

Abstract
Accurately predicting regulatory sequences and enhancers in entire genomes is an important but difficult problem, especially in large vertebrate genomes. With the advent of ChIP-seq technology, experimental detection of genome-wide EP300/CREBBP bound regions provides a powerful platform to develop predictive tools for regulatory sequences and to study their sequence properties. Here, we develop a support vector machine (SVM) framework which can accurately identify EP300-bound enhancers using only genomic sequence and an unbiased set of general sequence features. Moreover, we find that the predictive sequence features identified by the SVM classifier reveal biologically relevant sequence elements enriched in the enhancers, but we also identify other features that are significantly depleted in enhancers. The predictive sequence features are evolutionarily conserved and spatially clustered, providing further support of their functional significance. Although our SVM is trained on experimental data, we also predict novel enhancers and show that these putative enhancers are significantly enriched in both ChIP-seq signal and DNase I hypersensitivity signal in the mouse brain and are located near relevant genes. Finally, we present results of comparisons between other EP300/CREBBP data sets using our SVM and uncover sequence elements enriched and/or depleted in the different classes of enhancers. Many of these sequence features play a role in specifying tissue-specific or developmental-stage-specific enhancer activity, but our results indicate that some features operate in a general or tissue-independent manner. In addition to providing a high confidence list of enhancer targets for subsequent experimental investigation, these results contribute to our understanding of the general sequence structure of vertebrate enhancers.
Enhancers are gene regulatory sequences that can control transcriptional activities at a distance, independent of their position and orientation with respect to affected genes (Banerji 1981). Enhancer activity is modulated by interactions between sequence specific DNA binding proteins and sequence elements in the enhancer. Since individual transcription factor binding sites (TFBSs) can be relatively short and degenerate, TFBSs tend to be clustered to achieve precise temporal and developmental specificity (Kadonaga 2004). Factors bound to these sequences often interact with common coactivators, which, in turn, recruit the basal transcription machinery (Blackwood and Kadonaga 1998; Carter et al. 2002).
Identifying the sequence elements and the combinatorial rules that determine enhancer function is necessary to fully understand how enhancers direct the spatial and temporal regulation of gene expression. Experimentally identified enhancers with similar functions can be a good starting point for in-depth study of the underlying rules encoded in the regulatory DNA sequence. However, the systematic functional identification of such enhancers has been limited due to the fact that they are often distant from the genes they regulate, requiring the interrogation of large amounts of potential regulatory sequence. Most investigations make use of two complementary approaches to detect putative regulatory regions: comparative genomics, which identifies enhancers by their sequence conservation across related species; and functional genomics, which identifies enhancers by the common binding of transcriptionally associated factors or marks (for review, see Noonan and McCallion 2010).
Comparative genomics is based on the generally accepted hypothesis that functionally important regulatory sequences are under purifying selection. As a result, conserved noncoding sequences (CNSs) are natural candidates for putative enhancers. Early studies used CNSs to detect putative enhancers and test their activity in zebrafish or mouse reporter assays (Woolfe et al. 2004; Pennacchio et al. 2006; Visel et al. 2008). Although these conservation-based approaches achieve some success, limitations also exist. The function and spatio-temporal specificity of CNSs cannot be determined by conservation alone and, therefore, requires additional experimentation. More importantly, several studies have shown that noncoding sequences that apparently lack conservation (as assessed by sequence alignment) may still contain functional regulatory elements (Fisher et al. 2006; ENCODE Project Consortium 2007; McGaughey et al. 2008).
Functional genomics is an experimentally driven approach that utilizes recently developed techniques of microarray hybridization or massively parallel sequencing in combination with chromatin immunoprecipitation (ChIP) on specific transcription factors (Johnson et al. 2007; Robertson et al. 2007), chromatin signatures (Heintzman et al. 2007, 2009), or coactivators (Visel et al. 2009; Kim et al. 2010). Specifically, some chromatin signatures or coactivator association (such as monomethylation of lysine 4 of histone H3, acetylation of lysine 27 of histone H3, and binding by coactivators EP300/CREBBP) are predictive markers of enhancer activity (Heintzman et al. 2007, 2009). The transcriptional coactivators EP300 (also known as P300) and CREBBP (also known as CBP) have proven to be useful for enhancer identification because of their general roles as cofactors in mammalian transcription. Through highly conserved protein-protein interactions, EP300/CREBBP are hypothesized to operate as coactivators in at least three ways: as a direct bridge between sequence-specific transcription factors (TFs) and RNA Polymerase II, as an indirect bridge between sequence specific TFs and other coactivators which recruit RNA Pol II, or by modifying chromatin structure via intrinsic acetyl-transferase activity (Chan and La Thangue 2001). Several studies have reported genome-wide mapping of EP300/CREBBP-bound enhancers in different contexts, for example, tissue-specific activity in dissected mouse tissue (Visel et al. 2009) and environment-dependent activity in neurons (Kim et al. 2010). Visel et al. validated that 90% of the EP300 enhancers tested recapitulated the expected spatial and temporal activity in vivo in a transgenic mouse enhancer assay. Functionally identified EP300-bound regions thus provide a robust starting point for further investigation of enhancers and their sequence properties.
In principle, a complete understanding of enhancer mechanism would include a description of specific internal sequence features and how they contribute to enhancer function. Previous studies that have attempted to predict enhancers from sequence have typically used sequence conservation, colocalization of previously characterized TFBSs [from databases such as TRANSFAC (Matys et al. 2003) or JASPAR (Bryne et al. 2008)], or a combination of the two. Many of these existing approaches were assessed by Su et al. (2010), who found that some were successful in identifying enhancers in Drosophila but that few generalized to mammalian systems. The most successful method in mammalian enhancer prediction used a combination of conservation and low-order Markov models of sequence features (Elnitski et al. 2003; King et al. 2005). In more recent work, Leung and Eisen (2009) used word frequency profile similarity between pairs of sequences to detect novel enhancers, but training on small numbers of enhancers can be susceptible to noise. Another notable recent computational approach uses combinations of known TFBSs and de novo position weight matrices (PWMs) to detect enhancers (Narlikar et al. 2010).
In this paper, we present a discriminative computational framework to detect enhancers from DNA sequence alone that does not rely on conservation or known TF binding specificities. We use a support vector machine (SVM) to differentiate enhancers from nonfunctional regions, using DNA sequence elements as features. SVMs (Boser et al. 1992; Vapnik 1995) have been successfully applied in many biological contexts (for review, see Schölkopf et al. 2004; Ben-Hur et al. 2008): cancer tissue classification (Furey et al. 2000); protein domain classification (Karchin et al. 2002; Leslie et al. 2002, 2004); splice site prediction (Rätsch et al. 2005; Sonnenburg et al. 2007); and nucleosome positioning (Peckham et al. 2007). In our case, because of the potentially diverse mechanisms which direct EP300 and CREBBP binding, we use a complete set of DNA sequence features to capture combinations of binding sites active in different tissues and times of development. To study these distinct modes of regulation, we investigate EP300/CREBBP binding in mouse embryos (Visel et al. 2009), activated cultured neurons (Kim et al. 2010), and embryonic stem (ES) cells (Chen et al. 2008). Our analysis will initially focus on Visel's data set, where several thousands of EP300-bound DNA elements were collected by ChIP-seq in dissected mouse embryo forebrain, midbrain, and limb. We evaluate our method by predicting enhancers vs. random sequence and between EP300/CREBBP ChIP-seq data sets. These comparisons reveal a diversity of predictive sequence features, both within and across data sets. Supplemental Table S1 provides an outline of the analyses performed in this paper.
We show that sequence features in the experimentally identified enhancer set are sufficient to accurately discriminate enhancers from random genomic regions. We also show that the most predictive sequence elements are related to biologically relevant transcription factor binding sites. Notably, our method also finds that some sequence elements are significantly absent in the enhancers (those with large negative SVM weights). For example, we find that binding sites for the zinc finger E-box binding homeobox (ZEB) transcription factor family is depleted in the forebrain enhancers, consistent with its biological role as a transcriptional repressor (Vandewalle et al. 2008). In addition, we provide evidence that enriched sequence elements are positionally constrained within the enhancers and that they are more evolutionarily conserved than less predictive elements in the enhancers, reflecting the combinatorial structure of tissue-specific enhancers.
We further apply our SVM method to predict putative enhancers in both the mouse genome and the human genome from DNA sequence alone. Many of these novel enhancers overlap with regions enriched in EP300 ChIP-seq reads, exhibit greatly increased hypersensitivity to DNase I in the mouse brain, and are proximal to biologically relevant genes. All of these assessments exclude the original EP300 training set enhancers from the analysis. The successful identification of tissue-specific DNase I hypersensitive sites provides powerful independent evidence for the validity of our approach.
Results
Enhancers can be accurately predicted from DNA sequence
Our primary concern in this paper is to identify which sequence features are specific to enhancers and to investigate the degree to which we can identify functional enhancer regions in a mammalian genome using only DNA sequence features in these regions. We initially focus on recent genome-wide experiments that identified EP300 binding sites by ChIP-seq (Visel et al. 2009) in three different tissues (forebrain, midbrain, and limb) at embryonic day 11.5 in mice. Cross-linking in dissected tissue at a particular time point during development can identify tissue-specific enhancers, even when the developmental regulators that mediate EP300 binding are unknown. While EP300 ChIP may not detect all the enhancers active under these conditions, we initially analyze this data set to identify sequence features responsible for EP300 binding in these tissues.
