). Together, these results demonstrate that each type of element implicated by our computational analyses is indeed important for RP promoter activity.
Conclusions and Discussion
In summary, we devised an experimental system for accurately measuring promoter activities that is based on direct fusion of promoter sequences to fluorescent reporters, which can detect activity differences that are as small as ∼10%. We used our system to generate promoter fusions for most of the ribosomal protein (RP) genes in yeast, resulting in the largest library of natural promoter fusions in any eukaryote to date.
Despite the importance of their coordinate regulation for controlling cell growth, little is known about how regulation of RP genes is encoded within their promoters, and our results provide insights into both the logic and potential role of this encoding. Notably, we found that most of the RP promoters whose corresponding gene exists in a single copy in the yeast genome are among the promoters with the highest activities. This suggests that proper RP stoichiometry is encoded, in part, within RP promoters, thereby providing insight into the longstanding conundrum of how proper RP stoichiometry in yeast is achieved in light of the copy-number differences that exist between its single-copy and duplicated RP genes.
On a broader scale, the high sensitivity of our experimental system makes it suitable for studying the mapping between DNA sequence and transcriptional output, and our analyses provide several insights into this mapping in the case of RP promoters. For two RP transcriptional activators, Fhl1 and Sfp1, the mapping appears to be simple, because we found that, on average, promoters with binding sites for these factors have higher promoter activities, suggesting that their presence leads to direct binding of their cognate factor and to more transcription. Intriguingly, sites for these factors exhibit a strong spatial preference to ∼200 bp upstream of the TrSS, further suggesting that their effect depends on their distance from the transcription start site. Promoters with TATA boxes, the canonical binding site for the transcriptional machinery, also tend to have high activities, suggesting that they too have a simple mapping, whereby their presence leads to increased binding of the transcriptional machinery and, consequently, increased transcription. The positive contribution of TATA boxes to transcription is also likely to depend on their orientation and appearance within a close ∼100–200-bp distance to the TrSS.
For Rap1, we found significant differences between its organization in promoters of high and low activities, but here, a simple mechanism cannot explain the contribution of Rap1 to transcription. Thus, for Rap1, our results highlight a gap in our understanding of the mapping between DNA sequence and transcription. We found that, on average, high-activity promoters tend to have Rap1 sites, followed by a region with low nucleosome affinity. In contrast, promoters with lower activities have, on average, Rap1 sites that are located ∼60 bp further upstream compared with their location in the high-activity promoters, and are followed by a region with high nucleosome affinity. Rap1 is clearly critical for RP transcription, since deleting its sites (Woudt et al. 1986; Mencia et al. 2002; Zhao et al. 2006), and even changing the orientation of its sites (Woudt et al. 1986) in RP promoters significantly reduces RP transcription. Our finding of different organizations of Rap1 sites between promoters with high and low activities further suggests that its contribution to transcription depends on its exact organization within the promoter, but it is unclear whether the effect depends on the exact location of the Rap1 site and/or on the presence of the nucleosome between the Rap1 site and the transcription start site. Regardless of which feature of the organization contributes most, the mechanism by which it contributes differentially is not clear. Since both Fhl1 and Sfp1 are bound in ChIP experiments to many more promoters (Harbison et al. 2004; Marion et al. 2004; Wade et al. 2004) than those in which we find their sites, Rap1 is also likely to be important for recruiting these factors to RP promoters, but here too, no simple mechanism can explain how differences between this recruitment in the high- and low-activity promoters may lead to differential promoter activities.
Our results also suggest that the intrinsic (DNA-encoded) nucleosome organization of RP promoters is a key contributor to their differential promoter activities. The contribution of the intrinsic nucleosome organization is likely mediated by the effect that it has on the accessibility of DNA to binding by both the transcriptional machinery and transcription factors, since RP promoters with higher activities have, on average, significantly reduced affinity to nucleosomes in the ∼300 bp upstream of the TrSS. Although several studies demonstrated that the intrinsic nucleosome organization determines many aspects of the nucleosome organization in vivo (Kaplan et al. 2009; Zhang et al. 2009), much less is known about the effect of the intrinsic organization on transcription. Our results thus suggest that intrinsic nucleosome organization may be an important component of the mapping between DNA sequence and transcription, and that changes in the DNA-encoded nucleosome organization of promoters may be a key genetic mechanism by which promoter activity changes are tuned during evolution (Field et al. 2009).
