

Abstract
The injured mammalian heart is particularly susceptible to tissue deterioration, scarring, and loss of contractile function in response to trauma or sustained disease. We tested the ability of a locally acting insulin-like growth factor-1 isoform (mIGF-1) to recover heart functionality, expressing the transgene in the mouse myocardium to exclude endocrine effects on other tissues. supplemental mIGF-1 expression did not perturb normal cardiac growth and physiology. Restoration of cardiac function in post-infarct mIGF-1 transgenic mice was facilitated by modulation of the inflammatory response and increased antiapoptotic signaling. mIGF-1 ventricular tissue exhibited increased proliferative activity several weeks after injury. The canonical signaling pathway involving Akt, mTOR, and p70S6 kinase was not induced in mIGF-1 hearts, which instead activated alternate PDK1 and SGK1 signaling intermediates. The robust response achieved with the mIGF-1 isoform provides a mechanistic basis for clinically feasible therapeutic strategies for improving the outcome of heart disease.
Keywords: cardiac muscle, insulin-like growth factor-1, regeneration, wound healing
The insulin/insulin-like growth factor signaling pathway arose early in the evolution and is highly conserved among invertebrates and vertebrates.1 Mammalian IGF-1 acts predominately as a growth, survival, and differentiation factor. The pleiotropic functions of IGF-1 are reflected in the complicated structure and regulation of Igf-1 gene.2 The products include variable amino-terminal signal peptides and different carboxy-terminal E peptides, the precise function of which is still unclear. Injury of mammalian tissues induces transient production of locally acting IGF-1 isoforms that control growth, survival, and differentiation.3 By contrast, high levels of circulating IGF-1, produced by the liver, has been implicated in the restriction of lifespan1 and predisposition to neoplasia.4
When expressed as transgenes, different IGF-1 isoforms have contrasting effects on the mouse heart. Transgenic mice generated with a minor human IGF-1 cDNA (IGF-1Eb) under the control of the rat α-myosin heavy chain (α-MHC) promoter showed no striking differences in size and cell volume when compared with control mice, but harbored an increased number of cardiomyocytes, coupling IGF-1 over-expression with myocyte proliferation.5 The hearts of these animals responded to coronary ligation with attenuated diastolic wall stress, cardiac weight, ventricular dilatation, and hypertrophy, attributable mainly to a prevention of cardiac cell death.6 In another report, cardiac expression of a modified human IGF-1 cDNA produced no hyperplasia but instead induced physiologic, then pathologic, cardiac hypertrophy in transgenic mice.7
Here, we have used the mIGF-1 isoform, comprising a Class 1 signal peptide and a C-terminal Ea extension peptide.8 This isoform is expressed at high levels in neonatal tissues and in the adult liver, but decreases during aging in extrahepatic tissues, where its expression is activated transiently in response to local damage.9 The regenerative properties of the mIGF-1 isoform and its dramatic promotion of cell survival and renewal in senescent muscle have been extensively documented,10 making it an attractive candidate for possible enhancement of cell-based regenerative therapies in injured postmitotic organs. We tested the potential of the mIGF-1 isoform to repair the injured heart and analyzed the signaling pathways induced by transgenic mIGF-1 under physiological and pathological conditions. By challenging the hearts of mice carrying a cardiac-restricted mIGF-1 transgene with 2 different modes of injury, we show that this isoform restores cardiac function by blocking scar formation and enabling myocardial reconstruction.
Materials and Methods
An expanded Materials and Methods section can be found in the online supplement, available at http://circres.ahajournals.org.
Generation of α-MHC/mIGF-1 Transgenic Mice
Transgenic FVB mice carrying a rat mIGF-1 cDNA driven by the mouse α-MyHC promoter were generated by standard methods and selected by positive PCR analysis of tail DNA. αMyHC/mIGF-1 transgenic mice were maintained as heterozygotes. All animals were housed in a temperature-controlled (22°C) room with a 12:12 hour light-dark cycle. All analyses were performed on male mice.
Isolation of Cardiac Cells
Adult mouse cardiomyocytes were isolated and cultured following the instructions of www.signaling-gateway.org.
Statistics
All comparisons between WT and TG mice were performed by means of paired Student t tests. A significant difference was considered when P<0.05, set as a double side value.