To model DNA sequence features, we use a support vector machine framework. In brief, an SVM finds a decision boundary that maximally distinguishes two sets of data, here a positive (enhancer) and negative (random genomic) sequence set. The basic approach is outlined in Figure 1A, and full details can be found in Methods. Weights, wi, determine the contribution of each feature to this boundary. Once the set of sequence features, xi, is specified, the weights are optimized to maximize the separation between the two classes. We use as sequence features the full set of k-mers of varying length (3–10 bp). While other authors have successfully used databases of experimentally characterized TFBSs as sequence features (Gotea et al. 2010), because the binding specificity of many transcription factors (TFs) has yet to be determined, we prefer k-mers (oligomers of length k) because they are an unbiased, general, and complete set of sequence features. An advantage of this framework is that the SVM can be subsequently used to scan the genome for novel enhancers not in the original training set. The results of scanning a well-studied region near Dlx1/2 is shown in Figure 1B and detects novel and experimentally confirmed enhancers, as discussed in detail below.
Figure 1.

Overview of our methodology. (A) k-mer frequencies are calculated for each of the EP300-bound and negative genomic training sequences. These feature vectors (x1,…,xn) are used to find SVM weights, w, which most accurately separate the positive (enhancer) and negative (genomic) training sets. (B) These weights are used to predict genome-wide enhancers (light green), based on their SVM score. (Brown) positive, (blue) negative. A well-studied region around Dlx1 and Dlx2 is shown here, both known to be expressed in the forebrain. While the predicted enhancers often overlap the training EP300 set (blue), novel enhancers are also predicted and often identify previously experimentally verified enhancers (red) absent from the EP300 training set. The predicted enhancers also preferentially occur in conserved nonexonic regions (dark green) and regions enriched in EP300 signal (dark blue).
To evaluate classification performance, we use a fivefold cross validation method. Initially, the data set to be classified is randomly partitioned into five subsets. One subset is then reserved as a test data set, and the SVM weights are trained on sequences in the remaining four subsets. The SVM is then used to predict the reserved test data set to assess its accuracy. This process is repeated five times so that every sequence element is classified in one test set. Because there is a trade-off between specificity (the accuracy of positively classified enhancers) and sensitivity (the fraction of positive enhancers detected), we measure the quality of the classifier by calculating the area under the ROC curve (auROC), as shown for several cases in Figure 2. We ultimately average the five test set auROCs to give a summary statistic of the SVM performance; these five test sets generate the error bars in Figure 2.
Figure 2.

Classification results on each tissue-specific enhancer set. (A) Classification of forebrain enhancers vs. random genomic sequences. (B) Classification of midbrain enhancers vs. random genomic sequences. (C) Classification of limb enhancers vs. random genomic sequences. Each graph in A, B, and C compares an SVM trained on the full set of 6-mers (solid), the top 100 selected 6-mers (dashed), and an alternative Naive Bayes classifier (dotted). Each curve is an average of five cross-fold validations on a reserved test set; error bars denote one standard deviation over the five cross-fold validation sets. Numbers in parentheses indicate the area under each ROC curve (auROC) for overall comparison. Both the full SVM and SVM with selected features perform very well and significantly better than Naive Bayes. Individually, each tissue-specific set can be accurately discriminated from nonenhancer genomic sequences. (D) Classification of specific tissues vs. other tissues. Forebrain (fb) and midbrain (mb) can be accurately discriminated from limb (lb) but not from each other (fb vs. mb), indicating common or overlapping modes of regulation. (E) Classification ROC curves for forebrain enhancers vs. random genomic sequences for larger negative set sizes. (F) Precision-recall curves for forebrain enhancers vs. random sequences corresponding to the ROC curves and negative sets in E; numbers in parentheses are auPRC. (G) Classification of EP300 forebrain enhancers, neuronal stimulus-dependent enhancers (CREBBP neuron), and mouse embryonic stem cell enhancers (EP300 ES) vs. random genomic sequence. Although the embryonic stem cell data set is somewhat less accurately classified, our SVMs successfully discriminate EP300 or CREBBP bound regions from random sequences. (H) Classification of EP300 fb, CREBBP neuron, and EP300 ES data sets vs. each other is also robust.
To test sensitivity to various assumptions in our SVM construction, we repeated these cross-validation experiments on each tissue-specific enhancer set using SVM classifiers with different types of kernels: spectrum kernels (Leslie et al. 2002), mismatch spectrum kernels (Leslie et al. 2004), and Gaussian kernels. The Gaussian kernel and spectrum kernel vary the functional form by which features contribute to the overall decision boundary, while the mismatch spectrum kernel retains the linear contribution of the features but uses a different set of features by allowing a certain number of base pair mismatches to a given k-mer (see Methods). In addition, we tested a commonly used alternative approach, the Naive Bayes classifier, which learns the parameters for each feature independently (the SVM learns parameters for all features at the same time). Despite this assumption of independence, the Naive Bayes classifier has performed very well on a broad range of machine learning applications.
Our main result, perhaps surprising, is that many SVMs can successfully distinguish enhancers from random genomic sequences with auROC > 0.9, regardless of: the types of kernels, the types of tissues, or the length of the k-mers (Fig. 2; Supplemental Fig. S1A). In general, larger k-mers achieved superior performance (Supplemental Fig. S1A), but predictive power begins to decrease when k is greater than six because of overfitting (the feature vector becomes sparse). On the other hand, Naive Bayes classifiers are significantly less accurate in discriminating enhancers from random genomic sequences (auROC < 0.79), indicating that the assumption of conditional independence between k-mers in the Naive Bayes model impairs its performance. Figure 2A–C shows summaries of comparison between ROC curves of SVM (solid) and Naive Bayes (dotted). Because of its robust performance (auROC = 0.94) and ease of interpretation, we adopt the 6-mer spectrum kernel as our standard model for the remainder of the paper.
Besides distinguishing individual enhancer sets from random genomic sequences, we next tested whether our SVM method could also distinguish between enhancers in different tissues (forebrain, midbrain, limb). Since some enhancers are active in two or more tissues, these overlapping regions were removed from both sets before analysis. With the full set of 6-mers, forebrain and midbrain enhancers can be discriminated from limb enhancers with a reasonable auROC of ∼0.84–0.86. However, the SVM failed to successfully discriminate forebrain and midbrain enhancers (Fig. 2D). This indicates that the compositions of TFBSs enriched in forebrain and midbrain enhancers may be similar to each other but are sufficiently different from those in limb-specific enhancers to permit classification. Significant overlap between the forebrain and midbrain enhancer sets in the original data set supports this interpretation (48.7% of midbrain enhancers are also in the forebrain set).
When comparing against random genomic sequence, we have the freedom to choose the size of the negative sequence set. The genomic ratio of enhancers to nonenhancer sequence is very large (we estimate that enhancers comprise 1%–2% of the genome in a given cell-type), and ideally we would compare alternative prediction methods using a very large negative set. However, some of the computational methods we compared could not handle such large amounts of sequence due to memory constraints. To compare between data sets, we used the same ratio between positives and negatives. To test the scaling with negative set size, we used three negative sets (roughly balanced, 1×, 50× larger, and 100× larger than the positive enhancer set). Although auROC is a standard metric, when the positive and negative sets are unbalanced, the precision-recall (P-R) curve is a more reliable measure of performance than the ROC curve. Precision is the ratio of true positives to predicted positives, and recall is identical to the true positive rate in the ROC curve. The P-R curves can be quantified by the area under the precision-recall curve (auPRC), or average precision. For the classification of EP300 forebrain (fb), limb (lb), and midbrain (mb) enhancers from genomic sequence, auROC is unaffected by the size of the negative set (Fig. 2E), but auPRC drops (Fig. 2F) as n becomes large and the high-scoring tail of the negative sequences becomes competitive with the true positive sequences. However, the trends of auROC and auPRC are usually consistent. Comparison of auROC and auPRC for the negative set size scaling for all positive data sets is shown in Supplemental Figure S3.
Most predictive sequence elements are known transcription factor binding sites
We next investigated which subsets of sequence features allowed the SVM to successfully discriminate enhancers from random sequence. The SVM discriminant function is defined as the sum of weighted frequencies of k-mers in the case of the k-spectrum kernel, and the classification is determined by the sign of the discriminant function (see Methods). Therefore, k-mers with large positive and negative SVM weights indicate predictive sequence features: k-mers with large positive weights are sequence features specific to enhancer sequences, and k-mers with large negative weights are sequences that are present in random genomic sequence but depleted in enhancers. We conducted the SVM classification again, using only the subset of k-mers with largest positive and negative SVM weights (Supplemental Fig. S1). The SVM using fifty 6-mers with the largest positive weights and another fifty 6-mers with the largest negative weights achieves auROC of 0.90 for the forebrain enhancer data set. This demonstrates that the largest weight k-mers predict enhancers with similar accuracy, although the auROC does decrease somewhat compared to the result with all k-mers (Fig. 2A–C). Interestingly, the most frequently observed k-mers do not always have the largest SVM weights or vice versa. We find only a weak correlation between SVM weights and k-mer frequencies (Supplemental Fig. S4). The most predictive single k-mer (auROC = 0.65) is AGCTGC, which is present in 60% of the true positive forebrain enhancers, but it is also present in 34% of the negative genomic regions. By combining many k-mers, the full SVM and the SVM with the 100 top k-mers achieve greater accuracy than single k-mers. The SVM's outperformance of the Naive Bayes classifier, which assumes feature independence, indicates that these features contribute cooperatively.
Significantly, many of the most predictive k-mers, (those with the largest positive weights) are recognizable as binding sites for TFs known to be involved in embryonic nervous system development. We systematically scored each of the predictive k-mers with PWMs for known motifs available in public databases [JASPAR (Bryne et al. 2008), TRANSFAC (Matys et al. 2003), and UniPROBE (Newburger and Bulyk 2009)] using the TOMTOM package (Gupta et al. 2007). Because the databases contain many PWMs from families of TFs with similar specificity, many PWMs often score highly for a given k-mer, so we report for each k-mer the family of matched TFs with q-value < 0.1 (Storey and Tibshirani 2003), and list representative high scoring TFs within that family. This mapped known TFBS to 85% of the most predictive k-mers, while only 24% of all k-mers match a known TFBS (Binomial test P-value = 1.5 × 10−8). Table 1A shows the fifteen 6-mers with the largest positive SVM weights. The full lists of SVM weights used in our analysis are provided in the Supplemental Material. The elements that positively contribute to EP300 binding include many k-mers with TAAT or ATTA cores, which are bound by the homeodomain family (Berger et al. 2008). Several homeodomain protein genes have restricted expression in the embryonic mouse forebrain and are required for proper forebrain development, such as Otx and Dlx (Bulfone et al. 1993; Matsuo et al. 1995; Zerucha et al. 2000). Other predictive factors include the members of the basic helix-loop-helix (bHLH) family, which bind variations of E-box elements (CANNTG). Some bHLH factors are known to be crucial regulators of neural and cortical development (Lee 1997; Bertrand et al. 2002; Ross et al. 2003) and are also known to interact with the coactivator EP300/CREBBP (Chan and La Thangue 2001).
