

Abstract
With the increasing popularity of whole-genome shotgun sequencing (WGSS) via high-throughput sequencing technologies, it is becoming highly desirable to perform comparative studies involving multiple individuals (from a specific population, race, or a group sharing a particular phenotype). The conventional approach for a comparative genome variation study involves two key steps: (1) each paired-end high-throughput sequenced genome is compared with a reference genome and its (structural) differences are identified; (2) the lists of structural variants in each genome are compared against each other. In this study we propose to move away from this two-step approach to a novel one in which all genomes are compared with the reference genome simultaneously for obtaining much higher accuracy in structural variation detection. For this purpose, we introduce the maximum parsimony–based simultaneous structural variation discovery problem for a set of high-throughput sequenced genomes and provide efficient algorithms to solve it. We compare the proposed framework with the conventional framework, on the genomes of the Yoruban mother–father–child trio, as well as the CEU trio of European ancestry (both sequenced by Illumina platforms). We observed that the conventional framework predicts an unexpectedly high number of de novo variations in the child in comparison to the parents and misses some of the known variations. Our proposed framework, on the other hand, not only significantly reduces the number of incorrectly predicted de novo variations but also predicts more of the known (true) variations.
High-throughput–next-generation sequencing (NGS) technologies are reducing the cost and increasing the world-wide capacity for sequence production at an unprecedented rate. Large-scale projects based on NGS aim to sequence 2000 (1000 Genomes Project Consortium 2010) or 10,000 individual genomes (International Cancer Genome Consortium et al. 2010) and analyze genomic variation at a population scale. Genomic variation, especially structural variation (involving insertion, deletion, duplication, translocation, and transposition events) detection through NGS, promises to be one of the key diagnostic tools for cancer and other diseases with genomic origin (Feuk et al. 2006; Stankiewicz and Lupski 2010). One recent study (Leary et al. 2010), for example, demonstrates that patient-specific structural variants identified in blood samples could be used as personalized biomarkers for monitoring tumor progression and response to cancer therapies. The main potential use of NGS in clinical applications, however, would be the identification of genomic variants including the structural ones as recurrent biomarkers in patient subgroups that are scarcely observed in healthy tissues. Some recent studies on specific cancer types, on the other hand, have not been able to identify recurrent structural biomarkers (e.g., Clark et al. 2010; Mardis 2010). Although it is possible that such genomic signals simply do not exist in the cancer types studied, another likely explanation is that the computational tools used in these studies were not sufficiently accurate to correctly identify and/or prioritize recurrent structural variants. The emerging area of personalized genomics will surely benefit from computational tools that can correctly identify recurrent structural variants among a collection of genomes and transcriptomes.
To identify genomic variations with much higher accuracy than what is currently possible, we propose to move from the current model of (1) detecting genomic variations in individual next-generation sequenced (NGS) donor genomes independently, and (2) checking whether two or more donor genomes, indeed, agree or disagree on the variations—we will call this model the “independent structural variation detection and merging” (ISV&M) framework. As an alternative, we introduce a new model in which genomic variation is detected among multiple genomes simultaneously.
Our new model can be likened to multiple sequence alignment methods that were introduced to overcome the limitations of pairwise sequence aligners—the primary source of sequence analysis in the early days of genomics. Pairwise sequence alignment methods implicitly aim to match identical regions among two input sequences that are not interrupted by mismatches or indels. They achieve this under the maximum parsimony principle that suggests to minimize the (probabilistically weighted) number of single nucleotide insertions, deletions, and mismatches in an alignment. Unfortunately, the most likely alignment is many times incorrect. Accuracy in sequence alignment can be improved significantly by the use of multiple sequence aligners, provided that several related sequences are available for use. Today, at least for the purposes of identifying genomic variants at a single-nucleotide scale, multiple alignment is the “technology” of choice.
The main contribution of this study is a set of novel algorithms for identifying structural differences among multiple genomes through the use of multiple sequence comparison methods. Our algorithms will help better analyze vast amounts of publicly available genomic sequence data (e.g., the 1000 Genomes Project) (Mills et al. 2010; 1000 Genomes Project Consortium 2010), which include WGSS data from diverse populations (and members of the same population or even family).
Existing methods for structural variant (SV) discovery and their limitations
Available methods for SV discovery typically use paired-end sequencing: Inserts from a donor genome (from a tightly controlled length distribution) are read at two ends, which are later aligned to a reference genome. Provided that the mapping loci are correctly identified, an increase or decrease of the distance between the end reads indicates an insertion or a deletion. PEmer (Korbel et al. 2009), for example, maps each paired-end read to a unique location through the mapping software MAQ (Lee et al. 2008). A number of follow-up studies that use a similar “hard clustering” approach (Medvedev et al. 2009) include Pindel (Ye et al. 1999) and BreakDancer (Chen et al. 2009). All focus only on the “best mapping” of each read, provided by the mapping software in use. A survey (Medvedev et al. 2009) summarizes the basis of decision making for each of these methods and reports briefly on their performance. These methods typically work well on unique regions of the human genome; however, they naturally ignore potential multiple alignment locations in repeat regions, by either picking one arbitrary location among many possibilities or simply avoiding the use of reads that have multiple mapping locations. As a result, they cannot capture structural variations in repetitive regions of the human genome.
In a recent paper (Hach et al. 2010), it was demonstrated that ignoring possible mapping locations of a read may lead to significant loss of accuracy in structural variation detection. A 2 × 100-bp read provided by Illumina HiSeq2000 technology maps to more than 180 locations within six mismatches or indels. Picking an arbitrary location among these as the mapping location of a read naturally leads to both false positives and false negatives in SV discovery.
To address the above problem, a number of “soft clustering” techniques (Alkan et al. 2009; Hormozdiari et al. 2009; Lee et al. 2009) have been introduced in the past two years. Here, paired-end reads are mapped to all potential locations—through the use of the mapping algorithms such as mr and mrs FAST (Alkan et al. 2009; Hach et al. 2010). In soft clustering approaches, paired-end reads can have multiple mapping to the reference genome and thus suggest different variations. Each set of the discordant paired-end reads can be indicating a real structural variation or just be an artifact of the multiple mapping. These clusters of paired-end reads are denoted soft clusters (Medvedev et al. 2009). VariationHunter (Hormozdiari et al. 2009) is one of those soft-clustering methods that aims to resolve repetitive regions of the human genome through a combinatorial optimization framework for detecting insertion and deletion polymorphisms. A recent extension of VariationHunter (Hormozdiari et al. 2009) for mobile element insertion discovery (Hormozdiari et al. 2010) and a new computational pipeline, NovelSeq, for novel sequence insertion discovery (Hajirasouliha et al. 2010), also use soft-clustering techniques. MoDIL (Lee et al. 2009), as well as its follow-up MoGUL (Lee et al. 2010), evaluates the clusters of reads that seem to indicate a structural variant using a probabilistic framework, while Hydra (Quinlan et al. 2010) uses heuristics (based on the algorithmic strategies of VariationHunter) to detect structural variant breakpoints in the mouse genome. MoGUL (Lee et al. 2010) focuses on finding common insertion and deletion events in a pool of multiple low-coverage sequenced genomes.