Despite the distinct distributions of sequence features that we found between the high- and low-activity promoters, some RP promoters with low activities contain sequence features that appear predominantly in promoters with high activities, suggesting that we still have gaps in our quantitative understanding of how these sequence features affect transcription. Conversely, some RP promoters have high activities, but lack the sequence features that we find in many of the high-activity promoters, suggesting that we may still be missing sequence features or proteins that regulate RP transcription. In a search for one such potential regulator, we examined high-resolution ChIP measurements (Lavoie et al. 2010) of the high-mobility group protein Hmo1 across the RP promoters. Notably, we found that Hmo1 binding is significantly higher in RP promoters with intermediate activities (P < 10−4), and its binding within these promoters is preferentially localized just downstream from the Rap1 sites to the location of the strongly positioned nucleosome that exists in these promoters (Supplemental Fig. 19). Although Hmo1 was shown to bind most RP promoters in a Rap1-dependent manner (Hall et al. 2006), to our knowledge our results provide the first association between Hmo1 binding and promoter activities, suggesting that Hmo1 may affect RP promoter activities, although the mechanism by which it may do so is unclear. Aside from unidentified promoter elements, since we included the 5′ untranslated region of each RP gene in the promoter sequence that we fused to the YFP reporter, it is also possible that some of the unexplained variability in YFP expression results from differential post-transcriptional effects that these 5′ UTRs may have.
Overall, our results provide new insights into the transcriptional regulation of RP genes in yeast, and suggest that RP promoters have evolved in part to ensure proper RP stoichiometry. We unravel some of the mechanisms and sequence features by which RP promoters achieve their differential activities, and suggest that these activities are encoded by combinations and spatial distributions of a rich set of sequence elements that direct both transcription-factor binding and nucleosome organization. These results advance our general understanding of the mapping between DNA sequence and transcription, and also identify concrete examples where we still have gaps in this understanding. We propose that further progress in filling these gaps can be achieved by applying similar approaches to other native promoters and to large-scale promoter libraries with designed mutations, ultimately leading to a quantitative understanding of how transcriptional regulation is encoded by DNA sequence.
Methods
Constructing promoter strains
A construct of ADH1 terminator–mCherry–TEF2 promoter–YFP–ADH1 terminator–NAT1 (see sequence in Supplemental Data3) was inserted into the SGA compatible strain Y8205 at the his3 deletion location (the construct replaced chromosome 15, at base pairs 721987–722506). The resulting strain served as a master strain for the entire library. Desired promoters were lifted by PCR from the BY4741 yeast strain. Primers contained one part matching the ends of the lifted promoters, and a constant part at their 5′ end matching the first 25 bases of the YFP gene (for reverse primers) or a linker sequence (for forward primers; see all primer sequences in Supplemental Data3). Each promoter was linked to a URA3 selection marker (Linshiz et al. 2008) and then amplified such that its genomic integration sites increased to 45/50 bp. Integration into the genome was performed by homologous recombination as described in Gietz and Schiestl (2007). All steps were performed on 96 well plates, except for growing the final clones, which was performed on 6 well plates (2% Agar, SCD–URA). To validate the inserted promoter sequences, the insertions were lifted from each target strain by PCR and sequenced.
Constructing promoter strains with targeted mutations
To create a mutated promoter, we amplified it in two parts, which flank the desired mutation area. The left part was amplified using a reverse primer with a 35-bp tail at its 5′ end that contains the desired mutation, while the right part was amplified using a forward primer that also had a similar tail. The two new parts, both containing the desired mutation in an overlapping region of 35 bp were then connected, similar to the way in which we connected promoters to the URA selection marker. See Supplemental Figures 20 and 21 for a detailed description of the mutations performed.