Table 1.
Predictive 6-mers of EP300 forebrain

One of the distinguishing features of our approach is its ability to detect binding sites that are significantly absent or depleted in EP300 enhancers. The presence of k-mers with large negative weights in a sequence significantly decreases the likelihood that that sequence will be classified as an enhancer. Biologically, the presence of these binding sites would interfere with the operation of the enhancer in a specific tissue. We consistently observe that ZEB1-related k-mers have the largest negative weights in forebrain enhancers (Table 1B). For example, the ZEB1 binding k-mer CAGGTA is present in 29% of the negative sequences but only 18% of the forebrain enhancer sequences. Also known as AREB6, ZEB1 (zinc finger E-box binding homeobox 1) is a member of the ZEB family of transcription factors, which play crucial roles in epithelial-mesenchymal transitions (EMT) in development and in tumor metastasis by repressing transcription of several epithelial genes including E-cadherin (Vandewalle et al. 2008). Although ZEB family members can work as both activators and repressors, their depletion in EP300-bound regions implies that ZEB1 binding can disrupt EP300 activation.
Although some negative weight k-mers are predictive (e.g., ZEB1), on average the positive weights in Table 1A are more predictive than the negative weights (Table 1B) for all data sets. The absolute values of most negative weight k-mers are significantly less than those of the positive weight k-mers, as shown in Figure 3 (discussed below), where each k-mer weight is plotted along the vertical axis. The asymmetry in SVM weights indicates that the predictive features are primarily identifying k-mers that are enriched in the enhancers rather than k-mers that are enriched in random genomic sequence (or equivalently, depleted in enhancers).
Figure 3.

Predictive SVM sequence features are more conserved. Scatter plot between SVM weights and conservation scores (phastCons scores) for 6-mers in forebrain enhancers. Two well-known TFBS, TAAT cores (red rectangles), and E-box elements (blue triangles) are highlighted. Three standard deviations above the mean (corresponding to P-value of ∼0.001) is denoted for each axis independently. The sequence of all 6-mers beyond three standard deviations above the mean is displayed.
Predictive sequence elements are evolutionarily conserved and positionally constrained within enhancers
In their previous analysis, Visel et al. showed that most EP300-bound regions are enriched in evolutionarily constrained noncoding regions (Visel et al. 2009). However, not all sequences in the EP300-bound regions (average length 750–800 bp) are conserved; rather, several more localized peaks of conservation (10–100 bp) within the EP300-bound regions are observed in most cases. These peaks of localized conservation probably identify the smaller functional regions within a more extended enhancer. We hypothesized that if the predictive k-mers reflect actual TFBSs, they would tend to be preferentially located within these evolutionarily conserved localized regions. To test this systematically, we measured the degree to which individual k-mers were present in conserved regions by averaging the phastCons conservation score (Siepel et al. 2005) over each instance of the k-mer (see Methods), and examined its correlation with SVM weight. Figure 3 shows that k-mers with large positive SVM weights are significantly more conserved than average. All but one (CCCCTC) of the 6-mers with large positive SVM weights (three or more standard deviations above the mean) have large conservation scores (at least one and a half standard deviation above the mean conservation score). While the most predictive k-mers are significantly more conserved, moderate correlation between the phastCons conservation scores and the SVM weights for all k-mers is also observed (Pearson correlation coefficient = 0.35). This evidence supports the idea that the predictive sequence features are more evolutionarily conserved than the less predictive regions within the enhancers.
Since conservation is found in narrow peaks within the enhancers, it follows that there might be additional positional constraints between the predictive elements. Mechanistically, these constraints are most likely indicative of a cooperative mechanism, either involving TF-TF interactions or spatially constrained activity of individual factors. Spatial constraints between TFBSs have been observed frequently in yeast (Beer and Tavazoie 2004). In Figure 4, we compare the distribution of minimum pairwise distances between the ten most predictive sequence elements in the forebrain enhancers (6-mers with the largest positive weights) to their distribution in the null sequences. The forebrain pairwise distance distribution is shifted to lower distances (they are closer to each other) compared to null sequences. To measure the statistical significance of this difference, we calculated the pairwise distance distribution for these 6-mers in 100 different negative sets. The standard deviations of these 100 negative sets are shown as dashed lines in Figure 4, and the forebrain distribution often deviates from the null distribution by several standard deviations, especially for small spacing. We can also measure the difference between the forebrain and null pairwise distance distributions by the two-sample Kolmogorov-Smirnov test, (P-value < 2.2 × 10−16), which further demonstrates the significant clustering of predictive sequence elements. More interestingly, if we concentrate on the small spacing end of this distribution (inset in Fig. 4), we observe periodic enrichments with characteristic spacing of 10–11 bp. The highest peak is around 11 bp, almost two times higher than the null distribution. These positional correlations suggest cooperative binding interactions in phase with the 10.5 bp DNA helix periodicity, consistent with previous observations (Erives and Levine 2004; Hallikas et al. 2006), and local physical interactions between the factors that bind these DNA sequence elements.
Figure 4.

Predictive SVM sequence features are spatially clustered. Distributions of minimum pairwise distances between the most predictive sequence features in forebrain enhancers vs. random genomic sequences. Ten 6-mers with the largest positive SVM weights (Table 1) are used. To measure the significance of these differences, we generated 100 distinct full negative genomic sequence sets (using our null model; see Methods). Each negative set has the same length, repeat fraction, and number of sequences as the EP300 forebrain enhancer training set. The predictive elements are significantly clustered in the forebrain enhancers compared to the random genomic sequences (the red distribution is significantly shifted toward smaller minimum distance). At higher resolution (inset), distinct peaks around 11 bp, 22 bp, etc., are observed, suggesting positioning in phase with the periodicity of the DNA helix. P-values are indicated: (*) <0.01, (**) <0.001, (***) <0.0001.
Genome-wide SVM predictions identify novel enhancers
To predict additional functional regions that were not determined to be EP300-bound from the ChIP-seq data, we scanned the entire genome systematically with our SVM. We segmented the mouse genome sequence into 1-kb regions with 0.5k-bp overlap, resulting in about 5.2 million overlapping sequence regions. To compare with the 2453 forebrain region “EP300 training set”, we followed Visel and removed centromeric regions, telomeric regions, and regions containing at least 70% repeats, (however, this filter had minimal impact on our predictions). We then scored all these 1-kb regions using the SVM with the k = 6 spectrum kernel for forebrain enhancers. An example of the continuous SVM score along the Dlx1/2 locus is shown in Figure 1B (“Raw SVM Score”). Dlx1 and 2 are expressed in the mouse forebrain (Bulfone et al. 1993; Ghanem et al. 2003; Wigle and Eisenstat 2008). Besides the sole EP300 training set element in this region (URE2) (labeled “EP300 ChiPseq” in Fig. 1B), two other enhancers within this locus have been experimentally validated (“Known Enhancers”) (labeled i12a and i12b) (Ghanem et al. 2003). These enhancers (i12a and i12b) were detected by our SVM but were not in the EP300 training set because their raw sequence read density was not above the stringent threshold used in Visel et al. (2009). Comparing the “Raw EP300 ChIPseq” track to our “Raw SVM score” in Figure 1B shows striking correlation: Most of our predicted high scoring SVM regions have raw EP300 ChIP-seq signal significantly above background but did not have sufficient read density to be included in the EP300 training set. To support this anecdotal evidence, we evaluated the genome-wide correlation between our SVM predicted regions and EP300 read density. In Supplemental Figure S5, we plot the EP300 ChIP-seq read density as a function of distance from the center of each of the top 1% SVM scoring regions. We find significant enrichment of EP300 ChIP-seq signal around the SVM predicted regions, indicating that many of these predicted loci are, indeed, bound to some extent by EP300 but fall somewhat below the read threshold used to determine the EP300 training set. Supplemental Figure S6 shows the correlation between SVM score and EP300 reads in all genomic 1-kb regions, showing again that there is a significant population of high scoring SVM regions enriched in EP300 signal but not in the EP300 training set.
To define a high confidence set of enhancer predictions, we chose an appropriate cutoff for the SVM score using more realistic large negative training set sizes (50× and 100× negative sequences), covering ∼6%–12% of the nonrepetitive genome. We can estimate our false discovery rate (the expected fraction of predicted positives which are false positives, FP/(FP+TP), from the P-R curves in Figure 2F. The precision is weakly dependent on negative set size when n is large, due to the fact that the positive and negative histograms of SVM scores have a similar shape for larger negative set sizes, as shown in Supplemental Figure S7. To trade off precision and recall, we choose a cutoff that corresponds to 50% recall, which at 1× is an SVM score of 1.0. For the large negative sets, precision is ∼50% when recall is 50%, and we therefore estimate our false discovery rate to be ∼50%. In other words, at this cutoff (SVM > 1.0) on the training set, we capture 50% of the EP300 training set regions and an equal number of negative regions.
In what follows we will be comparing the properties of our SVM predicted enhancer regions (SVM > 1.0), the EP300 training set regions, and nonenhancer genomic regions (SVM < 1.0). These three sets are all distinct, i.e., each genomic 1-kb region can only belong in one class. Any 1-kb region which overlaps a training set region by as little as 1 bp is excluded from the SVM sets and included in the EP300 training set. We will show that the EP300 training set and SVM predicted regions have similar properties, much different than the nonenhancer regions.