In this study, we demonstrate, for example, that on the well-known NGS genomes of the Yoruban family (involving a child, the mother, and the father—NA18506, NA18507, NA18508) Bentley et al. (2008), the independent application of VariationHunter (the only publicly available algorithm for Alu discovery on NGS genomes) predicts up to 410 de novo Alu inserts in the child! A careful inspection of the clusters obtained by VariationHunter on all three individuals, on the other hand, reveals that all of these 410 novel Alu inserts predicted are, indeed, false positives mostly due to single nucleic variations (SNVs) or varying read coverage, etc.
Note that soft-clustering strategies for SV detection between one donor genome and a reference genome do provide both false positives, as well as false negatives, due to SNV effects and others. However, the proportion of false positives among all positives predicted will be low because of the high number of actual SVs typically observed between a donor and the reference. On the other hand, when the goal is to identify structural differences between two highly related donors, i.e., donor 1 by (D1) and donor 2 (D2), by using the reference (R) as an intermediary, while the number of false positives (between D1 and R and between D2 and R) will be of similar scale, the proportion of false positives among all positives will be high, simply due to the low number of actual SVs that would be present between the donor genomes. Thus, although VariationHunter (and other soft-clustering strategies) may provide high levels of accuracy for SV detection between one donor and the reference genome, it may provide a low level of accuracy when finding the structural differences between two (or more) donor genomes.
The CommonLAW approach
For the purpose of addressing the above issues arising in soft-clustering techniques, we introduced the problem of simultaneous SV discovery among multiple paired-end NGS donor genomes—with the help of a complete reference genome. To solve this problem, we also introduced novel combinatorial algorithms, which we collectively call CommonLAW (Common Loci structural Alteration discovery Widgets). CommonLAW aims to predict SVs in several donor genomes by means of minimizing a weighted sum of structural differences between the donor genomes as well as one reference genome.5 The (pairwise) weights are a function of (1) the expected genomic proximity of the individual donors sequenced (see details in the Results section); and (2) the type, loci, and length of the individual structural alterations considered. The problem of minimizing (for example, sum-of-pairs) genomic alterations between multiple genomes is NP-hard. In this study, we describe a tight (i.e., asymptotically the best possible) approximation algorithm for the general simultaneous SV discovery problem—this algorithm is at the heart of the CommonLAW package. In addition, CommonLAW includes several efficient algorithms and heuristics for some special cases of the problem.
We have tested CommonLAW on the genomes of three Yoruban (YRI) individuals (mother–father–child trio) sequenced by the Illumina Genome Analyzer with ∼30× coverage (i.e., 3.24 × 1011 bp of sequencing data), for the purpose of predicting deletions and Alu insertions. We compare the deletion predictions with the validated deletions reported in the 1000 Genomes Project Consortium (2010) and Mills et al. (2010). We compare the Alu insertion predictions with Alu polymorphism loci reported in dbRIP (Wang et al. 2006). In both cases, we observe that CommonLAW provides a much higher level of accuracy in comparison to VariationHunter, the only publicly available computational method for Alu insertion discovery in NGS genomes.
In addition, we have tested CommonLAW on a high-coverage parent–offspring trio of European ancestry from Utah (CEU), recently sequenced and analyzed by the 1000 Genomes Project (1000 Genomes Project Consortium 2010). We demonstrate the predictive power of CommonLAW by comparing its calls with the validated deletions reported in the 1000 Genomes Project Consortium (2010) and Mills et al. (2010).
Methods
Simultaneous structural variation discovery among multiple genomes
Given a reference genome and a collection of paired-end sequenced genomes, G1,…,Gλ, the Simultaneous Structural Variation Discovery among Multiple Genomes (SSV-MG) problem asks to simultaneously analyze the genomes so as to predict structural differences between them and the reference genome. For solving the SSV-MG problem, notice that a paired-end read from a genome Gk with no concordant alignment on the reference genome suggests an SV event in Gk (Volik et al. 2003; Tuzun et al. 2005). Unfortunately, if the number of discordant alignment locations of a paired-end read is more than one, the paired-end read potentially supports several SV events. The crucial question we try to answer in this study is, among all potential SV events supported by a discordant paired-end read, which one is correct? In the presence of a single donor genome, one answer to this question was given by Hormozdiari et al. (2009) with the introduction of novel approximation algorithms for the “Maximum Parsimony Structural Variation” (MPSV) discovery problem. In Hormozdiari et al. (2009), an SV cluster is defined as a set of discordant (paired-end read) alignments that can support the same potential SV event; similarly, a maximal SV cluster is defined as an SV cluster to which no other alignments could be added (Bashir et al. 2008; Hormozdiari et al. 2009, 2010; Sindi et al. 2009; Hajirasouliha et al. 2010). A maximal SV cluster is considered to be a valid cluster if it satisfies a certain set of mathematical rules specifically defined for each SV event type (Hormozdiari et al. 2009, 2010; Hajirasouliha et al. 2010).
As defined in Hormozdiari et al. (2009), the MPSV problem for a single donor genome asks to compute a unique assignment for each discordant paired-end read to a maximal valid SV cluster such that the total number of implied SVs is minimized. The SSV-MG problem, which generalizes the MPSV problem to multiple donor genomes, also asks to identify a set of maximal SV clusters to which each discordant paired-end read can uniquely be assigned—under the maximum parsimony criteria. A solution of the SSV-MG problem is said to provide support for each SV cluster as a function of the discordant paired-end reads it assigns to the SV cluster. Intuitively, if the support comes from paired-end reads from a large number of—especially highly related—genomes (e.g., members of a family), the SV event is more likely to be “correct.” The maximum parsimony criteria we use are formulated to reflect this observation as follows: Each SV event in a solution to the SSV-MG problem is associated with a weight, which is a function of the set of the donor genomes on which the SV event is present (i.e., has at least one discordant paired-end read mapping that is assigned to the associated SV cluster). If an SV event is present among many donor genomes, its weight will be relatively small; on the other hand, an SV event that is unique to only one donor genome will have a larger weight. In this setting, the SSV-MG problem asks to identify a set of SV events whose total weight is as small as possible.