Results
The mIGF-1 Isoform Accelerates Cardiac Growth
We generated transgenic mice with a rat mIGF-1 cDNA driven by the mouse α-myosin heavy chain (α-MHC) promoter to restrict expression of mIGF-1 to the mouse myocardium and exclude possible endocrine effects on other tissues (Figure 1A). Cardiac-restricted mIGF-1 expression levels increased with age in all founders (F010, F022, and F018) tested, and reached a steady level at 1 month (Figure 1B). Expression of the α-MHC/mIGF-1 transgene in adult mice was restricted to the heart (Figure 1C) and was undetectable in other tissues using the rat IGF-1 probe. A single transgenic line was selected for further analysis (F018).
Figure 1.

Characterization of αMyHC/mIGF-1 transgenic mice. A, Schematic representation of the rodent Igf-1 gene. B and C, Northern blot analysis of total RNA from wild-type (WT) and transgenic (mIGF-1) hearts at different ages (B) and different tissues (C) using a rat Igf-1 32P labeled probe. Ethidium bromide (EtBr) was used to verify equal RNA loading. D, Histological analysis of WT and mIGF-1 transgenic (TG) hearts by Hematoxylin and Eosin staining. Lower panel shows adult heart weight/body weight (P<0.05). Values are the average of 6 independent analyses. E, Cell size differences in WT and TG hearts. F, RT-PCR analysis of the hypertrophic marker ANP in adult hearts. PCR values were normalized for β-actin content. Densitometric analysis was performed on 3 independent experiments. Asterisks indicate significant relative values (P<0.05).
Postnatal 2-month mIGF-1 transgenic hearts displayed accelerated cardiomyocyte hypertrophy, precociously attaining wild-type adult heart size (Figure 1D), attributable to a significant increase in cell size compared with wild-type hearts (Figure 1E and supplemental Figure I). Importantly, analysis of cardiomyocytes cross-sectional area (CSA) showed that cells overexpressing mIGF-1 had a comparable size at 2 and 4 months (supplemental Figure I), indicating that heart growth was no longer induced by transgene overexpression. Cardiac hypertrophy was related to higher expression levels of ANP at 1 and 2 months, without any further significant change (Figure 1F). Other markers underlining cardiac hypertrophy, such as BNP, α-skeletal actin, β-myosin heavy chain, and glutamate transporter 1, were not affected (data not shown). Assessment of cardiac function by echocardiography at 4 months showed that mIGF-1 induced a 20% concentric left ventricular hypertrophy (supplemental Table I). In transgenic male mice, echocardiography identified a small but significant decrease in cardiac contractility (13% decrease in ejection fraction [EF] and fractional shortening [FS]). Mildly compromised diastolic function was identified by the 21% decrease of the E/A ratio and the prolongation of the A wave duration (+14%). Nevertheless, cardiac output and chamber diameters remained at normal levels throughout postnatal stages (supplemental Table I).
The mIGF-1 Transgene Promotes Effective Myocardial Repair and Functional Restoration
The restorative capacity of mIGF-1 transgenic hearts was analyzed by ligation of the left coronary artery (LCA) of 4-month-old mice. In wild-type LCA induced infarcts characterized by progressive and extended fibrotic tissue formation (Figure 2A, upper panel), accompanied by functional impairment after 1 month that worsened after 2 months (Figure 2B and supplemental Figure IIA). Percent values of FS and EF were significantly decreased compared with sham operated mice (supplemental Table II and Figure IIB). In contrast, infarcted mIGF-1 transgenic hearts showed a moderate but significant decrease in the percentage of EF and FS after 1 month, with no significant changes after 2 months compared with mIGF-1 transgenic sham-operated mice and wild-type ligated mice (Figure 2B, supplemental Figure IIA and Table II). The mIGF-1–mediated blockade of the normal progressive impairment in infarcted heart function was accompanied by reduced scar formation (Figure 2A, lower panel), and a significantly smaller infarct size compared with control hearts (supplemental Figure IIC, lower panel). Recovery of cardiac function as well as morphological restoration of infarcted mIGF-1 transgenic hearts was confirmed by normal left ventricular motion in systolic and diastolic phases (supplemental Movie II) compared with mIGF-1 transgenic uninjured hearts (supplemental Movie I). In contrast, wild-type hearts (supplemental Movie IV) presented chamber enlargement and a significant decrease in wall motility near the infarct, when compared with uninjured wild-type hearts (supplemental Movie III).
Figure 2.

