

Abstract
The design, synthesis, and biological activity of fluorinated amino-derivatives of the sesquiterpene lactone, parthenolide, are described. A fluorinated aminoparthenolide analogue with biological activity similar to the parent natural product was discovered, and its X-ray structure was obtained. This lead compound was then studied using 19F NMR in the presence and absence of glutathione to obtain additional mechanism of action data, and it was found that the aminoparthenolide eliminates amine faster in the presence of glutathione than in the absence of glutathione. The exact changes in concentrations of fluorinated compound and amine were quantified by a concentration-reference method using 19F NMR; a major benefit of applying this strategy is that no deuterated solvents or internal standards are required to obtain accurate concentrations. These mechanistic data with glutathione may contribute to the conversion of the amino-derivative to parthenolide, the active pharmacological agent, in glutathione-rich cancer cells.
Introduction
The sesquiterpene lactones are a class of bioactive natural products that commonly have an α-methylene-γ-lactone substructure that reacts with nucleophilic sulfhydryl groups present in enzymes, proteins, and glutathione.1 These thiol additions may be reversible or irreversible and they define the mechanism of action of these natural products.1,2 However, many of these compounds are poorly soluble in water and this issue has limited their potential therapeutic use in humans. To address this shortcoming, amines have been added to the α-methylene-γ-lactone substructure as an efficient way to enhance the water solubility of the parent molecules and to retain their biological activity. Amino-adducts have been prepared from the sesquiterpene lactones, alantolactone,3 costunolide,4 parthenolide (1),5–7 α-santonin,8 helenalin,9 and ambrosin (Figure 1).10 A key amino-adduct of parthenolide (i.e. DMAPT or LC-1) has advanced into clinical studies in humans,11–13 despite a long-standing bias in medicinal chemistry against using molecules with a potential covalent mechanism of action in clinical trials.2 Also, the sesquiterpene lactones, sausseramines A–E, have a naturally occurring α-methylamino lactone substructure rather than an α-methylene-γ-lactone.14,15
Figure 1.

Structures of selected sesquiterpene lactones and the amino derivative, DMAPT.
It has been hypothesized that these α-methylamino lactone derivatives serve as prodrugs in which the amine is released and the enone is regenerated in the presence of the biological nucleophiles (Figure 2).3,7,10,14,15 Yoshikawa and co-workers have demonstrated that this retro-Michael reaction occurs with the sausseramines in 1% aqueous HCl solution at room temperature within 12 h.15 Subsequently, Hsieh and co-workers analyzed cell lysates by LC/MS/MS after treatment with an amino-parthenolide derivative and found only the presence of parthenolide.7 On the other hand, Crooks and coworkers have shown that less than 3% of the aminoparthenolide, DMAPT, degrades to parthenolide in cell culture buffer after a 24 h incubation.5 Based on these literature precedents, we aimed to design a derivative of parthenolide that could serve as a probe for this retro-Michael reaction in the presence of the biological thiol, glutathione. Such a mechanistic probe would have potential use with other sesquiterpene lactones and molecules with a covalent binding mechanism. A key requisite to accomplish this objective would be to selectively observe release of the amine from the parent molecule or prodrug.
Figure 2.

Conversion of α-methylamino-γ-butyrolactones to α-methylene-γ-butyrolactones and trapping with biological thiols.
The incorporation of fluorine onto molecules is a powerful strategy in medicinal chemistry to modulate hydrophobic character and decrease metabolism,16 and the derivatization of bioactive natural products with fluorine has been attracting considerable attention.17–19 Another powerful, yet somewhat less-utilized role for fluorine is as a tag for 19F NMR. Indeed, 19F NMR is an analytical tool that is ideal for probing biological systems with fluorinated compounds, because unlike the 1H and 13C nuclei, there is no background signal for 19F.20–27 Fluorinated molecules have served as valuable 19F NMR probes in high-throughput screening,20,26,27 drug metabolism,21 protein binding,20,22,24,26,27 and assessing gene expression.23,25 We hypothesized that the incorporation of fluorine onto the amine on an aminoparthenolide would not adversely affect biological activity. Additionally, we envisioned that the release of the fluorinated amine from the parent compound would be observed by 19F NMR, even at low concentrations, and additional mechanism of action data could be obtained with these 19F NMR probes (Figure 3). Recently, we have described a concentration-reference NMR method to determine the concentration of pharmaceutical agents without the use of deuterated solvents or internal standards.28 This method determines the concentration of analyte from solvent as a reference and applies the data to the calculation of partition coefficient, or log P. We now aim to extend the utility of this NMR technique to 19F NMR, because it will enable the direct determination of concentration of a fluorinated pharmaceutical agent in biological conditions where standards and deuterated solvents are not normally compatible. Herein, we report the design, synthesis, and biological activity of fluorinated amino-derivatives of the sesquiterpene lactone, parthenolide, and the subsequent application of the no-D concentration-reference NMR technique to determine the concentration of fluorinated compound and production of fluorinated amine using 19F NMR. Our biological data establish similarities between the fluorinated aminoparthenolide and parthenolide, and the 19F NMR data quantitatively demonstrate that elimination of the fluorinated amine from the parthenolide derivative occurs more rapidly in the presence of the biological thiol, glutathione, than without glutathione.
Figure 3.

Design strategy for 19F NMR probes using fluorinated α-methylamino-γ-butyrolactones and the respective elimination of fluorinated amines.
Results and Discussion
Chemistry
Our initial strategy was to prepare a series of fluorinated amino-derivatives of the sesquiterpene lactone, parthenolide (1), and to identify an analogue with biological activity most similar to the parent compound. Interest in parthenolide (1) has surged beyond other members of the sesquiterpene lactones, because it was shown in 2005 to selectively kill cancer stem cells isolated from patients with myelogenous leukemia.29 We have previously synthesized fluorinated derivatives of this key natural product30 and demonstrated the ability of the parent compound and its derivatives to selectively initiate apoptosis in multiple myeloma cancer stem cell populations.31 Using the prior report of Crooks to prepare amino-adducts,5 we focused our attention on pyrrolidine and piperidine derivatives of parthenolide and identified respective fluorinated amines that are commercially available. Our choice of fluorinated amines corresponds well with the recent studies from scientists at Merck.32 Additionally, we selected amines that contain a trifluoromethyl group, because this functional group can be easily visualized by 19F NMR. In other words, using 19F NMR with a low concentration of fluorinated compound, the sensitivity for a trifluoromethyl group is three-fold higher than that of a single fluorine atom. Using pyrrolidine, fluoro-substituted pyrrolidines, piperidine and fluoro-substituted piperidines, the parthenolide derivatives 2–12 were prepared using the literature protocol (Table 1).5 Good yields of the products were routinely obtained and each compound was a single diastereomer, except compounds 3 and 5 that are inseparable 1:1 mixtures of diastereomers due to the use of racemic 3-fluoro and 3-trifluoromethylpiperidine, respectively. In order to unambiguously assign the stereochemistry of the newly formed center on the lactone ring in compounds 2–12, we obtained an X-ray structure of compound 8 (Figure 4). As expected, the R-isomer at the C11 position is formed exclusively and this result is similar to literature precedent.6 The identity of the stereocenter and conformation of the macrocycle 8 correlate well with prior X-ray data from the aminoparthenolide, DMAPT.33
Table 1.