The algorithmic formulation of the SSV-MG problem
Given an NGS sequenced donor genome Gk, let the set of its discordant reads (i.e., the reads that do not have a concordant mapping) be 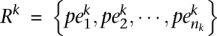 ; thus, nk denotes the number of discordant reads of Gk. Let
; thus, nk denotes the number of discordant reads of Gk. Let 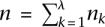 be the total number of discordant reads among all the donor genomes, and let R = R1 ∪ R2 ∪ … ∪ Rλ be the set of all discordant reads. For the algorithmic formulation of the SSV-MG problem, the donor genome Gk and all its discordant reads are said to be of “color” k.
be the total number of discordant reads among all the donor genomes, and let R = R1 ∪ R2 ∪ … ∪ Rλ be the set of all discordant reads. For the algorithmic formulation of the SSV-MG problem, the donor genome Gk and all its discordant reads are said to be of “color” k.
Note that each discordant read may have several alignment locations on the reference genome, thus, as we discussed above, the aim is to find a unique assignment of each discordant read in R to exactly one of the maximal SV clusters (and, hence, to one potential SV event). (For detailed definitions of discordant reads and multiple paired-end read alignments, please see Hormozdiari et al. 2009.)
Let 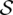 be the set of all maximal valid clusters. For each r ∈ R, let
be the set of all maximal valid clusters. For each r ∈ R, let 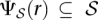 , denote the set of all maximum valid clusters “supported by” r, i.e., for which r has an associated alignment. For each possible subset of colors (i.e., donor genomes)
, denote the set of all maximum valid clusters “supported by” r, i.e., for which r has an associated alignment. For each possible subset of colors (i.e., donor genomes) 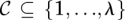 , we define a weight
, we define a weight 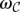 as a measure of genetic affinity between the donor genomes in this subset. For example, the weight of a subset of two donor genomes can be defined as the estimated ratio of the total number of SVs in the two genomes and the number of shared SVs in the genomes—i.e., the likelihood of an SV event being shared among the two donor genomes, rather than being present in only one donor genome. Then we can define the weight of an SV event (i.e., maximal valid cluster) s, denoted ws, as
as a measure of genetic affinity between the donor genomes in this subset. For example, the weight of a subset of two donor genomes can be defined as the estimated ratio of the total number of SVs in the two genomes and the number of shared SVs in the genomes—i.e., the likelihood of an SV event being shared among the two donor genomes, rather than being present in only one donor genome. Then we can define the weight of an SV event (i.e., maximal valid cluster) s, denoted ws, as 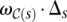 , where
, where 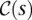 is the set of donor genomes sharing the SV event s and Δs is a measure of the likelihood of the SV event s, which depends only on the length and the type of s—as discussed in the introduction section.
is the set of donor genomes sharing the SV event s and Δs is a measure of the likelihood of the SV event s, which depends only on the length and the type of s—as discussed in the introduction section.
Based on these notions, the Simultaneous Structural Variation discovery among Multiple Genomes (SSV-MG) problem asks to assign each discordant read r ∈ R to one of the maximal valid SV clusters in 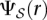 such that the following optimization function (COST) is minimized:
such that the following optimization function (COST) is minimized:
Here Is is an indicator variable equal to 1, if there if at least one discordant read assigned to s (i.e., s is selected); otherwise Is is equal to 0.
SSV-MG for two donor genomes
A special case of the SSV-MG problem is on comparing two donor genomes by the use of a reference genome as an intermediary. This case obviously applies to two highly related genomes such as those from healthy versus tumor tissues of an individual, for the purpose of identification of common and distinct SV events with respect to the reference genome, and, as a result, the structural differences between them.
We study this case through a combinatorial problem, namely, the Red-Black-Assignment problem where two colors, red and black are, respectively, associated with the two donor genomes (and their discordant paired-end reads). We call an SV event, which has assigned paired-end reads from both colors, multicolor and call an SV event that has assigned paired-end reads from a color red/black, respectively, red or black. Clearly, a multicolor SV event indicates no structural difference between the two genomes, whereas a red or a black SV event indicates a structural difference.
Let 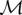 be the set of multicolor SV events,
be the set of multicolor SV events, 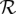 be the set of red events, and
be the set of red events, and  be the set of black events. The SSV-MG problem for this particular case asks to find a solution that minimizes the following cost function:
be the set of black events. The SSV-MG problem for this particular case asks to find a solution that minimizes the following cost function:
Clearly, a lower value of ω{red,black} in comparison to ω{red} or ω{black} asks for a more conservative estimate of the structural differences between the two genomes.
In the next section we show that the SSV-MG problem is NP-hard to solve exactly. In fact, it is also NP-hard to solve within an approximation factor of
This is the case even when Δs = 1 for all SV events s. Note that n is the total number of discordant reads, and ωmax and ωmin are the maximum and minimum possible weight for of an SV event, respectively. (Intuitively, ωmin is the weight of a multicolor SV event that has assigned reads from all different colors, and wmax is the weight of an SV event that has only assigned reads from one specific color.)
Hardness of approximating the general SSV-MG problem
We use an approximation preserving reduction from the well-known set cover problem. The set cover problem is defined as follows: Given a universe U with n elements and a family S of subsets of U (i.e., S = {S1,…,Sm}), we want to find the minimum number of sets in S whose union is U. Raz and Safra (1997) proved that there exists a constant d such that the set cover problem cannot be approximated within d log n unless P = NP. Alon et al. (2006) showed that the similar complexity result also holds with a smaller constant. We use this complexity result to prove that the SSV-MG problem cannot be approximated within a constant times 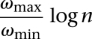 .
.
Lemma 1. There exists a constant d such that the set cover problem cannot be approximated within d log n even in the case where the size of the optimal solution for the problem is already known.
Proof. Given a set cover instance, we define OPT as the size of its optimal solution. Note that OPT is always an integer smaller than or equal to m, where m is the size of the family of subsets of S. We show that if there exists a black-box which finds a solution with a size d log n · OPT for the case where OPT is already known, the set cover problem can also be approximated within the same factor. This reduction would be in contradiction to the complexity result of Alon et al. (2006). Assume there exists such a black-box that finds an approximated solution with at most d log n · OPT in polynomial time. For each integer i ∈ {1,…,m}, we can now guess the value of OPT to be equal to i and execute m different black-boxes (i.e., for each i) in parallel. Next, we verify the outputs of those black-boxes terminated in polynomial time and find an approximated solution within the same factor for the general set cover problem.
Theorem 2. There exists a constant c such that SSV-MG has no approximation factor smaller than 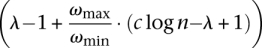 , unless P = NP.
, unless P = NP.