Enhanced cardiac repair and functions in mIGF-1 transgenic mice after myocardial infarction. A, Whole mount and histological analysis of sham-operated control (WT) heart (left) and LCA WT and TG hearts (right) 2 months after operation. Arrows indicate fibrotic tissue. LA indicates left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle. Histological analysis by trichrome staining is shown for each treatment. B, Functional recovery of mIGF-1 transgenic mice after LCA. Mean percentage values of FS (upper panel) and EF (lower panel) are representative of 3 readings on each animal and averaged among groups. Asterisks (*) show significant values (P<0.05) between uninjured and injured hearts in WT (yellow square) and TG (red square) mice. § shows significant values between WT and TG injured hearts. C, Histological analysis (trichrome) of WT and TG hearts at 48 hours, 1 week, and 1 month after CTX injection in the left ventricular wall. Comparable results were obtained with similar analyses on 6 different groups. D, Functional recovery of mIGF-1 transgenic mice 1 month after CTX injection. Mean percentage values are representative of 3 readings on each animal and averaged among groups. Asterisk (*) indicates significant values decreasing in WT compared with TG hearts (P<0.05). E, Real time PCR of mIGF-1 transcript in physiological conditions and 24 hours after CTX injection, using IGF-1Ea Taqman probe (Applied Biosystem). PCR values were normalized for GAPDH content in each sample. Asterisk (*) indicates significant increasing values compared with WT uninjured hearts, whereas § represents significant decreasing values compared with TG uninjured hearts.
An alternate injury method involving direct cardiotoxin (CTX) injection into the cardiac left ventricle wall produced similar results to those obtained with LCA, although the CTX model produces a well-delineated transmural lesion and reduces the risk of ventricular fibrillation.11 This technique was used in subsequent experiments. Single CTX injections in 4-month wild-type and mIGF-1 transgenic hearts induced reproducible, small, and localized damage with evident cell death and marked inflammation after 48 hours and 1 week (Figure 2C). To test whether CTX injection reduced the variability of the extent of myocardial injury normally inherent to LCA models, we injected mice with Evans Blue dye (EBD), as vital stain of myocytes permeability and membrane-associated fragility12 (supplemental Figure IIIA). We found an equivalent increase in myocardial damage in wild-type and mIGF-1 hearts, indicating that the extent of initial injury is similar in both animals tested (supplemental Figure IIIB). In contrast to the characteristic progression of scar formation in wild-type hearts after 1 month (Figure 2C), mIGF-1 transgene expression induced repair of the injured tissue with minimal scar formation (Figure 2C), and a significant reduction of infarct size (supplemental Figure IIC, upper panel).
Although the overall cardiac function of wild-type and transgenic injured hearts was not dramatically affected by a single shot of CTX injection, analysis of mean values of EF and FS by high-resolution echocardiography were moderately but significantly impaired in wild-type hearts when compared with transgenic hearts (EF 61%±7% compared with 78%±3%; FS 34%±4% compared with 47%±3%; Figure 2D and supplemental Figure IIB), indicating that mIGF-1 induced both morphological and functional repair.
Variations in the level of IGF-1 transcript induced by CTX injection in both wild-type and transgenic hearts were evaluated by real time PCR with a Taqman probe recognizing both endogenous mouse IGF-1Ea and the rat transgene (Figure 2E). mIGF-1 is 35-fold higher in transgenic hearts compared with wild-type hearts. After CTX injection (24 hours), the level of the transgene significantly decreased (30%), likely attributable to CTX-induced downregulation of the mouse αMHC promoter (Figure 2E). Interestingly, this IGF-1 isoform increased significantly in wild-type hearts after CTX injection (19%), as reported in previous analysis.9
mIGF-1 Mediates Heart Repair by Modulation of the Inflammatory Response
The early events characterizing postmyocardial infarction include complement activation, free radical generation, che-mokine upregulation, and activation of cytokine cascades.13 Real time PCR analysis showed that at 24 hours after CTX injection, proinflammatory IL6 and IL1β transcript levels moderately increased or remained unchanged, respectively, in transgenic hearts (Figure 3A). Contrarily, wild-type hearts showed a significant increase of both interleukins (Figure 3A). Other cytokines involved in the inflammatory response, such as IL12A and B, IFNγ, TGFβ, and MCP1 were not affected in wild-type and mIGF-1 transgenic hearts by CTX-induced injury (supplemental Figure IV), highlighting a selective role for certain cytokines in the injured heart. In contrast, antiinflammatory IL4 transcript levels were down-regulated in wild-type hearts after CTX injection (42% after 24 hours, and 32% after 1 week), but were significantly increased in transgenic hearts 1 week after injury (50% compared with wild-type; Figure 3B, left panel). Transcripts encoding IL10, another antiinflammatory cytokine that suppresses injury and blocks scar formation,14 were rapidly increased in mIGF-1 transgenic hearts 24 hours after injury (90% compared with wild-type), and to a greater extent at 1 week (113% compared with wild-type; Figure 3B, right panel). Interestingly, the Cdk inhibitor p21WAF1/CIP1 was up-regulated up to 1 week after injury in the mIGF-1 transgenic hearts (Figure 3C), opening a novel and so far unexpected role for this Cdk inhibitor in the initial stages of cardiac repair induced by mIGF-1.