At an SVM score threshold of 1.0, we predict 33,232 1-kb regions in the genome (outside of the EP300 training set), or 26,920 enhancers after merging overlapping regions, and we expect about 13,460 of these to be true enhancers. This threshold appears to be a good tradeoff between detecting many biologically significant enhancers with an acceptable false discovery rate. The full lists of SVM scores for these regions are included as Supplementary Material. We also established the robustness of these top SVM scoring regions by training separate SVMs with independent random null sequence sets as the negative class. There is extensive overlap between the top scoring regions using these different SVMs (Supplemental Table S2), and the correlation of individual SVM scores between two different SVMs is high (Pearson correlation coefficient = 91.5%), as shown in Supplemental Figure S8. That the SVM classifier identifies many more sequence regions than the EP300 training set may be due to several factors: (1) As discussed above, these predicted regions may be false positive enhancers; (2) they may be true positive enhancers that were undetected in the ChIP experiments because of an overly stringent cutoff for defining the EP300 training set; (3) they may be true positive enhancers that are not EP300-bound in this tissue at the developmental stage of the experiment but may be EP300-bound in other tissues or times; or (4) they may be true positive enhancers that operate independently of EP300 but share some similar sequence features. All but the first possibility are potentially biologically interesting.
To assess the validity of these genome-wide predictions with independent experimentation, we quantified the DNase I hypersensitivity of the high scoring forebrain SVM regions with experiments in embryonic mouse whole brain provided by the mouse ENCODE project (data available from http://genome.ucsc.edu/ENCODE/; J. Stamatoyannopoulos, in prep), using methods described in John et al. (2011). DNase I hypersensitivity measurements detect open or accessible chromatin, including promoters and enhancers, independent of EP300 binding. Although these DNase I experiments are not strictly specific to forebrain and were 3 d later in development, enrichment in brain hypersensitivity strongly corroborates our predictions as tissue-specific enhancers. In Figure 5, we split the predicted 1-kb regions from the EP300 fb trained SVM into four classes (SVM < 0.5, red; 0.5 < SVM < 1.0, gray; 1.0 < SVM < 1.5, cyan; and SVM > 1.5, blue) and one EP300 training set class (EP300-bound regions, green). We plot the distributions of average intensity of DNase I hypersensitivity of the different SVM scoring classes in Figure 5A, which shows a dramatic increase in DNase I signal in E14.5 brain only for high scoring SVM regions. There is no enrichment of DNase I signal for the same regions in other tissues; for example adult kidney is shown in Figure 5B as a negative control. Because the DNase I hypersensitive regions include promoters and other open regions, the converse is not true, i.e., while almost all high-scoring SVM regions have a high DNase I signal, not all high-signal DNase I regions have a high SVM score (data not shown). With this understanding, we can evaluate the precision and specificity with which our SVM detects DNase I sensitive enhancers. Because the SVM score and DNase I signals are continuous, we consider DNase I signal > 10 to be positive (open chromatin), and DNase I < 2 to be negative (not open) for purposes of quantification, consistent with the distributions in Figure 5A,B. Then, regions with DNase I > 10 and SVM > 1.0 are true positive predictions, and DNase I < 2 and SVM > 1.0 regions are false positive predictions. Table 2 shows the number of 1-kb genomic regions in each class. The precision is TP/(TP+FP), or the accuracy of the predicted positives. The sensitivity is 1−FPR (false positive rate), or the fraction of negatives that we predict to be positive. As shown in Table 2, SVM > 1.0 predictions have a 56.3% precision, and more stringent SVM > 1.5 predictions have a 74.5% precision. These results are consistent with our above estimate that 50% of our novel predictions are true enhancers functioning in mouse brain.
Figure 5.

SVM-predicted regions are hypersensitive to DNase I in the relevant context. To independently confirm our predictions with DNase I measurements in the embryonic mouse brain, we plot the distributions of the average intensity of DNase I hypersensitivity of different forebrain SVM scoring regions. (A) DNase I hypersensitivity measured in E14.5 wholebrain. (B) DNase I hypersensitivity measured in an adult 8-wk kidney, as a negative control. We observe significant enrichments only in high-scoring SVM-predicted regions in the brain.
Table 2.
Precision and sensitivity of detecting DNase I hypersensitive enhancers

To further support the biological significance of these novel SVM-predicted enhancers, we examined their proximity to forebrain-expressed genes. Microarray experiments (Visel et al. 2009) identified 885 (495) genes overexpressed (underexpressed) in the forebrain at E11.5. We examined the intergenic distance between the EP300 training set regions and the transcription start site (TSS) of the nearest overexpressed genes. We also found the distance between our SVM-predicted enhancer regions and the overexpressed genes. All regions overlapping a training set region were omitted from the set of predictions. As shown in Figure 6, both the EP300 training set and our predicted enhancer regions are significantly enriched near (within 10 kb of) the TSS of a forebrain overexpressed gene. Notably, the SVM predicted regions with the more stringent SVM cutoff score (SVM > 2.0) are even more enriched within 10 kb of the overexpressed genes than the EP300 training set, further evidence that the SVM is capturing functional regions with spatial and temporal specificity. In comparison, randomly chosen genomic regions show no such enrichment. While the EP300 training set is not enriched near forebrain underexpressed genes, our SVM predicted regions are significantly enriched within 10 kb of forebrain underexpressed genes (Fig. 6). What is a potential role of these predicted regions near underexpressed genes? Because the EP300 bound regions are not enriched near the underexpressed genes, it is unlikely that EP300 is acting as a transcriptional repressor here. It seems more likely that the SVM is predicting enhancers that are bound by EP300 in other tissues or at other times in development. These enhancers could activate the neighboring genes relative to their expression level at E11.5 in the forebrain, which would appear indistinguishable from forebrain repression. This hypothesis is supported by the fact that several of the underexpressed genes with nearby SVM-predicted enhancers play roles in nervous system development, including many Hox genes known to function in A-P axis patterning.
Figure 6.

SVM-predicted enhancers are preferentially located near transcript start sites (TSSs) of forebrain-expressed genes. Here we plot the distribution of the distance between the EP300 and SVM-predicted regions and the nearest forebrain-expressed gene [as assessed by the microarray experiments of Visel et al. (2009)]. Any region which overlapped a training set region was excluded from the analysis. Both the EP300 (red) and SVM-predicted regions are preferentially located within 10 kb of the TSS of a forebrain-overexpressed gene (above the axis). This is true whether we use a cut-off of SVM > 1.5 (green) or a more restrictive SVM > 2.0 (blue) to define the enhancer set. As a null set, we compare to the average of 100 randomized genomic positions, with a 95% confidence interval shown (gray). Interestingly, when we calculate the same distributions for the distance between a EP300 or SVM-predicted region and the nearest forebrain-underexpressed gene (below the axis), only the SVM-predicted regions show significant clustering toward the TSS, relative to the randomized control. Although the EP300 data preferentially identifies activating enhancers in the forebrain, the SVM may be detecting common sequence features shared in enhancers, which are repressive in the forebrain but are activating in other contexts.
SVM also predicts human enhancers
We next assessed the ability of our SVM to predict human enhancers. We found human orthologous regions (hg18) of the mouse EP300 training set with the liftOver utility from the UCSC genome browser (Karolchik et al. 2008). With 70% or greater identity, 2205 of the 2453 forebrain enhancers were successfully mapped onto the human genome. We discarded 13 mapped sequences longer than 3 kb. We then trained SVMs to discriminate this positive human training set from an equal number of human random sequences generated by our null model and achieved reasonably high auROC = 0.87 (Supplemental Fig. S9). We also tested more stringent orthology cutoffs (requiring 90% and 95% identity instead of 70%) and found that the overall performance was very similar (Supplemental Fig. S9). Thus, an SVM trained on human sequence homologous to the mouse EP300 training set sequences is able to predict test set enhancers with only slightly reduced accuracy relative to mouse.
In addition, we predicted human enhancer regions with a SVM trained on the mouse data set, which does not require sequence alignment to identify orthologous regions. This approach might be valuable in situations where it is difficult or impossible to obtain similar data sets in each species. It also provides further information about the conservation of predictive k-mers between the two species. We first compared these two raw SVM scores (one trained on the human homologous set, the other on the mouse data set) on the human genome around Otx2, observing very similar SVM score patterns. Moreover, an experimentally verified enhancer (Kurokawa et al. 2004) is captured by both SVMs (Supplemental Fig. S10). We then systematically analyzed the entire genome to assess how many top SVM-scoring regions overlap each other (Supplemental Table S3). Although the overlaps are not as significant as scores using only different negative sets (Supplemental Table S2), a large fraction of top SVM-scoring regions are still shared between the two SVMs, so to a large degree, an SVM trained on mouse can be used to successfully predict human enhancers. This result is in general agreement with in vivo experimental results (Wilson et al. 2008) where human DNA transplanted into mice was shown to bind mouse TFs (HNF1A, HNF4A, HNF6) in a pattern virtually indistinguishable from their binding patterns in human, indicating that variations in genomic TF binding between human and mouse are due to local DNA sequence differences, not due to evolutionary divergence of individual TF binding specificities between the two species.