Proof. We use a reduction from the set cover problem where the size of its optimal solution is already known. For simplicity, we call this problem Set Cover Optimal Known (SC-OK) throughout this proof. Suppose we are given an SC-OK instance with U = {x1,…,xn} and S = {S1,…,Sm} as its universe and family of subsets, respectively, and let OPT be the size of its optimal solution. We construct an instance of the SSV-MG problem as follows: For each color ℓ(1 ≤ ℓ ≤ λ − 1) and for each j(1 ≤ j ≤ OPT), we introduce a new element yℓ,j with the color ℓ. The color of the elements in U is set to λ. Let Y = {yℓ,j |1 ≤ ℓ ≤ λ − 1, 1 ≤ j ≤ OPT} be the set of all these new elements. We define U′ = U ∪ Y, as a new universe for the instance of SSV-MG, and construct its family of subsets S′ as follows: Corresponding to each Si ∈ S, we have a subset 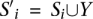 in S′. In other words, all the subsets in the family will share all of the new (λ − 1)OPT elements. It can be seen that an optimal solution for SC-OK gives an optimal solution for SSV-MG with a cost equal to ωmin · OPT since all the selected subsets can have all different λ colors assigned to them. Furthermore, any feasible solution for SC-OK with k ≥ (λ − 1)OPT subsets gives a solution with the cost of at least ωmax · [k − (λ − 1)OPT] + ωmin · (λ − 1)OPT for SSV-MG. We have (λ − 1)OPT new elements with colors from 1 to λ − 1 (i.e., other than λ) and even if (1) we assign these new elements to (λ − 1)OPT different subsets, and (2) ωmin is equal to the weight of an SV event with assigned paired-end reads from two colors, the cost of SSV-MG cannot become less than ωmax · [k − (λ − 1)OPT] + ωmin · (λ − 1)OPT.6
in S′. In other words, all the subsets in the family will share all of the new (λ − 1)OPT elements. It can be seen that an optimal solution for SC-OK gives an optimal solution for SSV-MG with a cost equal to ωmin · OPT since all the selected subsets can have all different λ colors assigned to them. Furthermore, any feasible solution for SC-OK with k ≥ (λ − 1)OPT subsets gives a solution with the cost of at least ωmax · [k − (λ − 1)OPT] + ωmin · (λ − 1)OPT for SSV-MG. We have (λ − 1)OPT new elements with colors from 1 to λ − 1 (i.e., other than λ) and even if (1) we assign these new elements to (λ − 1)OPT different subsets, and (2) ωmin is equal to the weight of an SV event with assigned paired-end reads from two colors, the cost of SSV-MG cannot become less than ωmax · [k − (λ − 1)OPT] + ωmin · (λ − 1)OPT.6
We claim that if there exists an algorithm which gives an approximate solution within a factor of 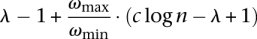 for the SSV-MG instance, then we can also give an approximate solution within a factor of c log n for SC-OK. As discussed earlier, the optimal solution for this SSV-MG instance has a cost ωmin · OPT and if the algorithm guarantees the desired factor, the cost of the solution would be at most
for the SSV-MG instance, then we can also give an approximate solution within a factor of c log n for SC-OK. As discussed earlier, the optimal solution for this SSV-MG instance has a cost ωmin · OPT and if the algorithm guarantees the desired factor, the cost of the solution would be at most 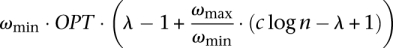 . Now we will show that, in this case, the total number of subsets in the solution returned will become less than c log n · OPT which contradicts the result of Alon et al. (2006) for a small constant c.
. Now we will show that, in this case, the total number of subsets in the solution returned will become less than c log n · OPT which contradicts the result of Alon et al. (2006) for a small constant c.
Assuming k is the total number of subsets in the solution, we have:
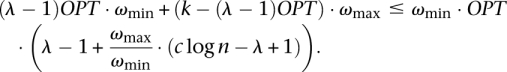 |
Thus,
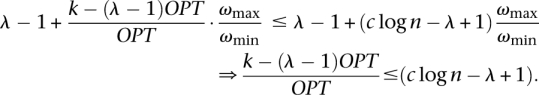 |
Thus,
So these k subsets will give a feasible solution within c log n to SC-OK which contradicts the complexity result of Alon et al. (2006), for a sufficiently small constant c.
A simple approximation algorithm for the SSV-MG problem
It is possible to obtain an approximate solution to the SSV-MG problem within an approximation factor matching the lower bound mentioned above, when Δs = 1 for all SV events s, in near-linear time. For that we adopt the greedy algorithm for approximating the well-known set cover problem (Vazirani 2001) to obtain a solution within O((ωmax/ωmin)log n) factor of the optimal solution for the SSV-MG problem. (Again, ωmax and ωmin are the maximum and minimum possible weights among all SV clusters.) The resulting algorithm, which we call the Simultaneous Set Cover method (SSV), selects sets iteratively: At each iteration, it selects the set that contains the largest number of elements not previously covered. For a given instance of the SSV-MG problem and its corresponding set cover instance, we denote the respective sizes of their optimal solution by OPTSSV and OPTSC. It is easy to see that OPTSSV ≥ ωmin · OPTSC, since at least OPTSC subsets have to be selected in SSV-MG to cover all the elements of the universe, and each of those subsets has a weight of at least ωmin. The greedy solution for the set cover problem gives a solution of at most log n · OPTSC, hence, the same solution for SSV-MG will have a cost of at most ωmax · log n · OPTSC. Thus, the SSC method gives an O((ωmax/ωmin)log n) approximation for the SSV-MG problem—which matches the lower bound indicated for the SSV-MG problem above.
A maximum flow-based update for Red–Black Assignment
Although the SSV method described above is very fast, the results it provides could be far from optimal. However, it is possible to improve the results of the SSV method through an additional (post-processing) step as follows: The SSV method picks a collection of valid clusters such that each discordant read is assigned to exactly one cluster. Each cluster is either multicolor or is considered Red or Black depending on whether it contains reads from both colors (i.e., donor genomes) or from a single color. The additional step we describe here does not change the clusters and the SV events they support. Rather, assuming that the clusters are “correct,” the additional step reassigns the discordant reads to the clusters so as to maximize the number of multicolored clusters. As a result of this assignment, we may end up with empty clusters; we simply discard these clusters and return the non-empty clusters as an (improved) solution to the SSV-MG problem. Note that the additional step is guaranteed to return a solution that is at least as good as the one returned by the SSV method; in many cases, the solution will be much better. Unfortunately, it can only be applied to the SSV-MG problem when the number of colors (i.e., donor genomes) is exactly two. Even for three colors, the problem of maximizing multi-colored clusters (i.e., those clusters with reads coming from all three donor genomes) is NP-hard (this is one of the first 21 NP-complete problems discovered by Karp 1972).
The additional step formulates the reassignment problem (of discordant reads to clusters) as a maximum flow problem as follows: Consider an instance of the Red–Black-Assignment problem and let SSELECTED = {S1,…,Sk} be the subsets of the family 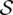 that are already selected in a solution. Let
that are already selected in a solution. Let 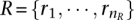 be the set of red elements and
be the set of red elements and 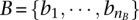 be the set of black elements, where nR + nB = n (i.e., the number of elements in the universe). We construct a network
be the set of black elements, where nR + nB = n (i.e., the number of elements in the universe). We construct a network  as follows: For 1 ≤ i ≤ k, each Si is represented by a vertex in the network and corresponding to every element in the universe, we have a vertex in the network in
as follows: For 1 ≤ i ≤ k, each Si is represented by a vertex in the network and corresponding to every element in the universe, we have a vertex in the network in 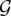 . For every pair (ri,Sp) such that ri is a member of Sp, we have an edge with a capacity equal to one and for every pair (Sq,bj) such that bj is a member of Sq, we have an edge (Sq,bj) with a capacity one. A source vertex SOURCE is connected to all vertexes in R, and all vertexes in B are connected to a sink vertex SINK. All the internal vertices (i.e., all vertices except the sink and the source) have capacity one as well.