Library measurements
Cells were inoculated from stocks of −80°C into SCD (180 uL, 96 well plate) and left to grow at 30°C for 48 h, reaching complete saturation. Next, 8 uL were passed into fresh medium (180 uL) according to the desired condition (e.g., SCD, Ethanol, heat shock). Measurements were carried out every ∼20 min using a robotic system (Tecan Freedom EVO) with a plate reader (Tecan Infinite F500). Each measurement included optical density (filter wavelengths 600 nm, bandwidth 10 nm), YFP fluorescence (excitation 500 nm, emission 540 nm, bandwidths 25/25 nm, accordingly) and mCherry fluorescence (excitation 570 nm, emission 630 nm, bandwidths 25/35 nm, accordingly). Measurements were carried out using a total of eight different conditions. In all experiments, yeast cells were grown on SC (6.9 g/L YNB, 1.6 g/L amino acids complete). Four conditions used different 2% sugar growth media: SC-Glucose, SC-Galactose, SC-Ethanol, and SC-Glycerol. The other four conditions used SC-Glucose with an additional stress factor: Rapamycin (40 ug/mL), amino acid starvation (no amino acids except Histidine and Leucine), heat shock (39°C), and osmotic stress (750 mM KCl). Every strain was measured in three biological replicates for each condition. Most of the data analysis was performed on data from growth on SC-Glucose (without stress), which was measured in five replicates.
Exponential growth phase detection
We developed an automated procedure that detects the exponential growth phase of a yeast culture growing in 96-well plates. To this end, we found that our OD measurements have four main growth phases and devised a procedure to detect them. In the first phase, known as the lag phase, the OD is relatively steady. In the second, the exponential phase, the cells grow at a constant rate and, hence, the OD increases exponentially. In the third phase, the growth rate decreases, possibly due to exhaustion of nutrients or to physical density, and the OD grows linearly (we refer to this stage as the linear phase). In the final stationary phase, the cells stop growing and the OD is relatively constant. Based on these observations, we devised a simple estimate OD', for the OD curve:
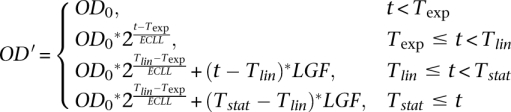 |
where OD0 is the initial OD value, Texp, Tlin, and Tstat are the timepoints at which the culture enters the exponential, linear, and stationary growth phase, respectively, ECCL is the cell cycle length of the exponential phase, and LGF is the slope of the OD curve during the linear phase. We fit the above model parameters using the optimization toolbox of Matlab, and verified that the quality of the fit for each plate was satisfactory. To avoid errors due to small inaccuracies in detecting the start and end time of the exponential growth phase, we ignored in all of our computations the first 10% and last 10% of the phase time period.
Estimating promoter activities from promoter strain measurements
The measurements were done in 96-well plates with a different promoter strain in each well, and consist of OD (optical density, indicative of cell population size), YFP, and mCherry measurements collected every 20 min. Every measured plate is subjected to several quality control steps. First, since all strains should have the same growth curves, strains with an abnormal growth curve are removed from further analysis. To identify such outlier strains we compute, at each timepoint, the Z-score of the OD measurement of every strain relative to the OD measurement of all other strains in that timepoint. We then sum the Z-scores of every strain across all timepoints and compute the mean and standard deviation of these summed Z-scores across all strains. Strains whose summed Z-score is more than three standard deviations above the mean of all strains are then removed. Next, to remove background levels from each YFP and mCherry measurement at every timepoint, we subtract the YFP and mCherry measurement of a strain that has no YFP gene (for YFP measurements) and no mCherry gene (for mCherry measurements). Similarly, from the OD measurement at each timepoint, we subtract the OD of a well that only contains the growth media. Finally, we discard individual OD, mCherry, and YFP measurements that deviate considerably (more than two standard deviations) from the average value of their neighboring timepoints. We note, however, that <2% of the individual timepoint measurements are removed this way.