Figure 3.

Inflammatory markers are repressed early in mIGF-1–induced cardiac amelioration. A, Real-time PCR analysis of the inflammatory interleukins IL6 and IL1β 24 hours after CTX injection in WT and TG hearts. Real-time PCR was normalized by GAPDH content in each sample. In the left and right panels asterisks indicate significant increasing values compared with uninjured WT or TG hearts in 3 independent experiments. B, Real-time PCR analysis of the antiinflammatory cytokines IL4 and IL10 in TG and WT hearts 24 hours and 1 week after CTX injection. PCR was normalized by GAPDH content in each sample. C, Western blot analysis of p21 protein content in WT and TG hearts 24 hours and 1 week after CTX injection. β-actin was used to normalize equal protein loading.
mIGF-1 Activates Survival Signaling Pathway in Myocardial Repair
Many of the cellular growth responses attributed to IGF-1 are mediated by activation of the PI3K/Akt/mTOR phosphorylation cascade, leading to upregulation of the translational machinery15 and to cardiac prosurvival signaling in vivo.16 Notably, Akt acts as protective agent in cardiomyocyte survival and function,17,18 although its activation may not be sufficient for long-term cardioprotection and may even have adverse chronic effects.19
To dissect the intracellular signaling induced by supplemental mIGF-1, we performed phosphoprotein profiling on wild-type and mIGF-1 transgenic heart lysates. As seen in supplemental Table III, the canonical Akt phosphorylation cascade was not activated in mIGF-1 transgenic hearts, and phosphorylation levels of downstream mTOR and p70S6K intermediates were downregulated and unaffected, respectively, compared with wild-type littermates. Nevertheless, mIGF-1 transgenic expression sustained S6 ribosomal protein phosphorylation at 2, 4, and 6 months compared with the wild-type hearts (Figure 4A). The relative activation of S6 in mIGF-1 transgenic hearts appeared to be independent of canonical Akt signaling, which decreased postnatally in both wild-type and mIGF-1 transgenic hearts (Figure 4B and supplemental Table III).
Figure 4.

mIGF-1 induces interaction of PDK1 with SGK1 but not with Akt to phosphorylate S6. A and B, Western blot analysis of Akt and S6 ribosomal protein phosphorylation. Each analysis is representative of 3 independent experiments with no significant variation (data not shown). C andD, IP-Western analysis of PDK1 interaction with SGK1 and Akt isoforms. No PDK1/Akt interaction was observed. TTE indicates total tissue extract. Additional bands present in Akt1 and Akt2 blots are attributable to antibody cross-reaction and to nonspecific interaction with IgG. E, Western blot analysis of S6 ribosomal protein phos-phorylation after CTX cardiac injury. Note persistence of pS6 levels in TG hearts at 1 week after injury. F and G, IP-Western analysis of PDK1 interaction with SGK1 and Akt isoforms after CTX cardiac injury. No PDK1/Akt interaction was observed.
PI3K also signals through PDK1, a master regulator of the AGC kinase family. PDK1 can directly phosphorylate p70S6K independently of Akt,20 and promotes cell survival through other intermediates, such as SGK1, a PI3K-dependent kinase that is highly expressed in the heart and promotes survival in cardiomyocytes.21 In the heart, PDK1 regulates glucose uptake and glycogen synthase through Akt and GSK3α/GSK3β intermediates.22 Increased levels of phosphorylated PDK1 were present in mIGF-1 transgenic hearts, although no corresponding increase in GSK3α or GSK3β activation was detected (supplemental Table III). Coimmunoprecipitation analysis showed that PDK1 complexed with SGK1 in mIGF-1 transgenic hearts, whereas in wild-type hearts this interaction was much less apparent (Figure 4C). No interaction between PDK1 and any of the Akt isoforms was detected in mIGF-1 transgenic hearts (Figure 4D), indicating that the cardiac signaling cascade induced by mIGF-1 is independent of Akt and p70S6K and preferentially uses the PDK1/SGK1 pathway to increase protein synthesis and growth.