. For every pair (ri,Sp) such that ri is a member of Sp, we have an edge with a capacity equal to one and for every pair (Sq,bj) such that bj is a member of Sq, we have an edge (Sq,bj) with a capacity one. A source vertex SOURCE is connected to all vertexes in R, and all vertexes in B are connected to a sink vertex SINK. All the internal vertices (i.e., all vertices except the sink and the source) have capacity one as well.
Our additional post-processing step computes the maximum (integral) flow from s to t and identifies all edges (ri,Sℓ) and (Sℓ,bj) in 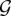 with unit flow in the network, and reassigns the elements
with unit flow in the network, and reassigns the elements 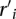 and
and 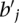 to the subset Sℓ. Observe that a solution to this maximum flow problem will maximize the number of multi-color subsets. Figure 1 demonstrates an example of how the network is constructed and how a solution to the maximum flow problem reassigns the discordant reads to clusters.
to the subset Sℓ. Observe that a solution to this maximum flow problem will maximize the number of multi-color subsets. Figure 1 demonstrates an example of how the network is constructed and how a solution to the maximum flow problem reassigns the discordant reads to clusters.
Figure 1.

The set R = {r1,r2,r3,r4} represents the red elements and B = {b1,b2,…,b7} represents the black elements. The selected subsets are S1 = {r1,r2,b1}, S2 = {r3,b1,b3}, S3 = {r4,b1,b4,b5,b6}, S4 = {b6}, and S5 = {r4,b6,b7}. All the edges and vertices have capacity one, and the maximum flow is shown in dark blue. As can be seen here, the maximum flow solution reassigns the reads so that three sets/clusters, s1, s2, s3, become multicolor and one set/cluster, s4 becomes empty. Thus, its associated potential SV event will not be among the predicted SV events by our method.
An 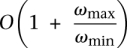 approximation algorithm for limited read mapping loci
approximation algorithm for limited read mapping loci
It is possible to further improve the algorithms presented above for the special case in which each discordant read maps to a small number of loci on the genome. For simplicity, we present the limited case that each discordant read maps to exactly two locations; i.e., in Red–Black-assignment problem terms, each element is a member of exactly two subsets. The generalization of this case to a more general one, in which each read can be present in at most f clusters, is not very difficult and is omitted.7
The special case, which we denote as the Red-Black-Assignment-F2 problem, has a graph-theoretical formulation similar to the vertex-cover problem. Let G be a simple graph for which there is a vertex si corresponding to each subset Si in the family 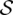 and there is an edge e = (si,sj) corresponding to each element e in U—provided e is in both Si and Sj. The edges of G are labeled with the color of their corresponding elements (either red or black). To solve the Red-Black-Assignment-F2 problem, all we need to do is to set an orientation to each edge: the vertex (corresponding to a cluster) to which a given edge (corresponding to a read) is pointing gives the cluster to which a read is assigned. The Red-Black-Assignment-F2 problem thus reduces to setting an orientation to the edges in this graph such that αωmin + βωmax is minimized: Here α is the number of vertices to which edges of both colors are pointing, and β is the number of vertices to which edges of only one color are pointing.8
and there is an edge e = (si,sj) corresponding to each element e in U—provided e is in both Si and Sj. The edges of G are labeled with the color of their corresponding elements (either red or black). To solve the Red-Black-Assignment-F2 problem, all we need to do is to set an orientation to each edge: the vertex (corresponding to a cluster) to which a given edge (corresponding to a read) is pointing gives the cluster to which a read is assigned. The Red-Black-Assignment-F2 problem thus reduces to setting an orientation to the edges in this graph such that αωmin + βωmax is minimized: Here α is the number of vertices to which edges of both colors are pointing, and β is the number of vertices to which edges of only one color are pointing.8
Let OPT be the minimum number of vertices required to cover all the edges (i.e., the size of a minimum vertex cover). It is easy to see that ωmin · OPT is a lower bound for Red-Black-Assignment-F2, and the simple greedy algorithm of the vertex cover problem (Vazirani 2001) gives a 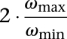 approximation.9 The following algorithm achieves a smaller (in fact, the best possible) approximation factor.
approximation.9 The following algorithm achieves a smaller (in fact, the best possible) approximation factor.
Denote the instance of orientation setting problem (to which the Red-Black-Assignment-F2 problem is reduced) by H. It is possible to compute a maximal matching (of vertices) in this graph in polynomial time; let M denote this matching. Suppose M has p edges with red labels and q edges with black labels, where p + q = |M|. Let R = {r1,r2,…,rp} be the set of red edges and B = {b1,b2,…,bq} be the set of black edges in the maximal matching.
Consider an edge eM in this matching and suppose that the optimal solution to the orientation setting problem points eM ∈ M to a multicolor vertex; also suppose that the other vertex to which eM is incident is not pointed by any other edge. Thus, in the optimal solution, the “cost” of covering each of the edges eM in the maximal matching m is at least ωmin. Our algorithm covers each such edge eM by selecting both vertices to which it is incident, incurring a cost of ωmax + ωmin.
If the optimal solution covers eM with a unicolor vertex to which it is incident, our algorithms cover it with a cost of at most 2ωmax, again by picking both vertices.
Provided the two objectives above are achieved, our algorithm guarantees an approximation factor of 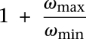 . We explain how these objectives are achieved, and thus a proof for the following claim, in the Supplemental Material.
. We explain how these objectives are achieved, and thus a proof for the following claim, in the Supplemental Material.
Theorem 3. The Red-Black-Assignment-F2 problem can be approximated within a factor of 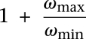 .
.
Efficient heuristic methods for the SSV-MG problem
In addition to the approximation algorithms given above, we provide two heuristics for solving the SSV-MG problem efficiently. The first heuristic uses the weights ωs to calculate a cost-effectiveness value for each cluster s, while the second heuristic deploys the concept of conflict resolution (introduced in Hormozdiari et al. 2010) to obtain more accurate results in diploid genomes.
Simultaneous Set Cover with Weights (SSC-W)
The first heuristic is a greedy method similar to the weighted set cover algorithm (Vazirani 2001) with one major difference. Here the weight ws of each subset s is not fixed throughout the algorithm, but rather is dependent on the elements that are assigned to that subset—more precisely, the weight is a function of how closely related the colors (i.e., donor genomes) assigned to that subset are. Because during the execution of the method, the colors assigned to each subset can change, so can the weight of that subset.