Synthesis of Fluorinated Amino-Derivatives of Parthenolide
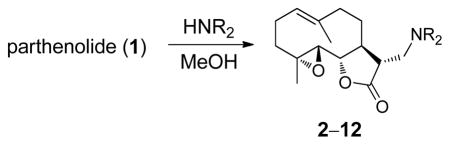 | ||
|---|---|---|
| HNR2 | product | yielda |
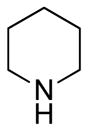
|
2 | 82% |
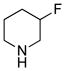
|
3 | 79% |
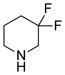
|
4 | 89% |
|
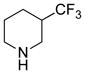
5 | 64% |
|
6 | 89% |
|
7 | 75% |
|
8 | 80% |
|
9 | 50% |
|
10 | 83% |
|
11 | 86% |
|
12 | 78% |
Isolated yields.
Figure 4.
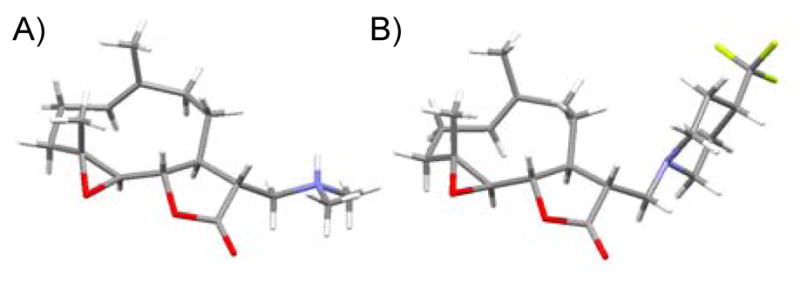
X-ray structures of aminoparthenolide derivatives: (A) (11R)-13-dimethylammonio-11,13-dihydro-parthenolide salt (semi-fumarate counterion removed for clarity),33 (B) compound 8.
Antiproliferative Activity
The biological activity exhibited by each of the parthenolide derivatives 2–12, as well as parthenolide (1), was determined using an antiproliferative assay with HL-60 (human promyelocytic leukemia) cells, because the major application of parthenolide and its derivatives is as an anti-leukemic agent.34 To ensure that the fluorinated lead compound most resembled the biological activity of the parent compound in our assays, a fine resolution cancer cell growth inhibition assay was developed and implemented. Typically, the NCI60 human tumor cell line anticancer drug screen is a significant tool in cancer research; it has solved technical barriers to in vitro drug screening35 and has generated orthogonal benefits in the form of multi-tissue systems biology studies that would not have been possible prior to its existence.36–40 However, the NCI60 screen was designed for applications in a high throughput manner for hundreds of thousands of compounds that manifest varying activity.34 Therefore, each compound is screened with only five 10-fold dilution steps, and a linear interpolation analytical method is used that avoids fitting a model to estimate GI50, TGI and LC50. While this strategy reduces the time and resources needed to characterize many compounds, fitting a parametric model to dose response data allows the benefit of pair-wise statistical comparison of GI50, TGI and LC50 across compounds.41 The design of the HL-60 assay employed for compounds 1–12 has its explicit goal as the ability to characterize the antiproliferative properties of sets of synthetic analogues to explore structural variation of a parent molecule. In this case, quantitative shifts in GI50, TGI or LC50 should be discernable among structural variants even though such shifts may occur within a relatively narrow dosage range considered to be pharmaceutically relevant (the nanomolar to low micromolar range). Individual compounds 1–12 were tested in quadruplicate across 72 h, and GI50 and TGI values were calculated along with the 95% confidence interval (Table 2). The LC50 values for 1, 2, 5–10, and 12 were also determined. Specifically, a sigmoid inhibitory dose-response model was fit to the data for each compound using nonlinear regression via a least squares fit with propagation of error throughout normalization calculations. For the best-fit values of GI50, TGI or LC50 for any two compounds, an extra sum-of-squares F test is used to determine whether a better fit is achieved via a single model or separate models for the two compounds, p value < 0.05.42,43
Table 2.
Antiproliferative Assay in HL-60 Cellsa
| compd | GI50 (95% CI) μM | TGI (95% CI) μM | LC50 (95% CI) μM |
|---|---|---|---|
| 1 | 0.52(0.19–1.42) | 1.36(0.65–2.85) | 1.88(0.72–4.90) |
| 2 | 0.84 (0.19–3.79) | 3.99(1.03–15.4) | 10.1 (0.78–130) |
| 3 | 1.13(0.64–20.1) | 12.2(1.98–75.7) | nd |
| 4 | 16.9(4.68–61.3) | nd | nd |
| 5 | 1.69(0.46–6.20) | 7.29(1.64–32.5) | 21.3(0.33–1400) |
| 6 | 1.77(0.52–6.05) | 5.70 (2.24–14.5) | 8.07(2.70–24.1) |
| 7 | 0.69 (0.07–7.12) | 5.58(1.18–27.3) | 17.7(0.45–700) |
| 8 | 1.00(0.57–1.75) | 2.14(1.10–4.15) | 3.11 (1.19–8.15) |
| 9 | 0.75(0.41–1.40) | 1.74(0.97–3.12) | 2.54(1.13–5.70) |
| 10 | 0.58(0.33–1.01) | 1.18(0.68–2.03) | 1.66(0.76–3.62) |
| 11 | 24.7(11.9–51.1) | 46.2(31.3–68.1) | nd |
| 12 | 8.32 (4.21–16.5) | 18.3(8.17–41.1) | 23.8(9.17–61.7) |
Antiproliferative assays were conducted in the HL-60 (human promyelocytic leukemia) cell line. nd is not determined.
Parthenolide (1) displays activity in our assay (GI50 = 0.52 μM) similar to previous reports in HL-60 cells.34 Amino-derivatives 2, 3, and 5–10 manifest similar GI50 values in the assay compared with 1. Indeed, most amino-derivatives of the sesquiterpene lactones produced biological activity very similar to the parent compound across many types of assays.3–10 Notable exceptions in this series of molecules are the 3,3-difluoro-piperidine analogue 4, the (S)-3-fluoropyrrolidine analogue 11, and the 3,3-difluoro-pyrrolidine analogue 12. A rationale for the comparison of the more potent activity of 4,4-difluoro-piperidine 7 (GI50 = 0.69 μM) to the less potent 3,3-difluoropiperidine 4 (GI50 = 16.9 μM) may be the attenuated basicity of the piperidine nitrogen on the later compound. This relationship between fluorination and pKa for 3,3-difluoropiperidine and 4,4-difluoropiperidine has been elegantly described by Diederich, Kansy, and Müller.44 The biological activity (i.e., GI50 values) across compounds 10–12 is intriguing, because the (S)-3-fluoropyrrolidine analogue 11 is almost 50-fold less potent than the (R)-3-fluoropyrrolidine analogue 10. Furthermore, the 3,3-difluoropyrrolidine 12 displays activity that can be envisioned to be 1:1 mixture of both (R)-10 and (S)-11. Analysis of the LC50 values distinguishes aminoparthenolide analogues 2, 3, and 5–7 from parthenolide (1) and 8–10. Even though many of the parthenolide derivatives inhibit cell growth, only a subset of compounds (i.e., 8–10) eliminate the HL-60 cell population at a level similar to the parent compound, and only two fluorinated compounds (i.e., 8 and 10) exhibit this effect. After this analysis of the biological data, we selected aminoparthenolide 8 for further studies with 19F NMR, because this compound has the requisite trifluoromethyl group and maintains activity similar to the parent compound (1). Side-by-side plots of the growth inhibition curves for parthenolide 1 and lead compound 8 show the high level of similarity between the biological activities in treated HL-60 cells (Figure 5). The slight divergence between the GI50 values was not found to be statistically significant.
Figure 5.