Comparison between different EP300/CREBBP ChIP-seq data sets reveals sequence elements important for pluripotency
The success of our SVMs in predicting EP300 binding in mouse embryonic brain and limb motivated a comparison with other EP300/CREBBP ChIP-seq data sets. We first looked at the overlap between Visel's in vivo data set (EP300 forebrain, midbrain, and limb) and two other data sets: CREBBP-bound regions in activated cultured mouse cortical neurons (Kim et al. 2010), and EP300-bound regions in cultured mouse embryonic stem cells (Chen et al. 2008). We will refer to these as “CREBBP neuron” and “EP300 ES” in the following discussion. We were interested in these data sets because they share similar ChIP-seq methodology, because it would help us address the overlap between activation mediated by the close homologs EP300 and CREBBP, and to address differences in EP300 binding in different tissues and cell populations. CREBBP neuron enhancers only overlap significantly with EP300 forebrain enhancers (not midbrain or limb) (Supplemental Table S4A). EP300 ES enhancers do not significantly overlap with any other set (fb, mb, lb, or CREBBP neuron) (Supplemental Table S4B). This indicates that EP300-mediated embryonic neuronal development is linked to CREBBP-mediated neural activity dependent transcription via extensively shared common regulatory regions. We indeed observe that several predictive k-mers with large positive weights, such as homeodomain binding sites (TAAT core) and bHLH domain binding sites (E-box, CANNTG), are shared between the two data sets (Table 1A; Supplemental Table S5A), which further indicates common modes of regulation.
Figure 2G shows ROC curves discriminating CREBBP neurons (auROC = 0.93) and EP300 ES (auROC = 0.77) from random genomic sequences. The lower EP300 ES auROC is partly due to the relatively smaller number of regions bound in the EP300 ES positive set. Also, the EP300 ES data set contains a larger fraction of repeat sequences, indicating that this data set may be less specific for functional EP300 binding. Nonetheless, SVMs still can extract informative k-mers from this data set and can largely discriminate the EP300 ES set from random genomic sequences. Alternatively, instead of comparing to random genomic sequence, we can also successfully classify these sets (EP300 forebrain, CREBBP neuron, EP300 ES) against each other, as shown in Figure 2H. It is interesting to note that EP300 forebrain can be discriminated from CREBBP neuron with high auROC, even though they share many regions and have some common predictive k-mers (homeodomain, SOX, bHLH) when classified against random sequence (Table 1A; Supplemental Table S5A). However, when classified against each other, we observe that the predictive k-mers specific for EP300 forebrain remain homeodomain, SOX, and bHLH, but the k-mers predictive for CREBBP neurons become nuclear factor I (NFI), activator protein 1 (AP1), and cyclic AMP-responsive element-binding protein (CREB) binding sites (Supplemental Table S7). Therefore, homeodomain, SOX, and bHLH binding sites may play more prominent roles in neural developmental processes than in neural activity dependent transcription.
We also assessed the biological significance of the predictive k-mers in these new data sets. We find that most of the predictive k-mers can be related to known TFBSs (Supplemental Tables S5, S6), and that many of the identified TFBSs are involved in signaling pathways known to function in the relevant experimental conditions. For the CREBBP neuron data set, AP1 related 6-mers, GACTCA and TGACTC, the first and third largest weights respectively (Supplemental Table S5A), are the target of heterodimers of the regulators Fos and Jun, which play critical roles in neural activity-dependent transcription regulation (Flavell and Greenberg 2008). CREB, which directly interacts with CREBBP, is also essential for the activation of several genes in response to neural stimulation, and its binding site is ranked fourth in Supplemental Table S5A (Flavell and Greenberg 2008; Kim et al. 2010). Kim et al. noted that two other transcription factors, neuronal PAS domain-containing protein 4 (NPAS4) and serum response factor (SRF) as well as CREB, strongly colocalize with CREBBP binding regions. NPAS4 contains a bHLH domain, and its canonical binding sites, E-box elements, are ranked at second and sixth in Supplemental Table S5A. The SRF binding site is also known as a CArG box, whose consensus sequence is CCWTATAWGG (Bryne et al. 2008). A specific k-mer instance of the CArG box is ATATGG, ranked at 17th with w = 3.00, just below the top fifteen in Supplemental Table S5A. Therefore, all well-characterized TFBSs known to play a role in neuronal activation are successfully captured by our SVM. Interestingly, we discovered that two additional transcription factor families also score highly in the CREBBP neuron data set: homeodomain and NFI. These families have been discussed little in this context, although it is known that both NFI and homeodomain transcription factors are key regulators of central nervous system development (Wilson and Koopman 2002; Mason et al. 2008). We found only one relevant example of neural activity-dependent expression of a homeobox protein, LMX1B (Demarque and Spitzer 2010). There may be still unknown mechanisms involving NFI and homeodomain proteins in the context of neural activity-dependent transcriptional regulation, but broadly speaking, our results indicate significant pleiotropy between neuronal developmental pathways and neural activity- dependent signaling pathways.
Comparison of the EP300 ES data to CREBBP neuron and EP300 forebrain can address which binding sites and factors are responsible for maintaining a differentiated or pluripotent state. For the EP300 ES data set, our method identifies factors known to be crucial for maintaining ES identity: We find high scoring binding sites for NANOG-POU5F1(also known as OCT4)-SOX2 SOX-family factors (Supplemental Table S6A), essentially the same binding sites found in previous studies (Pavesi et al. 2001; Chen et al. 2008). We have used a uniform approach to map k-mers to TFBS in the databases, but there is substantial overlap in many TF specificities, and some reported matrices may score higher than the biologically relevant database entry. For instance, in Supplemental Table S6A, the high-scoring matrices (SOX17, POU2F1, and POU3F3) appear on the list instead of the relevant SOX2, POU5F1, and NANOG, which have nearly identical binding sites. SOX2, POU5F1, and NANOG bind a combination of the SOX2 (CATTGT) and POU5F1 (ATGCAAAT) consensus sites (Chen et al. 2008), and the 6-mer subsequences within the combined binding site (CATTGTYATGCAAAT) have high SVM weights. Supplemental Figure S11 shows how large weight k-mers tile across this extended known binding site. In addition, we also find positive weight binding sites for ESRRB and STAT3, which are known to be frequently located nearby the NANOG-POU5F1-SOX2 clusters assessed by ChIP-seq analysis (Chen et al. 2008). More interestingly, we find that many of the positive weight EP300 ES k-mers (ESRRB, RORA1/2, PPARG) are among the largest negative weights in CREBBP neuron (Supplemental Table S6B), indicating that binding sites for factors responsible for maintaining pluripotency are significantly absent from neuronal enhancers (CREBBP neuron), as would be expected given the developmental maturity of neurons.
SVM can predict other ChIP-seq data sets
Until this point we have applied our SVM method to classify and detect EP300/CREBBP-bound enhancers, but this approach is equally applicable to any data set which may be framed as a sequence classification: e.g., ChIP-seq, ChIP-chip, or DNase I hypersensitivity data sets. In these situations, the SVM can be used to identify primary binding sites in regions identified by transcription factor ChIP experiments and may also identify binding sites for secondary factors colocalized with the ChIPed TF or binding sites significantly depleted in the functionally occupied regions. We note that popular de novo motif-finding methods such as AlignACE (Hughes et al. 2000) or MEME (Bailey and Elkan 1994) have limited success when applied to data sets of this size. When run on the forebrain enhancer data set, AlignACE (when it converged) failed to report any meaningful motifs. While Chen et al. (2008) did successfully identify SOX2, POU5F1 (OCT4), and NANOG binding sites in the EP300 ES data with Weeder (Pavesi et al. 2001), the EP300 ES data set was the smallest and least diverse of the data sets we analyzed.
To directly assess the ability of our SVM to predict binding of individual transcription factors, we analyzed ChIP-seq results on the TF ZNF263. We chose ZNF263, a 9-finger C2H2 zinc finger which is predicted to have a binding site of ∼24 bp, to assess how well k-mers can represent extended degenerate binding sites. We used ChIP-seq data on ZNF263 in a K562b cell line (Frietze et al. 2010) which identified 1418 strongly bound regions. Predicting against a 50× random negative set yielded auROC = 0.938 and auPRC = 0.51 (Supplemental Fig. S12B,D). Many of the largest weight k-mers are subsequences within the large PWM found by de novo motif-finding tools applied to this data set (Frietze et al. 2010), and the SVM is combining k-mers which tile across the binding site to achieve high predictive accuracy. The k-mer GAGCAC also received a large weight. This indicates that our approach should have significant predictive value for a wide range of binding data.
Comparison to alternative approaches
As an alternative to k-mers, we also tried using known PWMs as features in an SVM. We used 811 PWMs from existing databases of known TF specificities [JASPAR (Bryne et al. 2008), TRANSFAC (Matys et al. 2003), and UniPROBE (Newburger and Bulyk 2009)]. When using these features, we used the highest PWM scores in each sequence for each matrix as the feature vector. This 811-PWM SVM was able to achieve auROC = 0.87 for forebrain enhancers (compared to auROC = 0.93 for k-mers), somewhat less predictive than our k-mer approach (Supplemental Fig. S12A), against a 50× random negative set. However, this translates into a significantly lower auPRC = 0.22 (compared to auPRC = 0.43 for k-mers) (Supplemental Fig. S12B). The optimal combined weighting of the known PWMs and 6-mers features (2080 + 811 total features) gives marginal improvement (auROC = 0.93 and auPRC = 0.49) over 6-mers alone. We also applied the 811-PWM SVM to the ZNF263 data set, which achieved auROC = 0.83 (compared to auROC = 0.94 for k-mers), reflecting the fact that accurate PWMs for ZNF263 were absent from the databases (Supplemental Fig. S12B,D). Again this seemingly small change in auROC corresponds to a large drop in auPRC = 0.14, compared to auPRC = 0.51 for k-mers. This demonstrates that using sequence features from an unbiased and complete set can be more valuable than using an incomplete set of more accurate features (PWMs). Using the set of known TF PWMs is less predictive than our k-mer SVM, but a more complete set of PWMs might perform better. Combining the predictive k-mers into a more general PWM via a method similar to positional oligomer importance matrices (POIMs) (Sonnenburg et al. 2008) might allow clearer identification of informative sequence features from within the k-mer SVM but would not affect predictive performance.