The method selects the SV clusters in an iterative greedy manner based on their cost-effectiveness value in each iteration. In a given iteration, the method selects the set with the highest cost-effectiveness value, based on the maximum number of colors that can be assigned to the set in that iteration. The cost-effectiveness of a SV cluster s in the i iteration is equal to 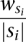 , where
, where 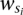 is the weight of the subset of s that is not yet covered (i.e., the reads in s that are not covered until the i iteration). Note that this greedy method will guarantee an approximation factor of
is the weight of the subset of s that is not yet covered (i.e., the reads in s that are not covered until the i iteration). Note that this greedy method will guarantee an approximation factor of 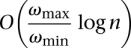 .
.
Simultaneous Set Cover with Weights and Conflict Resolution (SSC-W-CR)
The second heuristic uses the concept of Conflict Resolution and takes the diploid nature of the human genome into consideration. Hormozdiari et al. (2010) introduced a set of mathematical rules to prevent selecting SV events that cannot be happening simultaneously in reality in a haploid genome.10 The Conflict Resolution feature of this heuristic is based on those rules. Note that in Hormozdiari et al. (2010) we have modeled the conflicting SV events in a “conflict graph” in which each cluster is represented by a vertex. Two vertices are connected with an edge if the two SVs implied by the clusters are in conflict (for a detailed case study, see Hormozdiari et al. 2010). In SV detection in diploid genomes, a conflict-free set of SVs should not create a “triangle” in the conflict graph. In this heuristic, we extend the above notion from a single genome to multiple genomes, such that we are not allowed to assign the same color to three clusters (vertices) forming a triangle in the conflict graph. We have devised an iterative greedy method that selects clusters based on their cost-effectiveness: The cost-effectiveness of SV cluster s in iteration i is 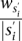 , where
, where 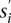 is the subset of paired-end reads in s that are not covered until this iteration and do not conflict (i.e., create a triangle) with previously selected SV clusters that have a common color. More formally, suppose that given the conflict graph
is the subset of paired-end reads in s that are not covered until this iteration and do not conflict (i.e., create a triangle) with previously selected SV clusters that have a common color. More formally, suppose that given the conflict graph 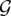 , for each of the sets picked prior to iteration i, a subset of λ colors has been assigned to them. Any paired-end read r ∈ s is considered to be a member of
, for each of the sets picked prior to iteration i, a subset of λ colors has been assigned to them. Any paired-end read r ∈ s is considered to be a member of 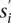 if:
if:
r is not covered by any of the i − 1 clusters picked prior to iteration i.
There is no pair of clusters q and p that have been picked in earlier iterations, such that q, p, and r form a triangle and both q and p include the color of r.
Results
We investigated the structural variation content of six human genomes in order to establish the benefits of Simultaneous Structural Variation discovery among Multiple Genomes (SSV-MG) compared with the Independent Structural Variation Discovery and Merging (ISV&M) strategy. The two data sets we investigate each constitutes a father–mother–child trio. The first trio is a Yoruba family living in Ibadan, Nigeria (YRI: NA18506, NA18507, NA18508) (Bentley et al. 2008). The second trio is a family from Utah with European ancestry (CEU: NA12878, NA12891, NA12892) sequenced with high coverage by the 1000 Genomes Project (1000 Genomes Project Consortium 2010). We aligned the downloaded paired-end reads to the human reference genome (NCBI Build 36) using mrFAST (Alkan et al. 2009). Statistics for each data set are provided in Table 1 (after removing low-quality paired-end reads).
Table 1.
Summary of the analyzed human genomes

We sought to establish whether simultaneous analysis of all three genomes (in each trio) would result in more accurate detection of structural variation events in comparison to the conventional two-step approach of ISV&M. For each of the trios, we analyzed the three genomes independently using the ISV&M-based approach and simultaneously using the SSV-MG framework; we then compared the results from each analysis.
For the ISV&M approach, we proceeded as follows:
The ISV step: We analyzed each genome independently, using VariationHunter (Hormozdiari et al. 2009) for deletions and extended VariationHunter for Alu insertions (Hormozdiari et al. 2010).11
The M step: To identify common structural variation among different genomes, we compared each data set and merged shared structural variation predictions. Two structural variation predictions were considered to be “shared” (i.e., they are the same variation in two different individuals) if the ends of each selected cluster were within 200 bp from each other. Finally, the support value of each shared SV is considered to be the total paired-end reads in the two (or more) individuals that support that shared SV.
In these experiments, we were purely interested in evaluating the added benefit of simultaneous analysis over independent analysis. We used VariationHunter (Hormozdiari et al. 2009), a maximum parsimony–based approach, for the ISV&M analysis since all the SSV-MG methods proposed here are also maximum parsimony–based methods. In addition, VariationHunter is one of the very few tools with capability to find mobile element insertions.
The experiments in this study focus on two types of structural variation:
Mobile element insertions (i.e., Alu insertions) on the YRI data set
Medium- and large-size deletions on the YRI and CEU data sets
Mobile element insertions
As we have described earlier (Hormozdiari et al. 2010, 2011), it is possible to use VariationHunter within the ISV&M framework for discovering mobile element insertions such as Alu elements on a WGSS donor genome with respect to a reference genome. Below we compare this approach, as a representative of the ISV&M framework, with the SSV-MG framework, more specifically SSC and SSC-W approximation algorithms.
We applied the ISV&M and the two SSV-MG approaches, namely, SSC and SSC-W algorithms (we set two potential Alu inserts that overlap highly as the same Alu; this implies that conflict resolution is not necessary for this case) to the discovery of Alu insertions in the YRI trio. The results from each analysis were then compared to Alu polymorphism loci reported in dbRIP (Wang et al. 2006)—which provides an estimate on the trade-off between the number of predictions made and the fraction of the known Alu insertions captured. Since the contents of dbRIP are curated from a variety of data sources, for a given Alu insertion prediction, a match in dbRIP is a good indicator that the prediction is a true positive. Note that we call a predicted Alu insertion, a match to an Alu insertion locus reported in dbRIP (Wang et al. 2006), if the reported locus in dbRIP is within 100 bp of the breakpoints predicted.
For each method, we calculated the number of Alu insertions with a dbRIP match for a range of thresholds for the read support for each Alu insertion. The fraction of dbRIP matching Alu insertions is consistently higher for SSC and SSC-W methods in comparison to that of the ISV&M framework for all threshold values for support; see Figure 2A.
Figure 2.

Alu insertion analysis of the YRI trio genomes. (A) Comparison of the Alu predictions made by the ISV&M, SSC, and SSC-W algorithms, which match Alu insertion loci reported in dbRIP (true positive control set). The x-axis represents the number of Alu insertions (with the highest support), while the y-axis represents the number of these insertions that have a match in dbRIP. (B–D) The number of common and de novo events in each genome as predicted by the ISV&M, SSC, and SSC-W algorithms, respectively (the top 3000 predictions were considered).