Comparison of growth inhibition of HL-60 cells between parthenolide (1) and compound 8 after three days. Data for parthenolide (1) are colored in black and compound 8 are colored in red. GI50 values for 1 and 8 are not statistically significantly different. The horizontal dotted line marks 100% Day 0 cell density (i.e. the original population size of the cells when they were first seeded into the wells).
19F NMR and Quantitative Studies
With the aminoparthenolide 8 in hand, we now aimed to use the compound as a 19F NMR probe to elucidate the role of its structural alteration in the presence of glutathione and to acquire additional mechanism of action data for aminoparthenolides. Higher levels of the key biological thiol, glutathione, have been observed in cancer cells over non-malignant cells,45,46 and these data have been recently exploited by Boger and co-workers for the preferential activation of prodrugs of duocarmycin-based anticancer agents in cancer cells.47 Glutathione is especially important in the environment of cancer cell populations, and Chmielewski,48 Finn,49 and others50 have described fluorescence probes for imaging cellular thiols, such as glutathione. 19F NMR probes for measuring the presence of glutathione are rare in the literature;51 however, an advantage for 19F labeling over fluorescent labeling is that structural data for the molecule and its derivatives can be gathered by 19F NMR. Moreover, our studies were planned to extend the use of fluorinated agents in gathering mechanism of action by measuring changes to the compound itself, rather than the presence/reactivity of the biological thiol. The key feature in compound 8 is that the trifluoromethyl group is present on the parent compound, but will also be present on the amine upon elimination (see Fig. 3). The change in signal will be recorded in the 19F NMR spectrum, and the change in concentration will be recorded using the no-D concentration-reference method (see below).28 Indeed, previous 19F NMR methods routinely require the addition of deuterated solvent,20–22,51 and the significance of our plan is that deuterated solvents are not needed. Also, 19F NMR data can be acquired rapidly with high sensitivity. Our initial studies were conducted at pH 7.4 (biological pH) using compound 8 (1 mM) in the presence of glutathione (13.5 mM) and in the absence of glutathione; common biological concentrations of glutathione are 1 to 15 mM.48 To ensure complete solubility of these components at biological pH, a 7:3 DMSO/H2O (v/v) solvent mixture was chosen; the major limiting factor was the solubility of 8 in aqueous solution. Even though the aqueous solubility of DMAPT was significantly enhanced by conversion to its fumarate salt,5 our attempts to convert 8 to its fumarate salt using this method were not successful because the amine salt would not crystallize from organic solvents (e.g. Et2O and EtOH). Although a high percentage of DMSO limits the direct translation for our results to aqueous biological environments, it ensured complete solubility throughout the experiments and has been routinely used as a co-solvent for observing Michael additions.2,49 Next, preliminary studies were conducted at 37 °C, 60 °C, 80 °C, and 100 °C to identify temperatures that would promote the elimination of the amine from 8. Ultimately, 80 °C was selected, because some elimination of the amine occurred after 24 h (Figure 6A) and no other by-products were observed in the full 19F NMR spectrum even after extended reaction times, such as three days (data not shown). Therefore, studies were conducted across a three day time-course (similar to the antiproliferative assay) and 19F NMR spectra were obtained at 24 h intervals at 80°C for samples with and without glutathione (Figure 6). Only 128 scans were required, and thus, the total acquisition time for each 19F NMR spectrum was 3 min 24 sec with a typical sensitivity of 40:1 orbetter. The data clearly display the production of 4-trifluoromethylpiperidine from compound 8 in the presence and absence of glutathione; however, the production of the free amine is nearly complete in the presence of glutathione whereas a much smaller amount is generated when glutathione is not present.
Figure 6.

19F NMR spectrum across three days depicting the production of 4-trifluoromethylpiperidine from aminoparthenolide 8 in the absence and presence of glutathione. A.) Time course of 1 mM compd 8 in 7:3 DMSO/H2O (pH 7.4) at 80 °C. B.) Time course of 1 mM compd 8 in the presence of 13.5 mM glutathione in 7:3 DMSO/H2O (pH 7.4) at 80 °C.
In order to quantify the concentration changes in the parent compound 8 and the 4-trifluoromethylpiperidine, we applied our concentration-reference method28 and carried out the two reactions side-by-side, each in triplicate (Figure 7). The concentrations were calculated as the average of the three trials, plotted across the 72 h time-course, and referenced to the first measurement (t = 0 h, 1 mM compound 8). Again, neither deuterated solvents nor internal standards were used. In the absence of glutathione, the production of 4-trifluoromethylpiperidine rose to nearly 167 μM and leveled off across three days. However, in the presence of glutathione, the concentration of 4-trifluoromethylpiperidine rose to nearly 0.7 mM, while the concentration of compound 8 continually decreased to nearly 170 μM (starting from 1.0 mM). This concentration-reference method can quickly quantify low micromolar concentrations of analyte in solution with high sensitivity. Overall, our 19F NMR data shows that elimination of the fluorinated amine from the parent aminoparthenolide derivative occurs more rapidly in the presence of glutathione than in the absence of glutathione. The implications of these findings are that if the amine is eliminated in the presence of a biological thiol, the parent compound (now with a Michael acceptor) will preferentially react with the thiol rather than re-combine with the amine. These data and assertions correlate well with the previous work of Finn and co-workers, which describes that biological thiols can easily outcompete amines in binding to Michael acceptors.49
Figure 7.

Graph illustrating the NMR quantification of the production of 4-trifluoromethylpiperidine from aminoparthenolide 8 during three days in the absence and presence of glutathione. The concentrations of 4-trifluoromethylpiperidine and compd 8 are given as the average values across three trials with error depicted as two standard deviations. Concentrations are referenced to the first measurement (t = 0 h, 1 mM). Concentrations for 4-trifluoromethylpiperidine are highlighted in red, and concentrations for compd 8 are highlighted in blue. A.) Concentrations from a 72 h time course starting with 1 mM compd 8 in 7:3 DMSO/H2O (pH 7.4) at 80 °C. B.) Concentrations from a 72 h time course starting with 1 mM compd 8 in the presence of 13.5 mM glutathione in 7:3 DMSO/H2O (pH 7.4) at 80 °C.
Conclusions
The design, synthesis, and biological activity of fluorinated amino-derivatives of the sesquiterpene lactone, parthenolide, were described. During the course of this work, we have identified an aminoparthenolide with biological activity similar to the parent natural product and obtained its X-ray structure. This lead compound was then studied using 19F NMR in the presence and absence of glutathione to obtain additional mechanism of action data, and it was found that a larger concentration of amine was observed from the aminoparthenolide in the presence of glutathione. The exact changes in concentrations were quantified by 19F NMR without the necessity of deuterated solvents or the addition of standards using a concentration-reference technique. We assert that the implications of the mechanistic data obtained from these studies are that if the amine is eliminated in the presence of a nucleophilic thiol, the addition of the thiol to the compound (now a Michael acceptor) will out-complete the re-addition of the amine. This action may contribute to the overall mechanism of action and cellular selectivity of the compound. For example, in populations of cancer cells with higher levels of glutathione than non-malignant cells, the conversion of an aminoparthenolide to parthenolide may be more prevalent. The main limitation of our 19F NMR studies is that a large portion of DMSO was required to maintain the solubility of the aminoparthenolide derivative, and this fact may limit the translational potential of these data. The preparation of highly water-soluble fluorinated amino-compounds is clearly the next objective as well as the direct observation and quantification of concentration changes of fluorinated compounds in cell culture and other biological systems. Also, the studies detailed here contribute to developing a general method of creating fluorinated amino-derivatives of compounds with covalent-binding modes for quantitative mechanism of action studies in living systems by 19F NMR. Additional studies to identify fluorinated amines with hydrophilic substituents to enhance the aqueous solubility of subsequent amino-adducts are currently underway.