Next, we identify the exponential growth phase (see above) and compute the average YFP (mCherry) promoter activity of every strain per cell per second over the exponential phase, by dividing the total amount of YFP (mCherry) produced during the exponential phase by the integral of the OD levels during this time interval. Since both the YFP and mCherry proteins are stable and long-lived, the difference in YFP (mCherry) level between the end and the beginning of the exponential phase corresponds to the amount of YFP (mCherry) produced during that time interval. Finally, to obtain a promoter activity estimate from multiple replicates (measurement plates), we first scale the values of each plate to equalize, across all plates, the average value of four technical replicates of the same promoter strain (RPL3) in each plate. For each strain, we then take its promoter activity to be the average rate of that strain across all measurement plates.
Computational model of cis-regulation
We developed a mechanistically motivated model that predicts promoter activities (TR) from DNA sequence and factor concentration alone. Our model defines the promoter activity of a promoter sequence to be the probability of polymerase binding in the region proximal to the TrSS (200 bp upstream of the TrSS), multiplied by the sum of interactions between polymerase and each potential bound transcription-factor binding site:
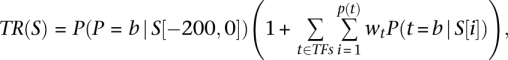 |
where S represents the promoter sequence, P(P=b|S[-200,0]) is the probability of polymerase binding to the 200 bp upstream of the TrSS, p(t) is the set of potential binding sites for transcription factor t, wt is a weight representing the interaction between transcription factor t and polymerase, and P(t=b|S[i]) is the probability that transcription-factor t binds its potential site at position i in the promoter sequence S. As the potential sites for each factor, p(t), we take all sites whose binding-site strength exceeds a site threshold score of Thrt, where Thrt is a free parameter and transcription-factor scores are computed based on their known sequence specificities (Basehoar et al. 2004; Badis et al. 2008; Zhu et al. 2009). In estimating the probability of transcription-factor binding, we model the competition between factor binding and nucleosome binding, such that the probability of factor binding is equal to the weight of the configuration in which the factor is bound, divided by the sum of the weight of that configuration, the weight of the configuration in which the DNA is unbound, and the weight of the configuration in which a nucleosome is bound, covering the site:
where 1 represents the weight of the empty configuration, [t] and [nuc] are the concentrations of transcription factor t and of nucleosomes, respectively, and At(S[i]) and Anuc(S[i]) represent the affinity of factor t and of nucleosomes to the binding site at position i, respectively. The transcription factor concentration, [t], is a free parameter estimated during the model training procedure. Our model assumes that all potential binding sites of a given factor have the same affinity, At(S[i])=At, and thus, in this formulation, At is redundant with [t] and is set to Thrt. The nucleosome concentration, [nuc], is taken from Kaplan et al. (2009). For Anuc(S[i]), we use an existing sequence-based nucleosome affinity model (Kaplan et al. 2009) and compute the average nucleosome occupancy of a DNA sequence, deriving Anuc(S[i]) as follows:
where P(nuc=b|S[i]) is the average nucleosome occupancy over the binding site that starts at position i, computed by the model of Kaplan et al. (2009).
Finally, in computing the probability of polymerase binding to the 200 bp upstream of the TrSS, P(P=b|S[-200,0]), we have a free polymerase concentration parameter, [pol]. We also take nucleosome binding into account, as for transcription factors, by defining the binding location of polymerase as the 10 bp with the lowest nucleosome occupancy within the 200-bp region. This makes the assumption that polymerase can bind with equal probability within the 200-bp upstream of the TrSS, and thus, under this assumption its most likely binding location is at the region of lowest nucleosome occupancy.