Since the two data sets we considered each involves members of a family, it is expected that many of the Alu insertions observed are common to all three genomes. However, the ISV&M framework predicted only 507 common Alu insertion loci common to all three individuals, in contrast to 1044 common inserts predicted by the SSC method and 1257 common inserts predicted by the SSC-W method (see Fig. 2B–D).
The rate of de novo Alu insertions is estimated to be one new Alu insertion per 20 births (Cordauxa et al. 2006). Thus, it is quite unlikely that the genome of the child (NA18506) in the YRI trio contains several Alu insertions that are not present in the parent genomes. However, the ISV&M framework based on VariationHunter (Hormozdiari et al. 2010) reported that among the top 3000 predicted loci,12 410 were de novo (that is, unique to the child). This number clearly is extremely high given our current knowledge of Alu insertion rates. Thus, the majority of these 410 events are likely to be misclassified as de novo events by the ISV&M framework. Interestingly, using the SSC algorithm, this number was reduced to only 20 de novo events among the top 3000 predictions (Fig. 2C). Furthermore, the SSC-W algorithm13 reduces the number of de novo Alu insertions to zero in the top 3000 Alu insertion predictions (see Fig. 2D).
Note that one of the Alu insertion loci predicted as a de novo insertion in NA18506 by both the SSC method and the ISV&M framework turned out to be a locus experimentally tested positive for an Alu insertion by a polymerase chain reaction (PCR) in the YRI trio (Hormozdiari et al. 2011). The result of PCR indicates that there is, indeed, an Alu insertion in the above locus in NA18506. However, it turned out that the insertion is not de novo but rather a transmission from the father (NA18507) to the child (NA18506). SSC-W, on the other hand, was able to correctly identify the Alu insertion in both NA18506 and NA18507 and thus was able to correctly classify the prediction as a transmitted event.
A similar analysis on the CEU trio also revealed similar results to those we obtained on the YRI trio (for details, see the Supplemental Material). Note that the number of Alu insertion predictions in the child, NA12878, is slightly more than what we expect. This is likely due to the sequence coverage imbalance among the genomes (32× for the child versus 13× for each parent).
Deletions
In this section, we compare the deletion calls made by algorithms proposed within the SSV-MG framework (i.e., SSC-W and SSC-W-CR) in comparison to those made by the ISV&M framework (i.e., VariationHunter) (Hormozdiari et al. 2009). The deletions considered here are medium- to large-scale events (>100 bp and <1 M bp) in both YRI and CEU trios. To verify (at least some of) the predictions made by the above algorithms, we used the validated SV events reported in the recent study by the 1000 Genomes Project Consortium (Mills et al. 2010). The results of this comparison are shown in Table 2. To make the comparison as thorough as possible, we considered various-sized subsets of calls for each method (obtained by varying the read support threshold on the predictions considered for each method).6 In comparison to the ISV&M framework, the SSV-MG algorithms consistently produce a higher fraction of validated predictions in both YRI and CEU trios (see Table 2). We also compared the deletion predictions made by each one of the methods considered here on each individual genome from the CEU trio, with the validated deletion calls on the same individual genome by the above-mentioned 1000 Genomes study (Mills et al. 2010). (Unfortunately, such a set of validated deletions does not exist for the YRI trio.) Table 3 provides the number of the validated deletion calls from each specific genome in the CEU trio among the best supported 5000 calls made by each one of the methods considered here. Note that the number of de novo deletions reported in the child genome of CEU trio (NA12878) should not be high—as per the Alu insertions—because each deletion is likely to have been inherited from one of the parents. Among the top 5000 deletion loci (on the child genome) predicted by the ISV&M framework, 84 were predicted to be de novo events. In contrast, among the top 5000 deletion loci (on the child) reported by the SSC-W algorithm, only 39 were predicted to be de novo events. This reduction of >50% on the number of misclassified deletions as de novo events demonstrates once again the improved predictive power of the SSV-MG framework over the ISV&M framework.
Table 2.
Comparison of deletions discovered in CEU and YRI trio against validated deletions

Table 3.
NA12878, NA12891, and NA12892 deletions discovered

Discussion
In this study, we demonstrate that analyzing a collection of high-throughput sequenced genomes jointly and simultaneously improves structural variation discovery, especially among highly related genomes. We focus on discovering deletions and Alu repeats among high-throughput paired-end sequenced genomes of family members and show that the algorithms we have developed for simultaneous genome analysis provide much lower false-positive rates in comparison to existing algorithms that analyze each genome independently. Our algorithms, which are collectively called CommonLAW (Common Loci structural Alteration Widgets), aim to solve the maximum-parsimony Structural Variation Discovery for Multiple Genomes problem optimally through a generalization of the maximum parsimony formulation and the associated algorithms introduced in Hormozdiari et al. (2009, 2010) for a single donor genome. We believe that the CommonLAW framework will help studies on multiple, highly related, high-coverage NGS genome sequences from members of a family or an ethnic group, tissue samples from one individual (e.g., primary tumor vs. metastatic tumor), individuals sharing a phenotype, etc., by identifying common and rare structural alterations more accurately. The YRI family on which we demonstrate the effectiveness of the CommonLAW framework in the context of de novo Alu repeat discovery or the CEU family whose genomes we analyzed for deletion discovery provides convincing evidence that the CommonLAW framework may make a significant difference in large-scale projects involving high-coverage NGS data. In addition, we believe that our methods can also be adopted to analyzing low-coverage NGS data for improving the accuracy provided by available software tools.
Data access
The CommonLaw package is currently available at http://compbio.cs.sfu.ca/strvar.htm and will be moved to its final destination at SourceForge (http://variationhunter.sourceforge.net/).
Acknowledgments
We thank C. Alkan for providing the initial mappings of the whole-genome shotgun (WGS) sequencing data and S. Alaei, H. Jowhari, and S. Oveis Gharan for fruitful discussions on the problem. We also thank L. Brunner for help preparing the manuscript. We thank the anonymous Yoruba and Utah CEPH families for contributing samples for research. This work was supported, in part, by the Natural Sciences and Engineering Research Council of Canada (NSERC), Bioinformatics for Combating Infectious Diseases (BCID) grants to S.C.S.; NSERC Alexander Graham Bell Canada Graduate Scholarships (CGS-D) to F.H. and I.H.; and U.S. National Institutes of Health (NIH) grant HG004120 to E.E.E. E.E.E. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Although it is easy to generalize the formulation we provide here to multiple reference genomes, we do not explore this problem here due to the lack of alternative, completely and independently assembled reference genomes.
For example, for the generalization to the case in which each element is in at most two subsets, observe that if an element is in only one subset, that subset must be included in any feasible solution.
Without loss of generality, we assume that ω{red} = ω{black} = ωmax.