Experimental Section
Chemistry
The synthesis of compounds 2–12 was conducted in a manner similar to the literature procedure.5 Characterization data for the new compounds 3–8 and 10–12 are listed below. Combustion analysis was conducted to establish a level of ≥ 95% purity.
(11R)-13-(3-Fluoropiperdine)-11,13-dihydroparthenolide (3)
79% yield. mp = 132–136 °C; 1H NMR (500 MHz, CDCl3) δ 5.20 (d, J = 12.0 Hz, 1H), 4.69–4.52 (m, 1H), 3.84 (t, J = 9.1 Hz, 1H), 3.83* (t, J = 9.1 Hz, 1H), 2.84* (t, J = 5.0 Hz, 1H), 2.82 (t, J = 5.0 Hz, 1H), 2.78–2.63 (m, 3H), 2.55–2.02 (m, 11H), 1.82 (m, 2H), 1.70 (s, 3H), 1.62 (m, 2H), 1.50 (m, 1H), 1.29 (s, 3H), 1.25 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 176.6, 176.5*, 134.7, 124.8, 87.8 (d, JCF = 171 Hz, 1C), 82.4, 66.7, 61.5, 58.4* (d, JCF = 22.1 Hz, 1C), 58.1 (d, JCF = 22.5 Hz, 1C), 56.8*, 56.0, 53.7*, 53.6, 48.1*, 47.4, 46.4*, 46.2, 41.1, 36.6, 30.2, 30.1*, 29.7 (d, JCF = 19.5 Hz, 1C), 29.6* (d, JCF = 19.8 Hz, 1C), 24.0, 22.0 (d, JCF = 6.4 Hz, 1C), 21.9 (d, JCF = 6.1 Hz, 1C), 17.2, 17.1; 19F NMR (282 MHz, CDCl3) δ −181.56 (m, 1F), −182.08* (m, 1F); IR (film) νmax 2928, 1769, 1200, 1112, 1073, 989 cm−1; HRMS (EI) m/z calcd for C20H30FNO3 (M+H)+ 352.2288, found 352.2284; Anal. Calcd for C20H30FNO3: C, 68.35; H, 8.60; N, 3.99. Found: C, 68.46; H, 8.48; N, 3.91. * denotes peak of diastereomer.
(11R)-13-(3,3-Difluoropiperdine)-11,13-dihydroparthenolide (4)
89% yield: mp = 182–186°C; 1H NMR (300 MHz, CDCl3) δ 5.19 (d, J = 11.8 Hz, 1H), 3.84 (t, J = 9.0 Hz, 1H), 2.84 (s, 1H), 2.82 (s, 1H), 2.80–2.00 (m, 13H), 1.95–1.72 (m, 4H), 1.69 (s, 3H), 1.60 (m, 1H), 1.28 (s, 1H), 1.24 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 176.4, 134.7, 124.7, 120.1 (t, JCF = 241 Hz), 82.5, 66.6, 61.5, 59.2 (t, JCF = 27.5 Hz), 55.3, 52.8, 47.4, 46.4, 41.0, 36.6, 32.0 (t, JCF = 22.9 Hz), 30.0, 24.0, 22.1 (t, JCF = 4.4 Hz, 1C), 17.2, 17.0; 19F NMR (282 MHz, CDCl3) δ − 100.5 (d, JFF = 236 Hz, 1F), − 101.8 (d, JFF = 236 Hz, 1F); IR(film) νmax 2925, 1766, 1172, 1117, 1003 cm−1; HRMS (ESI) m/z calcd for C20H29F2NO3 (M+H)+ 370.2194, found 370.12194; [α]22D −6.2° (c 1.39, CHCl3); Anal. Calcd for C20H29F2NO3·0.5H2O: C, 63.47 H, 7.99; N, 3.70. Found: C, 63.50; H, 7.61; N, 3.52.
(11R)-13-(3-(Trifluoromethyl)piperdine)-11,13-dihydroparthenolide (5)
64% yield. 1H NMR (300 MHz, CDCl3) δ 5.19 (d, J = 12.0 Hz, 1H), 3.84 (t, J = 9.1 Hz, 1H), 3.82* (t, J = 9.0 Hz, 1H), 3.03–2.67 (m, 5H), 2.48–1.89 (m, 12H), 1.78 (m, 1H), 1.70 (s, 3H), 1.65–1.42 (m, 3H), 1.30 (s, 3H), 1.25 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 176.3, 176.2*, 134.6, 126.8 (q, J = 278 Hz, 1C), 124.9, 82.3, 66.6, 61.5, 57.5*, 56.3, 54.5, 53.7*, 53.5*, 52.7, 48.7*, 47.6, 46.4, 46.3*, 41.2, 41.1 (q, JCF = 26 Hz, 1C), 36.6, 30.1, 30.0*, 24.3, 24.1, 23.9*, 22.9, 17.2, 17.0; 19F NMR (282 MHz, CDCl3) δ −72.15 (d, JHF = 27.9 Hz, 3F); IR (film) νmax 2927, 1770, 1257, 1124, 1093, 992 cm−1; HRMS (ESI) m/z calcd for C21H30F3NO3 (M+H)+ 402.2256, found 402.2260; Anal. Calcd for C21H30F3NO3·1.0EtOAc: C, 61.33; H, 7.82; N, 2.86. Found: C, 63.15; H, 7.82; N, 2.90. * denotes peak of diastereomer.
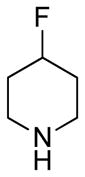
(11R)-13-(4-Fluoropiperdine)-11,13-dihydroparthenolide (6)
89% yield. mp = 142–144 °C; 1H NMR (300 MHz, CDCl3) δ 5.20 (d, J = 5.9 Hz, 1H), 4.66 (d, J = 9.3, 1H), 3.83 (t, J = 5.4 Hz, 1H), 2.81 (dd, J = 8.1, 2.9 Hz, 1H), 2.73–2.64 (m, 3H), 2,72 (d, J = 5.4 Hz, 1H), 2.44–2.35 (m, 4H), 2.29–2.04 (m, 6H), 1.96–1.78 (m, 4H), 1.70 (s, 3H). 1.64 (m, 1H), 1.30 (s, 3H), 1.23 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 176.4, 134.6, 124.9, 88.3 (d, JCF = 170 Hz, 1C), 82.2, 66.6, 61.5, 56.3, 50.3 (d, JCF = 6.4 Hz, 2C), 48.1, 46.5, 41.2, 36.6, 31.6 (d, JCF = 19.5 Hz, 2C), 30.1, 24.1, 17.2, 17.0; 19F NMR (282 MHz, CDCl3) δ −180.27 (m, 1F); IR (film) νmax 2929, 1770, 1174, 1039, 1006, 987 cm−1; HRMS (ESI) m/z calcd for C20H30FNO3 (M+H)+ 352.2288, found 352.2285; [α]28D −5.3° (c 0.60, CHCl3); Anal. Calcd for C20H30FNO3: C, 68.35; H, 8.60; N, 3.99. Found: C, 68.50; H, 8.70; N, 3.82.