In instantiating our model for RPs, we have four transcription factors (Rap1, Fhl1, and Sfp1, as well as TATA boxes that we consider as another factor [TBP]). Motivated by the spatial localization of sites for these factors (Fig. 5), we consider potential binding sites for these factors only within specific regions (for Rap1, 400 bp upstream of the TrSS; for Fhl1 and Sfp1, 300 bp upstream; and for TATA, 200 bp upstream). Thus, overall, our model has 13 free parameters: three for each of four factors (a concentration, [t], binding site threshold, Thrt, and polymerase interaction term, wt), and one for the polymerase concentration, [pol].
Acknowledgments
We thank Judith Berman, Ranit Kedmi, and Jon Widom for useful discussions and suggestions. We also thank Charlie Boon for supplying the yeast strain Y8205. This work was supported by grants from the European Research Council (ERC) and the National Institute of Health (NIH) to E.S. E.S. is the incumbent of the Soretta and Henry Shapiro Career Development Chair.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.119669.110.
References
- Badis G, Chan ET, van Bakel H, Pena-Castillo L, Tillo D, Tsui K, Carlson CD, Gossett AJ, Hasinoff MJ, Warren CL, et al. 2008. A library of yeast transcription factor motifs reveals a widespread function for Rsc3 in targeting nucleosome exclusion at promoters. Mol Cell 32: 878–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basehoar AD, Zanton SJ, Pugh BF 2004. Identification and distinct regulation of yeast TATA box-containing genes. Cell 116: 699–709 [DOI] [PubMed] [Google Scholar]
- Causton HC, Ren B, Koh SS, Harbison CT, Kanin E, Jennings EG, Lee TI, True HL, Lander ES, Young RA 2001. Remodeling of yeast genome expression in response to environmental changes. Mol Biol Cell 12: 323–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox RS 3rd, Surette MG, Elowitz MB 2007. Programming gene expression with combinatorial promoters. Mol Syst Biol 3: 145 doi: 10.1038/msb4100187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David L, Huber W, Granovskaia M, Toedling J, Palm CJ, Bofkin L, Jones T, Davis RW, Steinmetz LM 2006. A high-resolution map of transcription in the yeast genome. Proc Natl Acad Sci 103: 5320–5325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutschbauer AM, Jaramillo DF, Proctor M, Kumm J, Hillenmeyer ME, Davis RW, Nislow C, Giaever G 2005. Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics 169: 1915–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field Y, Fondufe-Mittendorf Y, Moore IK, Mieczkowski P, Kaplan N, Lubling Y, Lieb JD, Widom J, Segal E 2009. Gene expression divergence in yeast is coupled to evolution of DNA-encoded nucleosome organization. Nat Genet 41: 438–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO 2000. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11: 4241–4257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz J, Siggia ED, Cohen BA 2009. Analysis of combinatorial cis-regulation in synthetic and genomic promoters. Nature 457: 215–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH 2007. Microtiter plate transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2: 5–8 [DOI] [PubMed] [Google Scholar]
- Goncalves PM, Griffioen G, Minnee R, Bosma M, Kraakman LS, Mager WH, Planta RJ 1995. Transcription activation of yeast ribosomal protein genes requires additional elements apart from binding sites for Abf1p or Rap1p. Nucleic Acids Res 23: 1475–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DB, Wade JT, Struhl K 2006. An HMG protein, Hmo1, associates with promoters of many ribosomal protein genes and throughout the rRNA gene locus in Saccharomyces cerevisiae. Mol Cell Biol 26: 3672–3679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, et al. 2004. Transcriptional regulatory code of a eukaryotic genome. Nature 431: 99–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstege FC, Jennings EG, Wyrick JJ, Lee TI, Hengartner CJ, Green MR, Golub TR, Lander ES, Young RA 1998. Dissecting the regulatory circuitry of a eukaryotic genome. Cell 95: 717–728 [DOI] [PubMed] [Google Scholar]