The greedy algorithm selects at most 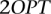 unicolor subsets.
unicolor subsets.
For example, two clusters that indicate a deletion and significantly overlap with each other are considered to be conflicting.
Note that any other method such as BreakDancer, MoDIL, or GASV could have been used in this step.
The 3000 loci with the highest number of paired-end read support.
The weights used for SSC-W were derived from the fraction of Alu insertions common between the individuals reported by the SSC results.
Note that, since ωmin is usually much smaller than the weight of events with two assigned colors and ωmax, we will get a much better bound in reality.
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.120501.111.
References
- Alkan C, Kidd JM, Marques-Bonet T, Aksay G, Antonacci F, Hormozdiari F, Kitzman JO, Baker C, Malig M, Mutlu O, et al. 2009. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet 41: 1061–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon N, Moshkovitz D, Safra S 2006. Algorithmic construction of sets for k-restrictions. ACM Trans Algorithms 2: 153–177 [Google Scholar]
- Bashir A, Volik S, Collins C, Bafna V, Raphael BJ 2008. Evaluation of paired-end sequencing strategies for detection of genome rearrangements in cancer. PLoS Comput Biol 4: e1000051 doi: 10.1371/journal.pcbi.1000051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. 2008. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456: 53–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Wallis J, McLellan M, Larson D, Kalicki J, Pohl C, McGrath S, Wendl M, Zhang Q, Locke D, et al. 2009. Breakdancer: An algorithm for high-resolution mapping of genomic structural variation. Nat Methods 6: 677–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MJ, Homer N, O'Connor BD, Chen Z, Eskin A, Lee H, Merriman B, Nelson SF 2010. U87mg decoded: The genomic sequence of a cytogenetically aberrant human cancer cell line. PLoS Genet 6: e1000832 doi: 10.1371/journal.pgen.1000832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordauxa R, Hedgesa DJ, Herkea SW, Batzer MA 2006. Estimating the retrotransposition rate of human alu elements. Gene 373: 134–137 [DOI] [PubMed] [Google Scholar]
- Feuk L, Carson AR, Scherer SW 2006. Structural variation in the human genome. Nat Rev Genet 7: 85–97 [DOI] [PubMed] [Google Scholar]
- Hach F, Hormozdiari F, Alkan C, Hormozdiari F, Birol I, Eichler EE, Sahinalp SC 2010. mrsFAST: a cache-oblivious algorithm for short-read mapping. Nat Methods 7: 576–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajirasouliha I, Hormozdiari F, Alkan C, Kidd JM, Birol I, Eichler EE, Sahinalp SC 2010. Detection and characterization of novel sequence insertions using paired-end next-generation sequencing. Bioinformatics 26: 1277–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F, Alkan C, Eichler EE, Sahinalp SC 2009. Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res 19: 1270–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F, Hajirasouliha I, Dao P, Hach F, Yorukoglu D, Alkan C, Eichler EE, Sahinalp SC 2010. Next-generation variationhunter: combinatorial algorithms for transposon insertion discovery. Bioinformatics 26: i350–i357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F, Alkan C, Ventura M, Hajirasouliha I, Malig M, Hach F, Yorukoglu D, Dao P, Bakhshi M, Sahinalp SC, et al. 2011. Alu repeat discovery and characterization within human genomes. Genome Res 21:840–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Cancer Genome Consortium, Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabé RR, Bhan MK, Calvo F, Eerola I, et al. 2010. International network of cancer genome projects. Nature 464: 993–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp RM 1972. Reducibility among combinatorial problems. In Complexity of computer computations (ed. Miller R), pp. 85–103 Springer, New York [Google Scholar]
- Korbel JO, Abyzov A, Mu XJ, Carriero N, Cayting P, Zhang Z, Snyder M, Gerstein MB 2009. PEMer: A computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome Biol 10: R23 doi: 10.1186/gb-2009-10-2-r23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leary R, Kinde I, Diehl F, Schmidt K, Clouser C, Duncan C, Antipova A, Lee C, McKernan K, De La Vega F, et al. 2010. Development of personalized tumor biomarkers using massively parallel sequencing. Sci Transl Med 2: p20ra14 doi: 10.1126/scitranslmed.3000702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Cheran E, Brudno M 2008. A robust framework for detecting structural variations in a genome. Bioinformatics 24: i59–i67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Hormozdiari F, Alkan C, Brudno M 2009. MoDIL: Detecting small indels from clone-end sequencing with mixtures of distributions. Nat Methods 6: 473–474 [DOI] [PubMed] [Google Scholar]
- Lee S, Xing E, Brudno M 2010. MoGUL: Detecting common insertions and deletions in a population. Res Comput Mol Biol 6044: 357–368 [Google Scholar]
- Mardis ER 2010. Cancer genomics identifies determinants of tumor biology. Genome Biol 11: 211 doi: 10.1186/gb-2010-11-5-211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev P, Stanciu M, Brudno M 2009. Computational methods for discovering structural variation with next-generation sequencing. Nat Methods 6: 13–20 [DOI] [PubMed] [Google Scholar]
- Mills R, Walter K, Stewart C, Handsaker R, Chen K, Alkan C, Abyzov A, Yoon S, Ye K, Cheetham R, et al. 2010. Mapping copy number variation by population-scale genome sequencing. Nature 470: 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1000 Genomes Project Consortium 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Clark RA, Sokolova S, Leibowitz ML, Zhang Y, Hurles ME, Mell JC, Hall IM 2010. Genome-wide mapping and assembly of structural variant breakpoints in the mouse genome. Genome Res 20: 623–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz R, Safra S 1997. A sub-constant error-probability low-degree test, and a sub-constant error-probability PCP characterization of NP. STOC 475–484 doi: 10.1145/258533.258641 [Google Scholar]
- Sindi SS, Helman E, Bashir A, Raphael BJ 2009. A geometric approach for classification and comparison of structural variants. Bioinformatics 25: i222–i230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR 2010. Structural variation in the human genome and its role in disease. Annu Rev Med 61: 437–455 [DOI] [PubMed] [Google Scholar]
- Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, et al. 2005. Fine-scale structural variation of the human genome. Nat Genet 37: 727–732 [DOI] [PubMed] [Google Scholar]
- Vazirani VV 2001. Approximation algorithms. Springer-Verlag, New York [Google Scholar]
- Volik S, Zhao S, Chin K, Brebner JH, Herndon DR, Tao Q, Kowbel D, Huang G, Lapuk A, Kuo WL, et al. 2003. End-sequence profiling: Sequence-based analysis of aberrant genomes. Proc Natl Acad Sci 100: 7696–7701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Song L, Grover D, Azrak S, Batzer M, Liang P 2006. dbRIP: A highly integrated database of retrotransposon insertion polymorphisms in humans. Hum Mutat 27: 323–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, Ning Z 1999. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25: 2865–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]