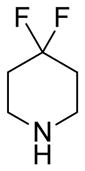
(11R)-13-(4,4-Difluoropiperdine)-11,13-dihydroparthenolide (7)
75% yield. mp = 148–152 °C; 1H NMR (300 MHz, CDCl3) δ 5.19 (d, J = 10.0 Hz, 1H), 3.84 (t, J = 9.1 Hz, 1H), 2.81 (qd, J = 13.7, 5.0 Hz, 2H), 2.71 (d, J = 8.9 Hz, 1H), 2.64 (t, J = 5.4 Hz, 4H), 2.47–1.90 (m, 12H), 1.70 (s, 3H), 1.63 (m, 1H), 1.29 (s, 3H), 1.26 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 176.2, 134.5, 125.1, 121.6 (t, JCF = 240 Hz, 1C), 82.2, 66.5, 61.5, 55.3, 50.8 (t, JCF = 4.9 Hz, 2C), 47.0, 46.7, 41.2, 36.6, 34.0 (t, JCF = 22.8 Hz, 2C), 30.0, 24.1, 17.2, 17.0; 19F NMR (282 MHz, CDCl3) δ −98.27 (m, 2F); IR νmax 2923, 1764, 1254, 1132, 1100, 991 cm−1; HRMS (ESI) m/z calcd for C20H29F2NO3 (M+H)+ 370.2194, found 370.2191; [α]24D −11.6° (c 0.56, CHCl3); Anal. Calcd for C20H29F2NO3: C, 65.02; H, 7.91; N, 3.79. Found: C, 64.66; H, 7.76; N, 3.75.
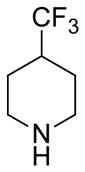
(11R)-13-(4-(Trifluoromethyl)piperdine)-11,13-dihydroparthenolide (8)
80% yield. mp = 182–184 °C; 1H NMR (300 MHz, CDCl3) δ 5.20 (d, J = 11.3 Hz, 1H), 3.83 (t, J = 9.0 Hz, 1H), 2.92 (d, J = 10.5 Hz, 2H), 2.80 (dd, J = 13.6, 4.9 Hz, 1H), 2.72 (d, J = 9.0 Hz, 1H), 2.70 (dd, J = 13.6, 4.9 Hz, 1H), 2.47–1.96 (m, 11H), 1.83 (m, 2H), 1.70 (s, 3H), 1.63–1.49 (m, 3H), 1.29 (s, 3H), 1.25 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 176.4, 134.6, 127.1 (q, JCF = 277 Hz, 1C), 125.0, 82.2, 66.6, 61.5, 56.5, 53.6, 52.7, 48.1, 46.4, 41.2, 40.1 (q, JCF = 27.2 Hz, 1C), 36.6, 30.1, 24.8, 24.7, 24.1, 17.2, 17.0; 19F NMR (282 MHz, CDCl3) δ −73.86 (d, JHF = 7.7 Hz 3F); IR (film) νmax 2930, 1769, 1254, 1137, 1083, 988 cm−1; HRMS (ESI) m/z calcd for C21H30F3NO3 (M+H)+ 402.2256, found 402.2260; [α]24D −2.6° (c 0.26, CHCl3); Anal. Calcd for C21H30F3NO3: C, 62.83; H, 7.53; N, 3.49. Found: C, 62.86; H, 7.45; N, 3.49.
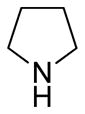
(11R)-13-((R)-(−)-3-Fluoropyrrolidine)-11,13-dihydroparthenolide (10)
83% yield. mp = 114–122 °C; 1H NMR (300 MHz, CDCl3) δ 5.23 (m, 1H), 5.18–5.04 (m, 1H), 3.83 (t, J = 8.7 Hz, 1H), 3.03–2.67 (m, 6H), 2.52–2.03 (m, 11H), 1.69 (s, 3H), 1.62 (m, 1H), 1.30 (s, 3H), 1.25 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 176.3, 134.7, 125.0, 93.2 (d, JCF = 175 Hz, 1C), 82.2, 66.5, 61.5, 61.3 (d, JCF = 22.5 Hz, 1C), 53.2, 53.1, 47.5, 47.4, 41.1, 36.6, 32.9 (d, JCF = 21.9 Hz, 1C), 30.0, 24.1, 17.2, 16.9; 19F NMR (282 MHz, CDCl3) δ −168.59 (dqu, JHF = 56.4, 28.8 Hz, 1F); IR (film) νmax 2923, 1770, 1252, 1153, 1072, 989 cm−1; HRMS (EI) m/z calcd for C19H28FNO3 (M+H)+ 338.2131, found 338.2132; [α]25D −9.8° (c 0.41, CHCl3); Anal. Calcd for C19H28FNO3·1.0MeOH: C, 65.02; H, 8.73; N, 3.79. Found: C, 67.06; H, 8.35; N, 3.60.
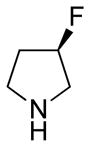
(11R)-13-((S)-(+)-3-Fluoropyrrolidine)-11,13-dihydroparthenolide (11)
86% yield. mp = 116–118 °C; 1H NMR (300 MHz, CDCl3) δ 5.22 (m, 1H), 5.18–5.05 (m, 1H), 3.83 (t, J = 8.7 Hz, 1H), 2.99–2.72 (m, 6H), 2.53–1.99 (m, 11H), 1.69 (s, 3H), 1.63 (m, 1H), 1.29 (s, 3H), 1.25 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 176.3, 134.6, 125.0, 93.5 (d, JCF = 175 Hz, 1C), 82.2, 66.5, 61.5, 61.3 (d, JCF = 22.6 Hz, 1C), 53.1, 52.9, 47.3, 47.2, 41.1, 36.6, 32.7 (d, JCF = 22.0 Hz, 1C), 30.0, 24.1, 17.2, 16.9; 19F NMR (282 MHz, CDCl3) δ −168.80 (dqu, JHF = 55.8, 28.8 Hz, 1F); IR (film) νmax 2919, 1771, 1252, 1176, 1073, 988 cm−1; HRMS (EI) m/z calcd for C19H28FNO3 (M+H)+ 338.2131, found 338.2133; [α]27D −19.6° (c 0.48, CHCl3); Anal. Calcd for C19H28FNO3: C, 67.63; H, 8.36; N, 4.15. Found: C, 67.45; H, 8.50; N, 3.98.
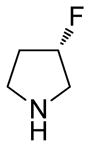
(11R)-13-(3-3-Difluoropyrrolidine)-11,13-dihydroparthenolide (12)
yield 78%. mp = 154–156 °C; 1H NMR (300 MHz, CDCl3) δ 5.20 (d, J = 11.6 Hz, 1H), 3.84 (t, J = 8.9 Hz, 1H), 3.01–2.88 (m, 4H), 2.81 (td, J = 7.0, 2.6 Hz, 2H), 2.73 (d, J = 8.9 Hz, 1H), 2.41–1.96 (m, 10H), 1.70 (s, 3H), 1.62 (m, 1H), 1.30 (s, 3H), 1.24 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 176.0, 134.5, 129.6 (t, JCF = 247 Hz, 1C), 125.2, 82.3, 66.4, 62.4 (t, JCF = 29 Hz, 1C), 61.5, 52.9, 52.8, 47.3, 47.0, 41.1, 36.6, 35.8 (t, JCF = 24 Hz, 1C), 29.9, 24.1, 17.2, 16.9; 19F NMR (282 MHz, CDCl3) δ −92.58 (dqu, JFF = 229 Hz, JHF = 14.4 Hz, 1F), −93.50 (dqu, JFF = 229 Hz, JHF = 14.4 Hz, 1F); IR (film) νmax 2927, 1772, 1264, 1109, 1073, 990 cm−1; HRMS (EI) m/z calcd for C19H27F2NO3 (M+H)+ 356.2037, found 356.2033; [α]24D −9.9° (c 0.86, CHCl3); Anal. Calcd for C19H27F2NO3·0.5H2O: C, 62.62; H, 7.74; N, 3.84. Found: C, 63.55; H, 7.56; N, 3.67.