We also compare our approach to alternative kernel methods. We applied the weighted degree kernel with shifts (WDS) (Rätsch et al. 2005) to the CREBBP neuron data set (as WDS requires input sequences of equal length) and found auROC = 0.83, compared to auROC = 0.93 for our k-mer SVM. A notable SVM based approach which incorporates positional information between general k-mer features (KIRMES) has been recently described (Schultheiss et al. 2009; Schultheiss 2010). We applied this package to the forebrain EP300 data set and found auROC = 0.90. In the current implementation of KIRMES, k-mers are selected by their relative frequency in the positive set, and it is likely that further optimization would make this approach comparable to our k-mer SVM result. Additionally, the periodic spatial distribution in Figure 4 suggests that a model based on difference in angle (similar to Hallikas et al. 2006) would be more appropriate than the Gaussian spatial dependence used in KIRMES. Another approach to predict promoters (Megraw et al. 2009) used PWMs and l1-logistic regression. We found little difference between logistic regression and SVM: Using our k-mer feature vectors in l1-logistic regression yielded auROC = 0.92 on the EP300 forebrain data set, using publicly available software (Koh et al. 2007).
Discussion
In this study, we have shown that a support vector machine can accurately predict regulatory sequences without any prior knowledge about transcription factor binding sites, using only general genomic sequence information. While the ROC and P-R curves demonstrate that the SVM is able to identify enhancers based on their sequence features, the biological relevance of the predicted enhancers is further supported by the following: (1) Most of the predictive sequence features identified by our methods are binding sites of previously characterized TFBSs known to play a role in the relevant context; (2) the enriched predictive sequence features are much more evolutionarily conserved within the enhancers than the less predictive sequence features, which suggests that the predictive features are under selection and comprise the functional subset of the larger enhancer regions; (3) these sequence features are significantly more spatially clustered in the enhancers than would be expected by chance, also a well-known characteristic of functional binding sites; (4) genomic regions with high forebrain SVM scores are strongly enriched in DNase I hypersensitivity signals in mouse brain but not in other tissues; (5) the predicted enhancers frequently overlap with regions of enhanced ChIP-seq signals but are somewhat below the signal cutoff necessary to be included in the original EP300 training set; and (6) these novel predicted enhancers are preferentially positioned near biologically relevant genes, and many have been experimentally verified in other studies, which further supports their biological relevance and functional roles.
When scanning the whole genome to predict putative enhancers, we predict that 50% of our 26,920 nonoverlapping enhancers with forebrain SVM scores above 1.0 are true positives. This is a conservative estimate of our ability to detect novel enhancers, since, when scanning the genome, we have scored 1-kb arbitrarily delimited chunks of sequence; more accurate predictions might be possible by varying the endpoints of the predicted regions. Nevertheless, this genome-wide scan discovers thousands of novel predicted enhancers that were not in the original experimental training set. We have shown that we can predict human enhancers based on these mouse enhancer experiments by measuring the overlap between human enhancers predicted by an SVM trained on the mouse sequence and comparing these predictions to an SVM trained on human sequence orthologous to the mouse enhancer sequences. Finally, by comparing between other EP300/CREBBP ChIP-seq data sets, we find sequence features that are able to differentiate between enhancers that operate in different tissues or at different developmental stages. Some of these sequence features are enriched in enhancers in one specific tissue or state, but other predictive elements are notably depleted in some classes of enhancers.
It is perhaps surprising that such a simple description of sequence features (k-mer frequencies) is able to classify enhancers and ChIP-seq data so well. The SVM is apparently combining k-mer features in a sufficiently flexible way to reflect combinations of binding sites and/or sequence signals which modulate chromatin accessibility. Developing an optimal sequence feature vector remains an area for future work; however, our results showing that the SVM is more accurate than Naive Bayes suggests that successful prediction requires the ability to combine features without evaluating them independently.
Several features of our results suggest ways that our method could be improved to make more accurate predictions. It is likely that incorporating positional constraints between the features would improve the accuracy of the predictions, consistent with our observation of nonrandom spatial distributions between predictive features in the SVM. Kernel approaches have been developed which incorporate positional information, but most have been developed in the context of positional constraints relative to a single preferred genomic location or anchor point. In application to other problems, positional information relative to a transcription start site (Sonnenburg et al. 2006b), to a splice site (Rätsch et al. 2005; Sonnenburg et al. 2007), or to a translational start site (Meinicke et al. 2004) has been implemented in SVM contexts. Positional preference relative to a mean anchor point has been incorporated in a de novo motif discovery method developed by Keilwagen et al. (2011). However, the aforementioned methods are not strictly appropriate to the biological problem of enhancer detection, because enhancers have no such preferred fixed location, and the relevant positional constraints are between sequence features within the enhancer. Many approaches have modeled clusters of known binding sites (for review, see Su et al. 2010) but have limited application to mammalian enhancer prediction.
Although we have provided evidence that our SVM-predicted regions are likely functional, to what degree are we predicting these enhancers accurately based on sequence features which are tissue-specific? Alternatively, we could be detecting sequence features which are general to larger classes of enhancers. These common features could allow access, could stabilize, or could be recognized by generic components of the enhanceosome (Thanos and Maniatis 1995; Maniatis et al. 1998), whose activity could be modulated by tissue-specific factors, much as Pol II operates generally. Ultimately this should be determined by individual experiments, but we here address this problem computationally by investigating overlaps between forebrain- and limb-specific predicted regions, which we then compare with the overlaps between EP300-enriched regions in forebrain and limb. For this comparison, we independently determined EP300-enriched regions from the raw data set using the same threshold criteria as the previous study (Visel et al. 2009) except that we have used fixed-length 1-kb regions, rather than the ChIP-seq-determined peak regions. With a 1% false discovery rate (FDR), we obtained 3390 EP300-enriched regions of forebrain and 2607 regions of limb. Visel's EP300-bound regions are highly tissue-specific; there are only 243 regions (7%–9%) shared by the two sets. For the SVM predictions, a significantly larger fraction of forebrain predicted regions (6104 out of 39,714, 15%) are found in 34% of the limb predicted regions (18,027). This suggests that our SVMs learn features that are generally enriched in enhancers, in addition to tissue-specific sequence features. As a result, two SVMs trained on entirely different data sets can predict common regions that have general enhancer function. Moreover, the 6104 regions predicted by both limb and forebrain SVMs overlap with small EP300 peaks that are somewhat below the conservative threshold (FDR < 0.01); almost 50% have peak in at least one tissue. This observation further supports our hypothesis that SVM-predicted regions are likely to be functional. A further complication is that individual tissues consist of heterogeneous populations of cell types, and enhancers predicted in distinct tissues may only be active in subsets of cell types. A detailed analysis of which sequence features impart tissue specificity and which are general is suggested as a focus for future investigations.
Methods
Data sets
As positive data sets, we initially used the genome-wide in vivo EP300 binding sites identified by ChIP-seq (Visel et al. 2009), composed of three different sets of tissue-specific enhancers (forebrain, midbrain, and limb) of embryonic day 11.5 mouse embryos. There were 2453, 561, and 2105 sites reported, respectively, and we directly use the entire sequences without modification. We also analyzed two other data sets (Chen et al. 2008; Kim et al. 2010). Chen et al. reported 524 EP300 binding sites in mouse embryonic stem cells, and Kim et al. reported ∼12,000 neural activity-dependent CREBBP binding sites in stimulated cultured mouse cortical neurons. Since both CREBBP data sets report only peaks of the ChIP-seq signals, we extended 100 bp (Fig. 2G) or 400 bp (Fig. 2H) in both directions from these peaks to obtain sequences for further analysis.
We generated negative sequence sets to match the distribution of sequence length and repeat element fraction of the corresponding positive sets (Supplemental Fig. S2). Repeat fractions were calculated using the repeat masked sequence data from the UCSC genome browser (Karolchik et al. 2008). We selected random genomic sequences from the mouse genome according to the following rejection sampling algorithm:
Sample a length l from the enhancer length distribution.
Sample a sequence of the length l, randomly from the genome.
Let x be the repeat fraction of the sampled sequence. Sample Y∼Bernoulli(αp(x)/q(x)), where p(x) is the probability that x occurs in the enhancers, q(x) is the probability that x occurs in the genomic sequence, α is the constant so that the maximum of p(x)/q(x) equals 1.
Accept the sequence if Y = 1; reject otherwise.
Repeat 1–4 until the desired number of sequences are sampled.
All positive and negative sequence data sets used for our analysis are available at http://www.beerlab.org/p300enhancer. We used the following negative set sizes—EP300 fb: n = 4000, 2453 (1×), 122,650 (50×), 245,300 (100×); EP300 mb: n = 4000, 561 (1×), 28,050 (50×), 56,100 (100×); EP300 lb: n = 4000, 2105 (1×), 105,250 (50×), 210,500 (100×); EP300 fb human: n = 2192 (1×); EP300 ES: n = 524 (1×), 5240 (10×), 26,200 (50×), 52,400 (100×); CREBBP neuron: n = 11,847 (1×), 592,350 (50×), 1,184,700 (100×); ZNF263: n = 1418 (1×), 70,900 (50×), 141,800 (100×).
Support vector machine
An SVM (Boser et al. 1992; Vapnik 1995) finds a decision boundary that separates the positive and negative training data. This decision boundary is a hyperplane which maximizes the margin between the two sets in the feature vector space. We have N labeled vectors 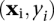 ,
, 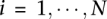 , where
, where 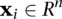 and
and 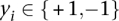 is the class label. For the linear case, the decision boundary is found by minimizing
is the class label. For the linear case, the decision boundary is found by minimizing  such that
such that 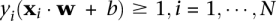 . In practice, the optimal solution is found by maximizing the dual form:
. In practice, the optimal solution is found by maximizing the dual form: 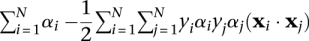 over
over 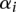 with the constraints,
with the constraints, 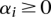 , and
, and 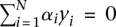 (Joachims 1999; Sonnenburg et al. 2006a). The SVM weight vector w can be constructed from the
(Joachims 1999; Sonnenburg et al. 2006a). The SVM weight vector w can be constructed from the 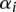 , using
, using 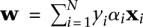 . The SVM discriminant function, or “SVM score,”
. The SVM discriminant function, or “SVM score,” 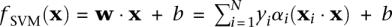 , represents the distance of any vector x from the decision boundary, and determines the predicted label of the vector x.