- Iyer V, Struhl K 1995. Poly(dA:dT), a ubiquitous promoter element that stimulates transcription via its intrinsic DNA structure. EMBO J 14: 2570–2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Q, Warner JR 1994. Ribosome synthesis during the growth cycle of Saccharomyces cerevisiae. Yeast 10: 151–157 [DOI] [PubMed] [Google Scholar]
- Kalir S, McClure J, Pabbaraju K, Southward C, Ronen M, Leibler S, Surette MG, Alon U 2001. Ordering genes in a flagella pathway by analysis of expression kinetics from living bacteria. Science 292: 2080–2083 [DOI] [PubMed] [Google Scholar]
- Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J, et al. 2009. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458: 362–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg RD, Lorch Y 1999. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 98: 285–294 [DOI] [PubMed] [Google Scholar]
- Lam FH, Steger DJ, O'Shea EK 2008. Chromatin decouples promoter threshold from dynamic range. Nature 453: 246–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie H, Hogues H, Mallick J, Sellam A, Nantel A, Whiteway M 2010. Evolutionary tinkering with conserved components of a transcriptional regulatory network. PLoS Biol 8: e1000329 doi: 10.1371/journal.pbio.1000329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, et al. 2002. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298: 799–804 [DOI] [PubMed] [Google Scholar]
- Li B, Vilardell J, Warner JR 1996. An RNA structure involved in feedback regulation of splicing and of translation is critical for biological fitness. Proc Natl Acad Sci 93: 1596–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieb JD, Liu X, Botstein D, Brown PO 2001. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat Genet 28: 327–334 [DOI] [PubMed] [Google Scholar]
- Ligr M, Siddharthan R, Cross FR, Siggia ED 2006. Gene expression from random libraries of yeast promoters. Genetics 172: 2113–2122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linshiz G, Yehezkel TB, Kaplan S, Gronau I, Ravid S, Adar R, Shapiro E 2008. Recursive construction of perfect DNA molecules from imperfect oligonucleotides. Mol Syst Biol 4: 191 doi: 10.1038/msb.2008.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson D, Raz T, Kieu A, Jones DR, Giladi E, Thayer E, Thompson JF, Letovsky S, Milos P, Causey M 2009. Quantification of the yeast transcriptome by single-molecule sequencing. Nat Biotechnol 27: 652–658 [DOI] [PubMed] [Google Scholar]
- Lucioli A, Presutti C, Ciafre S, Caffarelli E, Fragapane P, Bozzoni I 1988. Gene dosage alteration of L2 ribosomal protein genes in Saccharomyces cerevisiae: effects on ribosome synthesis. Mol Cell Biol 8: 4792–4798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIsaac KD, Wang T, Gordon DB, Gifford DK, Stormo GD, Fraenkel E 2006. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics 7: 113 doi: 10.1186/1471-2105-7-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion RM, Regev A, Segal E, Barash Y, Koller D, Friedman N, O'Shea EK 2004. Sfp1 is a stress- and nutrient-sensitive regulator of ribosomal protein gene expression. Proc Natl Acad Sci 101: 14315–14322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DE, Soulard A, Hall MN 2004. TOR regulates ribosomal protein gene expression via PKA and the Forkhead transcription factor FHL1. Cell 119: 969–979 [DOI] [PubMed] [Google Scholar]
- Mencia M, Moqtaderi Z, Geisberg JV, Kuras L, Struhl K 2002. Activator-specific recruitment of TFIID and regulation of ribosomal protein genes in yeast. Mol Cell 9: 823–833 [DOI] [PubMed] [Google Scholar]