Cell Culture.34
HL-60 (human promyelocytic leukemia) cells were obtained from ATCC. Cell culture was maintained in phenol red free RPMI-1640 supplemented with 10% fetal bovine serum and filter sterilized (hereafter referred to as “medium”). Cells were cultured at 37 °C in a humidified incubator under 5% CO2 in air.
Cell Proliferation Assay.34
The GI50, TGI, and LC50 values for HL-60 cell proliferation in the presence of parthenolide (1) and its amino-derivatives 2–12 were determined by MTT assay. On Day 0, HL-60 cells were seeded in 96-well plates at 5,000 cells per well in 100 μL medium (Day 0 plate) and 80 μL medium (Day 3 plate). “Day 0” plates contained alternating columns of wells with medium-only and medium + cells for the purpose of providing Day 0 cell density measurements and requisite background Ab 570 nm controls during plate reading. “Day 3” plates contained one column of medium-only wells as background light absorbance controls (wells A1–H1), one column of cells to receive vehicle control (0.5% DMSO in medium, wells A2–H2), and 10 columns of cells to receive dilution series for two compounds (compound 1 in wells E–H of each column and compound 2 in wells A–D of each column). Immediately after assembly, treatment wells of the Day 3 plate received 20 μL of a solution of test compound dissolved in medium + DMSO for a final concentration of 0.5% DMSO in growth medium. Each compound was tested at ten concentrations in quadruplicate. The cells in Day 3 plate column 2 received the vehicle control at this time. Immediately after assembly, the Day 0 plate was processed with an MTT protocol: 20 μL MTT solution (2.5 mg/mL) in serum-free RPMI-1640 was added to each well. The plate was then incubated for 4 h, after which each well received 100 μL acidic isopropanol (10% Triton X-100 plus 0.1 N HCl in anhydrous isopropanol). Formazan crystals were allowed to solubilize for 16 h. After complete solubilization of formazan crystals, Ab 570 nm was measured using a Spectramax 384 Plus plate reader (Molecular Devices). An identical MTT assay was performed with each Day 3 plate after 72 h incubation post-assembly. All plates were incubated at 37 °C in a humidified incubator under 5% CO2 in air. GI50, TGI, and LC50 values for HL-60 cell proliferation were calculated by GraphPad Prism 5 software. A full description of these calculations is provided in the Supporting Information.43
19F NMR and Quantitative Studies
A solution of glutathione (69 mg, 0.22 mmol) in water (4.5 mL) and DMSO (10.5 mL) was titrated to pH 7.4 (Fisher Scientific, Accumet Basic) using 1.0 M NaOH (220 μL). Next, glutathione solution (2.7 mL, pH 7.4) was treated with a 10 mM solution of compound 8 in DMSO (300 μL), and an aliquot of the resultant solution was immediately transferred to an NMR tube for 19F NMR analysis. The remaining solution of compound 8 and glutathione was heated to 80 °C in an oil bath, and additional aliquots were obtained at 24, 48, and 72 h for 19F NMR analysis. Side-by-side control experiments were performed without any glutathione using the same procedures and reaction conditions, and aliquots were also taken for 19F NMR analysis. The 1D 19F NMR spectra were acquired without lock on a Bruker DPX 300 MHz equipped with a QNP probe operating at rt. The sweep width was 253 ppm, acquisition time was 458 ms and the relaxation delay between scans was 1 s. For each fluorine spectrum, 128 scans were accumulated after 8 dummy scans. All FID were line-broadened by 3 Hz, Fourier transformed, manually phased and baseline corrected. Fluorine chemical shift was indirectly referenced by solvent (DMSO) proton at 2.5 ppm.52 Signals representing remaining compound 8 and amine were integrated, and the total compound amount at time 0 (mainly compound 8) was normalized to 1.28,53
Supplementary Material
Acknowledgments
We are grateful to Purdue University and a grant from the Indiana Elk Charities through the Purdue University Center for Cancer Research for financial support. We acknowledge the CCCB Cell Culture/Flow Cytometry Center, Purdue University as well as the Molecular Discovery and Evaluation Shared Resource, Purdue University Center for Cancer Research. Cancer cell growth inhibition assay development was performed with support from NCI T32CA09634-18 to Purdue University Center for Cancer Research, and the respective biological data were collected in the Purdue University Center for Cancer Research Molecular Discovery and Evaluation Shared Resource supported by NCI CCSG CA23168 to Purdue University Center for Cancer Research.
Abbreviations
- DMAPT
Dimethylaminoparthenolide
- HL-60
Human promyelocytic leukemia cells
- NCI60
National Cancer Institute 60 human cancer cell lines
- log P
Partition coefficient
Footnotes
Supporting Information Available: 1H and 13C NMR spectra and X-ray data. Also, a full description of the methods for the calculation of GI50, TGI, and LC50 values. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Kitson RRA, Millemaggi A, Taylor RJK. The Renaissance of α-Methylene-γ-butyrolactones: New Synthetic Approaches. Angew Chem Int Ed. 2009;48:9426–9451. doi: 10.1002/anie.200903108. [DOI] [PubMed] [Google Scholar]
- 2.Avonto C, Taglialatela-Scafati O, Pollastro F, Minassi A, Di Marzo V, De Petrocellis L, Appendino G. An NMR Spectroscopic Method to Identify and Classify Thiol-trapping Agents: Revival of Michael Acceptors for Drug Discovery? Angew Chem Int Ed. 2011;50:467–471. doi: 10.1002/anie.201005959. [DOI] [PubMed] [Google Scholar]
- 3.Lawrence NJ, McGown AT, Nduka J, Hadfield JA, Pritchard RG. Cytotoxic Michael-type Amine Adducts of α-Methylene Lactones Alantolactone and Isoalantolactone. Bioorg Med Chem Lett. 2001;11:429–431. doi: 10.1016/s0960-894x(00)00686-7. [DOI] [PubMed] [Google Scholar]
- 4.Srivastava SK, Abraham A, Bhat B, Jaggi M, Singh AT, Sanna VK, Singh G, Agarwal SK, Mukherjee R, Burman AC. Synthesis of 13-Amino Costunolide Derivatives as Anticancer Agents. Bioorg Med Chem Lett. 2006;16:4195–4199. doi: 10.1016/j.bmcl.2006.05.083. [DOI] [PubMed] [Google Scholar]
- 5.Neelakantan S, Nasim S, Guzman ML, Jordan CT, Crooks PA. Aminoparthenolides as Novel Anti-leukemic Agents: Discovery of the NF-κB Inhibitor, DMAPT (LC-1) Bioorg Med Chem Lett. 2009;19:4346–4349. doi: 10.1016/j.bmcl.2009.05.092. [DOI] [PubMed] [Google Scholar]