, represents the distance of any vector x from the decision boundary, and determines the predicted label of the vector x.
The inner product 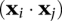 is a measure of the similarity of any two data points i and j in the feature space. The generality of the SVM arises from the fact that this term may be replaced by a more general measure of similarity, a kernel function
is a measure of the similarity of any two data points i and j in the feature space. The generality of the SVM arises from the fact that this term may be replaced by a more general measure of similarity, a kernel function 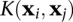 . Different kernels refer to different methods of measuring similarity. A very simple and general measure of sequence similarity is the k-spectrum kernel (Leslie et al. 2002), which describes the similarity of k-mer frequencies of two sequences. We have found that this kernel produces our best results, is easy to interpret, and can easily represent a combination of TF binding sites. To implement the k-spectrum kernel, we generate a k-mer count vector for the full set of distinct k-mers for each sequence. Then we normalize the count vector so that ‖x‖ = 1 to reduce the effect of the variable length of different enhancers. We loosely refer to this normed vector as the “k-mer frequency vector.” The kernel function is then just the inner product between two normalized frequency vectors. To reflect the fact that TFs bind double stranded DNA, the spectrum kernel function is slightly modified to account for both orientations. Instead of counting only an exact k-mer, its reverse complement is also counted, and then redundant k-mers are removed. For example, only one of AATGCT and AGCATT appears on the list of distinct k-mers. For 6-mers, there are 2080 distinct features after removing reverse complements; for 7-mers, there are 8192. This modification was applied to all kernel functions. The only difference between the k-spectrum kernel and the (k,m)-mismatch kernel is that the mismatch kernel allows m mismatches when counting k-mers (Leslie et al. 2004), reflecting the fact that some TFs bind degenerate sites. The Gaussian kernel uses the same feature vectors as the k-spectrum kernel but uses a nonlinear similarity measure via the kernel function
. Different kernels refer to different methods of measuring similarity. A very simple and general measure of sequence similarity is the k-spectrum kernel (Leslie et al. 2002), which describes the similarity of k-mer frequencies of two sequences. We have found that this kernel produces our best results, is easy to interpret, and can easily represent a combination of TF binding sites. To implement the k-spectrum kernel, we generate a k-mer count vector for the full set of distinct k-mers for each sequence. Then we normalize the count vector so that ‖x‖ = 1 to reduce the effect of the variable length of different enhancers. We loosely refer to this normed vector as the “k-mer frequency vector.” The kernel function is then just the inner product between two normalized frequency vectors. To reflect the fact that TFs bind double stranded DNA, the spectrum kernel function is slightly modified to account for both orientations. Instead of counting only an exact k-mer, its reverse complement is also counted, and then redundant k-mers are removed. For example, only one of AATGCT and AGCATT appears on the list of distinct k-mers. For 6-mers, there are 2080 distinct features after removing reverse complements; for 7-mers, there are 8192. This modification was applied to all kernel functions. The only difference between the k-spectrum kernel and the (k,m)-mismatch kernel is that the mismatch kernel allows m mismatches when counting k-mers (Leslie et al. 2004), reflecting the fact that some TFs bind degenerate sites. The Gaussian kernel uses the same feature vectors as the k-spectrum kernel but uses a nonlinear similarity measure via the kernel function 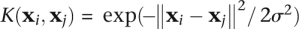 . Our implementation utilizes the Shogun machine learning toolbox (Sonnenburg et al. 2006a) and SVM light (Joachims 1999). The full lists of SVM weights are provided in the Supplemental Material, and python scripts are available from our website http://www.beerlab.org/p300enhancer.
. Our implementation utilizes the Shogun machine learning toolbox (Sonnenburg et al. 2006a) and SVM light (Joachims 1999). The full lists of SVM weights are provided in the Supplemental Material, and python scripts are available from our website http://www.beerlab.org/p300enhancer.
Acknowledgments
We thank Donavan Cheng, Mahmoud Ghandi, Rahul Karnik, Changhee Lee, and Andy McCallion for useful discussions and helpful comments. We also appreciate detailed suggestions from the anonymous reviewers who significantly improved the manuscript. We thank J. Stamatoyannopoulos and his lab for generating and allowing prepublication access to the mouse ENCODE DNase I data. M.B. was supported by the Searle Scholars Program and in part by NS062972 (NIH). R.K. was supported in part by NSF DBI-1845275.
Authors' contributions: D.L. conceived of the study as a final project in R.K.'s Foundations of Computational Biology course at JHU, D.L. and M.A.B. carried out the analysis, D.L. and M.A.B. wrote the paper, and all authors read and approved the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.121905.111.
References
- Bailey T, Elkan C 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36 [PubMed] [Google Scholar]
- Banerji J 1981. Expression of a β-globin gene is enhanced by remote SV40 DNA sequences. Cell 27: 299–308 [DOI] [PubMed] [Google Scholar]
- Beer MA, Tavazoie S 2004. Predicting gene expression from sequence. Cell 117: 185–198 [DOI] [PubMed] [Google Scholar]
- Ben-Hur A, Ong CS, Sonnenburg S, Schölkopf B, Rätsch G 2008. Support vector machines and kernels for computational biology. PLoS Comput Biol 4: e1000173 doi: 10.1371/journal.pcbi.1000173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Badis G, Gehrke AR, Talukder S, Philippakis AA, Peña-Castillo L, Alleyne TM, Mnaimneh S, Botvinnik OB, Chan ET, et al. 2008. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell 133: 1266–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand N, Castro DS, Guillemot F 2002. Proneural genes and the specification of neural cell types. Nat Rev Neurosci 3: 517–530 [DOI] [PubMed] [Google Scholar]
- Blackwood EM, Kadonaga JT 1998. Going the distance: A current view of enhancer action. Science 281: 60–63 [DOI] [PubMed] [Google Scholar]
- Boser BE, Guyon IM, Vapnik VN 1992. A training algorithm for optimal margin classifiers. In Proceedings of the Fifth Annual Workshop on Computational Learning Theory. Association for Computing Machinery (ACM), New York [Google Scholar]
- Bryne JC, Valen E, Tang ME, Marstrand T, Winther O, da Piedade I, Krogh A, Lenhard B, Sandelin A 2008. JASPAR, the open access database of transcription factor-binding profiles: New content and tools in the 2008 update. Nucleic Acids Res 36: D102–D106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulfone A, Puelles L, Porteus M, Frohman M, Martin G, Rubenstein J 1993. Spatially restricted expression of Dlx-1, Dlx-2 (Tes-1), Gbx-2, and Wnt-3 in the embryonic day 12.5 mouse forebrain defines potential transverse and longitudinal segmental boundaries. J Neurosci 13: 3155–3172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter D, Chakalova L, Osborne CS, Dai Y, Fraser P 2002. Long-range chromatin regulatory interactions in vivo. Nat Genet 32: 623–626 [DOI] [PubMed] [Google Scholar]
- Chan HM, La Thangue NB 2001. P300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci 114: 2363–2373 [DOI] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. 2008. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133: 1106–1117 [DOI] [PubMed] [Google Scholar]
- Demarque M, Spitzer NC 2010. Activity-dependent expression of Lmx1b regulates specification of serotonergic neurons modulating swimming behavior. Neuron 67: 321–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elnitski L, Hardison RC, Li J, Yang S, Kolbe D, Eswara P, O'Connor MJ, Schwartz S, Miller W, Chiaromonte F 2003. Distinguishing regulatory DNA from neutral sites. Genome Res 13: 64–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erives A, Levine M 2004. Coordinate enhancers share common organizational features in the Drosophila genome. Proc Natl Acad Sci 101: 3851–3856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher S, Grice EA, Vinton RM, Bessling SL, McCallion AS 2006. Conservation of RET regulatory function from human to zebrafish without sequence similarity. Science 312: 276–279 [DOI] [PubMed] [Google Scholar]
- Flavell SW, Greenberg ME 2008. Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu Rev Neurosci 31: 563–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frietze S, Lan X, Jin VX, Farnham PJ 2010. Genomic targets of the KRAB and SCAN domain-containing zinc finger protein 263. J Biol Chem 285: 1393–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey TS, Cristianini N, Duffy N, Bednarski DW, Schummer M, Haussler D 2000. Support vector machine classification and validation of cancer tissue samples using microarray expression data. Bioinformatics 16: 906–914 [DOI] [PubMed] [Google Scholar]
- Ghanem N, Jarinova O, Amores A, Long Q, Hatch G, Park BK, Rubenstein JLR, Ekker M 2003. Regulatory roles of conserved intergenic domains in vertebrate Dlx bigene clusters. Genome Res 13: 533–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotea V, Visel A, Westlund JM, Nobrega MA, Pennacchio LA, Ovcharenko I 2010. Homotypic clusters of transcription factor binding sites are a key component of human promoters and enhancers. Genome Res 20: 565–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble W 2007. Quantifying similarity between motifs. Genome Biol 8: R24 doi: 10.1186/gb-2007-8-2-r24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallikas O, Palin K, Sinjushina N, Rautiainen R, Partanen J, Ukkonen E, Taipale J 2006. Genome-wide prediction of mammalian enhancers based on analysis of transcription-factor binding affinity. Cell 124: 47–59 [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39: 311–318 [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. 2009. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459: 108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JD, Estep PW, Tavazoie S, Church GM 2000. Computational identification of Cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J Mol Biol 296: 1205–1214 [DOI] [PubMed] [Google Scholar]