- Moehle CM, Hinnebusch AG 1991. Association of RAP1 binding sites with stringent control of ribosomal protein gene transcription in Saccharomyces cerevisiae. Mol Cell Biol 11: 2723–2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KF, Balazsi G, Collins JJ 2007. Combinatorial promoter design for engineering noisy gene expression. Proc Natl Acad Sci 104: 12726–12731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320: 1344–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshlack A, Wakefield MJ 2009. Transcript length bias in RNA-seq data confounds systems biology. Biol Direct 4: 14 doi: 10.1186/1745-6150-4-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polach KJ, Widom J 1995. Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J Mol Biol 254: 130–149 [DOI] [PubMed] [Google Scholar]
- Raveh-Sadka T, Levo M, Segal E 2009. Incorporating nucleosomes into thermodynamic models of transcription regulation. In Proceedings of the 13th International Conference on Research in Computational Molecular Biology (RECOMB), Tucson, AZ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotenberg MO, Woolford JL Jr 1986. Tripartite upstream promoter element essential for expression of Saccharomyces cerevisiae ribosomal protein genes. Mol Cell Biol 6: 674–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotenberg MO, Moritz M, Woolford JL Jr 1988. Depletion of Saccharomyces cerevisiae ribosomal protein L16 causes a decrease in 60S ribosomal subunits and formation of half-mer polyribosomes. Genes Dev 2: 160–172 [DOI] [PubMed] [Google Scholar]
- Schawalder SB, Kabani M, Howald I, Choudhury U, Werner M, Shore D 2004. Growth-regulated recruitment of the essential yeast ribosomal protein gene activator Ifh1. Nature 432: 1058–1061 [DOI] [PubMed] [Google Scholar]
- Segal E, Widom J 2009. Poly(dA:dT) tracts: major determinants of nucleosome organization. Curr Opin Struct Biol 19: 65–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore D 1994. RAP1: a protean regulator in yeast. Trends Genet 10: 408–412 [DOI] [PubMed] [Google Scholar]
- Spahn CM, Beckmann R, Eswar N, Penczek PA, Sali A, Blobel G, Frank J 2001. Structure of the 80S ribosome from Saccharomyces cerevisiae–tRNA-ribosome and subunit-subunit interactions. Cell 107: 373–386 [DOI] [PubMed] [Google Scholar]
- Svaren J, Schmitz J, Horz W 1994. The transactivation domain of Pho4 is required for nucleosome disruption at the PHO5 promoter. EMBO J 13: 4856–4862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade JT, Hall DB, Struhl K 2004. The transcription factor Ifh1 is a key regulator of yeast ribosomal protein genes. Nature 432: 1054–1058 [DOI] [PubMed] [Google Scholar]
- Warner JR 1999. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci 24: 437–440 [DOI] [PubMed] [Google Scholar]
- Woudt LP, Smit AB, Mager WH, Planta RJ 1986. Conserved sequence elements upstream of the gene encoding yeast ribosomal protein L25 are involved in transcription activation. EMBO J 5: 1037–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassour M, Kaplan T, Fraser HB, Levin JZ, Pfiffner J, Adiconis X, Schroth G, Luo S, Khrebtukova I, Gnirke A, et al. 2009. Ab initio construction of a eukaryotic transcriptome by massively parallel mRNA sequencing. Proc Natl Acad Sci 106: 3264–3269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaslaver A, Mayo AE, Rosenberg R, Bashkin P, Sberro H, Tsalyuk M, Surette MG, Alon U 2004. Just-in-time transcription program in metabolic pathways. Nat Genet 36: 486–491 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Moqtaderi Z, Rattner BP, Euskirchen G, Snyder M, Kadonaga JT, Liu XS, Struhl K 2009. Intrinsic histone-DNA interactions are not the major determinant of nucleosome positions in vivo. Nat Struct Mol Biol 16: 847–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, McIntosh KB, Rudra D, Schawalder S, Shore D, Warner JR 2006. Fine-structure analysis of ribosomal protein gene transcription. Mol Cell Biol 26: 4853–4862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Byers K, McCord R, Shi Z, Berger M, Newburger D, Saulrieta K, Smith Z, Shah M, Radhakrishnan M, et al. 2009. High-resolution DNA binding specificity analysis of yeast transcription factors. Genome Res 19: 556–566 [DOI] [PMC free article] [PubMed] [Google Scholar]