- 6.Nasim S, Crooks PA. Antileukemic Activity of Aminoparthenolide Analogs. Bioorg Med Chem Lett. 2008;18:3870–3873. doi: 10.1016/j.bmcl.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 7.Hwang DR, Wu YS, Chang CW, Lien TW, Chen WC, Tan UK, Hsu JTA, Hsieh HP. Synthesis and Anti-viral Activity of a Series of Sesquiterpene Lactones and Analogues in the Subgenomic HCV Replicon System. Bioorg Med Chem. 2006;14:83–91. doi: 10.1016/j.bmc.2005.07.055. [DOI] [PubMed] [Google Scholar]
- 8.Klochkov SG, Afanas’eva SV, Pushin AN, Gerasimova GK, Vlasenkova NK, Bulychev YN. Synthesis and Cytotoxic Activity of α-Santonin Amino-derivatives. Chem Nat Compd. 2009;46:817–823. [Google Scholar]
- 9.Lee KH, Furukawa H, Huang ES. Antitumor Agents. 3. Synthesis and Cytotoxic Activity of Helenalin Amine Adducts and Related Derivatives. J Med Chem. 1972;15:609–611. doi: 10.1021/jm00276a010. [DOI] [PubMed] [Google Scholar]
- 10.Hejchman E, Haugwitz RD, Cushman M. Synthesis and Cytotoxicity of Water-soluble Ambrosin Prodrug Candidates. J Med Chem. 1995;38:3407–3410. doi: 10.1021/jm00017a025. [DOI] [PubMed] [Google Scholar]
- 11.Hassane DC, Sen S, Minhajuddin M, Rossi RM, Corbett CA, Balys M, Wei L, Crooks PA, Guzman ML, Jordan CT. Chemical Genomic Screening Reveals Synergism between Parthenolide and Inhibitors of the PI-3 Kinase and mTOR Pathways. Blood. 2010;116:5983–5990. doi: 10.1182/blood-2010-04-278044. [DOI] [PubMed] [Google Scholar]
- 12.Guzman ML, Rossi RM, Neelakantan S, Li X, Corbett CA, Hassane DC, Becker MW, Bennett JM, Sullivan E, Lachowicz JL, Vaughan A, Sweeney CJ, Matthews W, Carroll M, Liesveld JL, Crooks PA, Jordan CT. An Orally Bioavailable Parthenolide Analog Selectively Eradicates Acute Myelogenous Leukemia Stem and Progenitor Cells. Blood. 2007;110:4427–4435. doi: 10.1182/blood-2007-05-090621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghantous A, Gali-Muhtasib H, Vuorela H, Saliba NA, Darwiche N. What Made Sesquiterpene Lactones Reach Cancer Clinical Trials? Drug Discov Today. 2010;15:668–678. doi: 10.1016/j.drudis.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 14.Matsuda H, Toguchida I, Ninomiya K, Kageura T, Morikawa T, Yoshikawa M. Effects of Sesquiterpenes and Amino Acid-sesquiterpene Conjugates from the Roots of Saussurea lappa on Inducible Nitric Oxide Synthase and Heat Shock Protein in Lipopolysaccharide-activated Macrophages. Bioorg Med Chem. 2003;11:709–715. doi: 10.1016/s0968-0896(02)00471-6. [DOI] [PubMed] [Google Scholar]
- 15.Matsuda H, Kageura T, Inoue Y, Morikawa T, Yoshikawa M. Absolute Stereostructures and Syntheses of Saussureamines A, B, C, D and E, Amino Acid-Sesquiterpene Conjugates with Gastroprotective Effect, from the Roots of Saussurea lappa. Tetrahedron. 2000;56:7763–7777. [Google Scholar]
- 16.Hagmann WK. The Many Roles for Fluorine in Medicinal Chemistry. J Med Chem. 2008;51:4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- 17.Eustaquio AS, O’Hagan D, Moore BS. Engineering Fluorometabolite Production: Fluorinase Expression in Salinispora tropica Yields Fluorosalinosporamide. J Nat Prod. 2010;73:378–382. doi: 10.1021/np900719u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang P, Furuya T, Ritter T. Silver-Catalyzed Late-Stage Fluorination. J Am Chem Soc. 2010;132:12150–12154. doi: 10.1021/ja105834t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Erb J, Alden-Danforth E, Kopf N, Scerba MT, Lectka T. Combining Asymmetric Catalysis with Natural Product Functionalization through Enantioselective α-Fluorination. J Org Chem. 2010;75:969–971. doi: 10.1021/jo9024072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stockman BJ. 2-Fluoro-ATP as a Versatile Tool for 19F NMR-Based Activity Screening. J Am Chem Soc. 2008;130:5870–5871. doi: 10.1021/ja801588u. [DOI] [PubMed] [Google Scholar]
- 21.Keun HC, Athersuch TJ, Beckonert O, Wang Y, Saric J, Shockcor JP, Lindon JC, Wilson ID, Holmes E, Nicholson JK. Heteronuclear 19F–1H Statistical Total Correlation Spectroscopy as a Tool in Drug Metabolism: Study of Flucloxacillin Biotransformation. Anal Chem. 2008;80:1073–1079. doi: 10.1021/ac702040d. [DOI] [PubMed] [Google Scholar]
- 22.Papeo G, Giordano P, Brasca MG, Buzzo F, Caronni D, Ciprandi F, Mongelli N, Veronesi M, Vulpetti A, Dalvit C. Polyfluorinated Amino Acids for Sensitive 19F NMR-Based Screening and Kinetic Measurements. J Am Chem Soc. 2007;129:5665–5672. doi: 10.1021/ja069128s. [DOI] [PubMed] [Google Scholar]
- 23.Yu J, Mason RP. Synthesis and Characterization of Novel lacZ Gene Reporter Molecules: Detection of α-Galactosidase Activity by 19F Nuclear Magnetic Resonance of Polyglycosylated Fluorinated Vitamin B6. J Med Chem. 2006;49:1991–1999. doi: 10.1021/jm051049o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu L, Hajduk P, Mack J, Olejniczak E. Structural Studies of Bcl-xL/ligand Complexes using 19F NMR. J Biomol NMR. 2006;34:221–227. doi: 10.1007/s10858-006-0005-y. [DOI] [PubMed] [Google Scholar]
- 25.Yu J, Liu L, Kodibagkar VD, Cui W, Mason RP. Synthesis and Evaluation of Novel Enhanced Gene Reporter Molecules: Detection of β-Galactosidase Activity using 19F NMR of Trifluoromethylated aryl β-D-Galactopyranosides. Bioorg Med Chem. 2006;14:326–333. doi: 10.1016/j.bmc.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 26.Dalvit C, Ardini E, Flocco M, Fogliatto GP, Mongelli N, Veronesi M. A General NMR Method for Rapid, Efficient, and Reliable Biochemical Screening. J Am Chem Soc. 2003;125:14620–14625. doi: 10.1021/ja038128e. [DOI] [PubMed] [Google Scholar]
- 27.Dalvit C, Fagerness PE, Hadden DTA, Sarver RW, Stockman BJ. Fluorine-NMR Experiments for High-Throughput Screening: Theoretical Aspects, Practical Considerations, and Range of Applicability. J Am Chem Soc. 2003;125:7696–7703. doi: 10.1021/ja034646d. [DOI] [PubMed] [Google Scholar]
- 28.Mo H, Balko KM, Colby DA. A Practical Deuterium-free NMR Method for the Rapid Determination of 1-Octanol/Water Partition Coefficients of Pharmaceutical Agents. Bioorg Med Chem Lett. 2010;20:6712–6715. doi: 10.1016/j.bmcl.2010.08.145. [DOI] [PubMed] [Google Scholar]