- Joachims T 1999. Making large-scale support vector machine learning practical. In Advances in kernal methods, pp. 169–184 MIT Press, Cambridge, MA [Google Scholar]
- John S, Sabo PJ, Thurman RE, Sung M-H, Biddie SC, Johnson TA, Hager GL, Stamatoyannopoulos JA 2011. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet 43: 264–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B 2007. Genome-wide mapping of in vivo protein-DNA interactions. Science 316: 1497–1502 [DOI] [PubMed] [Google Scholar]
- Kadonaga JT 2004. Regulation of RNA polymerase II transcription by sequence-specific DNA binding factors. Cell 116: 247–257 [DOI] [PubMed] [Google Scholar]
- Karchin R, Karplus K, Haussler D 2002. Classifying G-protein coupled receptors with support vector machines. Bioinformatics 18: 147–159 [DOI] [PubMed] [Google Scholar]
- Karolchik D, Kuhn RM, Baertsch R, Barber GP, Clawson H, Diekhans M, Giardine B, Harte RA, Hinrichs AS, Hsu F, et al. 2008. The UCSC Genome Browser Database: 2008 update. Nucleic Acids Res 36: D773–D779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keilwagen J, Grau J, Paponov IA, Posch S, Strickert M, Grosse I 2011. De-novo discovery of differentially abundant transcription factor binding sites including their positional preference. PLoS Comput Biol 7: e1001070 doi: 10.1371/journal.pcbi.1001070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. 2010. Widespread transcription at neuronal activity-regulated enhancers. Nature 465: 182–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King DC, Taylor J, Elnitski L, Chiaromonte F, Miller W, Hardison RC 2005. Evaluation of regulatory potential and conservation scores for detecting cis-regulatory modules in aligned mammalian genome sequences. Genome Res 15: 1051–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh K, Kim S-J, Boyd S 2007. An interior-point method for large-scale l1-regularized logistic regression. J Mach Learn Res 8: 1519–1555 [Google Scholar]
- Kurokawa D, Kiyonari H, Nakayama R, Kimura-Yoshida C, Matsuo I, Aizawa S 2004. Regulation of Otx2 expression and its functions in mouse forebrain and midbrain. Development 131: 3319–3331 [DOI] [PubMed] [Google Scholar]
- Lee JE 1997. Basic helix-loop-helix genes in neural development. Curr Opin Neurobiol 7: 13–20 [DOI] [PubMed] [Google Scholar]
- Leslie C, Eskin E, Noble WS 2002. The spectrum kernel: A string kernel for SVM protein classification. Pac Symp Biocomput 7: 564–575 [PubMed] [Google Scholar]
- Leslie C, Eskin E, Cohen A, Weston J, Noble WS 2004. Mismatch string kernels for discriminative protein classification. Bioinformatics 20: 467–476 [DOI] [PubMed] [Google Scholar]
- Leung G, Eisen MB 2009. Identifying cis-regulatory sequences by word profile similarity. PLoS ONE 4: e6901 doi: 10.1371/journal.pone.0006901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T, Falvo JV, Kim TH, Kim TK, Lin CH, Parekh BS, Wathelet MG 1998. Structure and function of the interferon-β enhanceosome. Cold Spring Harb Symp Quant Biol 63: 609–620 [DOI] [PubMed] [Google Scholar]
- Mason S, Piper M, Gronostajski RM, Richards LJ 2008. Nuclear factor one transcription factors in CNS development. Mol Neurobiol 39: 10–23 [DOI] [PubMed] [Google Scholar]
- Matsuo I, Kuratani S, Kimura C, Takeda N, Aizawa S 1995. Mouse Otx2 functions in the formation and patterning of rostral head. Genes Dev 9: 2646–2658 [DOI] [PubMed] [Google Scholar]
- Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, et al. 2003. TRANSFAC(R): Transcriptional regulation, from patterns to profiles. Nucleic Acids Res 31: 374–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaughey DM, Vinton RM, Huynh J, Al-Saif A, Beer MA, McCallion AS 2008. Metrics of sequence constraint overlook regulatory sequences in an exhaustive analysis at phox2b. Genome Res 18: 252–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megraw M, Pereira F, Jensen ST, Ohler U, Hatzigeorgiou AG 2009. A transcription factor affinity-based code for mammalian transcription initiation. Genome Res 19: 644–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinicke P, Tech M, Morgenstern B, Merkl R 2004. Oligo kernels for datamining on biological sequences: A case study on prokaryotic translation initiation sites. BMC Bioinformatics 5: 169 doi: 10.1186/1471-2105-5-169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narlikar L, Sakabe NJ, Blanski AA, Arimura FE, Westlund JM, Nobrega MA, Ovcharenko I 2010. Genome-wide discovery of human heart enhancers. Genome Res 20: 381–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newburger DE, Bulyk ML 2009. UniPROBE: An online database of protein binding microarray data on protein-DNA interactions. Nucleic Acids Res 37: D77–D82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noonan JP, McCallion AS 2010. Genomics of long-range regulatory elements. Annu Rev Genomics Hum Genet 11: 1–23 [DOI] [PubMed] [Google Scholar]
- Pavesi G, Mauri G, Pesole G 2001. An algorithm for finding signals of unknown length in DNA sequences. Bioinformatics 17 (Suppl 1): S207–S214 [DOI] [PubMed] [Google Scholar]
- Peckham HE, Thurman RE, Fu Y, Stamatoyannopoulos JA, Noble WS, Struhl K, Weng Z 2007. Nucleosome positioning signals in genomic DNA. Genome Res 17: 1170–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, Minovitsky S, Dubchak I, Holt A, Lewis KD, et al. 2006. In vivo enhancer analysis of human conserved noncoding sequences. Nature 444: 499–502 [DOI] [PubMed] [Google Scholar]
- Rätsch G, Sonnenburg S, Schölkopf B 2005. RASE: Recognition of alternatively spliced exons in C. elegans. Bioinformatics 21 (Suppl 1): i369–i377 [DOI] [PubMed] [Google Scholar]
- Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, et al. 2007. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods 4: 651–657 [DOI] [PubMed] [Google Scholar]
- Ross SE, Greenberg ME, Stiles CD 2003. Basic helix-loop-helix factors in cortical development. Neuron 39: 13–25 [DOI] [PubMed] [Google Scholar]
- Schölkopf B, Tsuda K, Vert JP 2004. Kernel methods in computational biology. MIT Press, Cambridge, MA [Google Scholar]
- Schultheiss SJ 2010. Kernel-based identification of regulatory modules. In Computational biology of transcription factor binding (ed. Ladunga I.), Vol. 674, pp. 213–223 Humana Press, Totowa, NJ: [DOI] [PubMed] [Google Scholar]
- Schultheiss SJ, Busch W, Lohmann JU, Kohlbacher O, Rätsch G 2009. KIRMES: Kernel-based identification of regulatory modules in euchromatic sequences. Bioinformatics 25: 2126–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg S, Rätsch G, Schäfer C, Schölkopf B 2006a. Large scale multiple kernel learning. J Mach Learn Res 7: 1531–1565 [Google Scholar]
- Sonnenburg S, Zien A, Ratsch G 2006b. ARTS: Accurate recognition of transcription starts in human. Bioinformatics 22: e472–e480 [DOI] [PubMed] [Google Scholar]
- Sonnenburg S, Schweikert G, Philips P, Behr J, Ratsch G 2007. Accurate splice site prediction using support vector machines. BMC Bioinformatics 8: S7 doi: 10.1186/1471-2105-8-S10-S7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg S, Zien A, Philips P, Ratsch G 2008. POIMs: positional oligomer importance matrices—understanding support vector machine-based signal detectors. Bioinformatics 24: i6–i14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R 2003. Statistical significance for genomewide studies. Proc Natl Acad Sci 100: 9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J, Teichmann SA, Down TA 2010. Assessing computational methods of cis-regulatory module prediction. PLoS Comput Biol 6: e1001020 doi: 10.1371/journal.pcbi.1001020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanos D, Maniatis T 1995. Virus induction of human IFNβ gene expression requires the assembly of an enhanceosome. Cell 83: 1091–1100 [DOI] [PubMed] [Google Scholar]
- Vandewalle C, Roy F, Berx G 2008. The role of the ZEB family of transcription factors in development and disease. Cell Mol Life Sci 66: 773–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vapnik VN. The nature of statistical learning theory. Springer, New York: 1995. [Google Scholar]
- Visel A, Prabhakar S, Akiyama JA, Shoukry M, Lewis KD, Holt A, Plajzer-Frick I, Afzal V, Rubin EM, Pennacchio LA 2008. Ultraconservation identifies a small subset of extremely constrained developmental enhancers. Nat Genet 40: 158–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, Afzal V, et al. 2009. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457: 854–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigle JT, Eisenstat DD 2008. Homeobox genes in vertebrate forebrain development and disease. Clin Genet 73: 212–226 [DOI] [PubMed] [Google Scholar]
- Wilson M, Koopman P 2002. Matching SOX: Partner proteins and cofactors of the SOX family of transcriptional regulators. Curr Opin Genet Dev 12: 441–446 [DOI] [PubMed] [Google Scholar]
- Wilson MD, Barbosa-Morais NL, Schmidt D, Conboy CM, Vanes L, Tybulewicz VLJ, Fisher EMC, Tavare S, Odom DT 2008. Species-specific transcription in mice carrying human chromosome 21. Science 322: 434–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfe A, Goodson M, Goode DK, Snell P, McEwen GK, Vavouri T, Smith SF, North P, Callaway H, Kelly K, et al. 2004. Highly conserved noncoding sequences are associated with vertebrate development. PLoS Biol 3: e7 doi: 10.1371/journal.pbio.0030007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerucha T, Stühmer T, Hatch G, Park BK, Long Q, Yu G, Gambarotta A, Schultz JR, Rubenstein JLR, Ekker M 2000. A highly conserved enhancer in the Dlx5/Dlx6 intergenic region is the site of cross-regulatory interactions between Dlx genes in the embryonic forebrain. J Neurosci 20: 709–721 [DOI] [PMC free article] [PubMed] [Google Scholar]