- 29.Guzman ML, Rossi RM, Karnischky L, Li X, Peterson DR, Howard DS, Jordan CT. The Sesquiterpene Lactone Parthenolide Induces Apoptosis of Human Acute Myelogenous Leukemia Stem and Progenitor Cells. Blood. 2005;105:4163–4169. doi: 10.1182/blood-2004-10-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han C, Barrios FJ, Riofski MV, Colby DA. Semisynthetic Derivatives of Sesquiterpene Lactones by Palladium-Catalyzed Arylation of the α-Methylene-γ-lactone Substructure. J Org Chem. 2009;74:7176–7179. doi: 10.1021/jo901533e. [DOI] [PubMed] [Google Scholar]
- 31.Gunn EJ, Williams JT, Huynh DT, Iannotti MJ, Han C, Barrios FJ, Kendall S, Glackins CA, Colby DA, Kirshner J. The Natural Products Parthenolide and Andrographolide Exhibit Anticancer Stem Cell Activity in Multiple Myeloma. Leukemia Lymphoma. 2011;52:1085–1097. doi: 10.3109/10428194.2011.555891. [DOI] [PubMed] [Google Scholar]
- 32.Kerekes AD, Esposite SJ, Doll RJ, Tagat JR, Yu T, Xiao Y, Zhang Y, Prelusky DB, Tevar S, Gray K, Terracina GA, Lee S, Jones J, Liu M, Basso AD, Smith EB. Aurora Kinase Inhibitors Based on the Imidazo[1,2-a]pyrazine Core: Fluorine and Deuterium Incorporation Improve Oral Absorption and Exposure. J Med Chem. 2011;54:201–210. doi: 10.1021/jm1010995. [DOI] [PubMed] [Google Scholar]
- 33.Neelakantan S, Parkin S, Crooks PA. (11R)-13-Dimethylammonio-11, 13-Dihydro-4, 5-Epoxycostunolide Semifumarate. Acta Crystallogr Section E. 2009;65E:o1565. doi: 10.1107/S1600536809021941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakagawa Y, Iinuma M, Matsuura N, Yi K, Naoi M, Nakayama T, Nozawa Y, Akao Y. A Potent Apoptosis-inducing Activity of a Sesquiterpene Lactone, Arucanolide, in HL60 Cells: A Crucial Role of Apoptosis-inducing Factor. J Pharmacol Sci. 2005;97:242–252. doi: 10.1254/jphs.fp0040456. [DOI] [PubMed] [Google Scholar]
- 35.Shoemaker RH. The NCI60 Human Tumour Cell Line Anticancer Drug Screen. Nature Rev Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 36.Park ES, Rabinovsky R, Carey M, Hennessy BT, Agarwal R, Liu WB, Ju ZL, Deng WL, Lu YL, Woo HG, Kim SB, Cheong JH, Garraway LA, Weinstein JN, Mills GB, Lee JS, Davies MA. Integrative Analysis of Proteomic Signatures, Mutations, and Drug Responsiveness in the NCI 60 Cancer Cell Line Set. Mol Cancer Ther. 2010;9:257–267. doi: 10.1158/1535-7163.MCT-09-0743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P, Iyer V, Jeffrey SS, Van de Rijn M, Waltham M, Pergamenschikov A, Lee JCE, Lashkari D, Shalon D, Myers TG, Weinstein JN, Botstein D, Brown PO. Systematic Variation in Gene Expression Patterns in Human Cancer Cell Lines. Nature Genet. 2000;24:227–235. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]
- 38.Scherf U, Ross DT, Waltham M, Smith LH, Lee JK, Tanabe L, Kohn KW, Reinhold WC, Myers TG, Andrews DT, Scudiero DA, Eisen MB, Sausville EA, Pommier Y, Botstein D, Brown PO, Weinstein JN. A Gene Expression Database for the Molecular Pharmacology of Cancer. Nature Genet. 2000;24:236–244. doi: 10.1038/73439. [DOI] [PubMed] [Google Scholar]
- 39.Staunton JE, Slonim DK, Coller HA, Tamayo P, Angelo MJ, Park J, Scherf U, Lee JK, Reinhold WO, Weinstein JN, Mesirov JP, Lander ES, Golub TR. Chemosensitivity Prediction by Transcriptional Profiling. Proc Natl Acad Sci U S A. 2001;98:10787–10792. doi: 10.1073/pnas.191368598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Su G, Burant CF, Beecher CW, Athey BD, Meng F. Integrated Metabolome and Transcriptome Analysis of the NCI60 Dataset. BMC Bioinformatics. 2011;12(Suppl 1):S36. doi: 10.1186/1471-2105-12-S1-S36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baharith LA, Al-Khouli A, Raab GA. Cytotoxic Assays for Screening Anticancer Agents. Stat Med. 2006;25:2323–2339. doi: 10.1002/sim.2272. [DOI] [PubMed] [Google Scholar]
- 42.GraphPad Prism, 5.04. GraphPad Software; San Diego, California, U.S.A: 2010. [Google Scholar]
- 43.See Supporting Information for details.
- 44.Morgenthaler M, Schweizer E, Hoffmann-Röder A, Benini F, Martin RE, Jaeschke G, Wagner B, Fischer H, Bendels S, Zimmerli D, Schneider J, Diederich F, Kansy M, Müller K. Predicting and Tuning Physicochemical Properties in Lead Optimization: Amine Basicities. ChemMedChem. 2007;2:1100–1115. doi: 10.1002/cmdc.200700059. [DOI] [PubMed] [Google Scholar]
- 45.Meng Q, Peng Z, Chen L, Si J, Dong Z, Xia Y. Nuclear Factor-κB Modulates Cellular Glutathione and Prevents Oxidative Stress in Cancer Cells. Cancer Lett. 2010;299:45–53. doi: 10.1016/j.canlet.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Balendiran GK, Dabur R, Fraser D. The Role of Glutathione in Cancer. Cell Biochem Funct. 2004;22:343–352. doi: 10.1002/cbf.1149. [DOI] [PubMed] [Google Scholar]
- 47.Lajiness JP, Robertson WM, Dunwiddie I, Broward MA, Vielhauer GA, Weir SJ, Boger DL. Design, Synthesis, and Evaluation of Duocarmycin O-Amino Phenol Prodrugs Subject to Tunable Reductive Activation. J Med Chem. 2010;53:7731–7738. doi: 10.1021/jm1010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pires MM, Chmielewski J. Fluorescence Imaging of Cellular Glutathione Using a Latent Rhodamine. Org Lett. 2008;10:837–840. doi: 10.1021/ol702769n. [DOI] [PubMed] [Google Scholar]
- 49.Hong V, Kislukhin AA, Finn MG. Thiol-Selective Fluorogenic Probes for Labeling and Release. J Am Chem Soc. 2009;131:9986–9994. doi: 10.1021/ja809345d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim GJ, Lee K, Kwon H, Kim HJ. Ratiometric Fluorescence Imaging of Cellular Glutathione. Org Lett. 2011;13:2799–2801. doi: 10.1021/ol200967w. [DOI] [PubMed] [Google Scholar]
- 51.Potapenko DI, Bagryanskaya EG, Grigoriev IA, Maksimov AM, Reznikov VA, Platonov VE, Clanton TL, Khramtsov VV. Quantitative Determination of SH Groups using 19F NMR Spectroscopy and Disulfide of 2,3,5,6-Tetrafluoro-4-mercaptobenzoic acid. Magn Reson Chem. 2005;43:902–909. doi: 10.1002/mrc.1652. [DOI] [PubMed] [Google Scholar]
- 52.Harris RK, Becker ED, Cabral de Menezes SM, Goodfellow R, Granger P. NMR Nomenclature. Nuclear Spin Properties and Conventions for Chemical Shifts. Pure Appl Chem. 2001;73:1795–1818. doi: 10.1006/snmr.2002.0063. [DOI] [PubMed] [Google Scholar]
- 53.Mo H, Raftery D. Solvent Signal as an NMR Concentration Reference. Anal Chem. 2008;80:9835–9839. doi: 10.1021/ac801938j. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.