To determine whether the activation of the translational machinery and the interaction between PDK1 and SGK1 observed in physiological conditions were modulated or shifted to an Akt-dependent pathway in response to injury, we analyzed S6 ribosomal protein phosphorylation levels, as well as potential interactions between PDK1 and Akt or SGK1, in regenerating mIGF-1 hearts (Figure 4E through 4G). Phosphorylation of S6 was initially more pronounced in wild-type hearts 24 hours after injury, but by 1 week was higher in mIGF-1 transgenic hearts (Figure 4E), indicating the persistence of protein synthesis. As in uninjured mIGF-1 transgenic hearts, PDK1 was found complexed with SGK1 after CTX injury (Figure 4G) but not with any Akt isoform (Figure 4F), indicating that in both physiological and pathological conditions mIGF1 signals specifically through a PDK1/SGK1 intermediate pathway.
To test whether the PDK1-SGK1 signaling cascade in mIGF-1 transgenic hearts increased survival pathways, we analyzed the presence of TUNEL-positive nuclei around the area of injury 1 week after CTX injection. The amount of TUNEL-positive cells (Figure 5A, upper and lower panels and Figure 5B) increased significantly in wild-type hearts compared with mIGF-1 transgenic hearts. Expression levels of the proapoptotic proteins Bax and Bcl-xL were not affected (Figure 5C), although their activation by mitochondrial membrane translocation cannot be excluded.
Figure 5.

mIGF-1 transgenic expression protects against DNA damage and increases expression of the mitochondrial protein UCP1. A, TUNEL assay analysis of WT (upper panel) and TG hearts (lower panels) 1 week after CTX injection. White arrows in the WT heart indicate TUNEL-positive nuclei, whereas the TG heart showed a nonspecific signal. Red outline demarcates the border of the injury. B, Percentage amount of TUNEL-positive cells 1 week after CTX injection. Asterisk (*) indicates a significant decrease of apoptotic cells in TG hearts. C, Expression analysis of the pro-apoptotic markers Bax and Bcl-xL 6 hours, 24 hours, and 1 week after CTX injection. D, UCP1 RT-PCR of uninjured WT and TG hearts and injured WT and TG hearts 24 hours after CTX injection. Asterisk indicates a significant increase of UCP1 transcripts in TG hearts compared with WT hearts with or without CTX-induced infarct. § indicates UCP1 increasing levels compared with uninjured hearts. E, Affymetrix analysis of UCP1. The transcript is expressed as logarithmic scale of intensity in 2 TG and WT hearts. * indicates a significant increase of UCP1 transcripts in TG hearts.
To better elucidate the survival signaling regulated by mIGF-1 on cardiac damage we performed Affymetrix analysis 24 hours after CTX injection (supplemental Figure VA through VC). A specific gene expression profile, verified also by real time PCR (Figure 5D), revealed increased levels of the mitochondrial protein UCP1 (uncoupling protein 1) in transgenic hearts compared with injured wild-type hearts (Figure 5E). Interestingly, UCP1 transcripts are highly expressed in TG hearts in physiological conditions (Figure 5D), indicating that the transgene regulates UCP1 independently from cardiac damage.
Affymetrix analysis also revealed increased expression of adiponectin (also known as acrp30) and the antioxidant metallothionein 2 (supplemental Figure VD). These proteins are all involved in cardiac protection from ROS (reactive oxygen species) formation during apoptosis,23,24 as well as from ischemia-reperfusion.25 Taken together, these data confirm that supplemental mIGF-1 protects hearts from apoptotic death in the first week after CTX injection, limiting DNA damage and increasing the expression of ROS blocking proteins.
Increased Cell Proliferation in mIGF-1 Transgenic Hearts
To test whether mIGF-1 could induce a proliferative response on CTX injection, we assessed nuclear incorporation of continuously administered bromodeoxyuridine (BrdU), a marker of DNA synthesis, 48 hours, 1 week, and 1 month after CTX injection. No differences in BrdU incorporation were found in injured hearts 48 hours and 1 week after CTX injection (Figure 6B). At 1 month after infarct induction, however, total amount of BrdU-positive cells in the infarct border zone of the mIGF-1 hearts were significantly higher (35% compared with WT values) than in the wild-type hearts (Figure 6A and 6B), although a time-dependent increase of BrdU-positive nuclei has been observed in both wild-type and mIGF-1 transgenic hearts (Figure 6B). Frequent incorporation of BrdU in cardiomyocyte nuclei was seen in both cardiac tissue and individual cells after injury (Figure 6C left and middle panels, D and F), although abundant nonmuscle cells of diverse morphologies were also labeled in the vessels and surrounding myocardial tissue of mIGF-1 transgenic hearts (Figure 6C right panel, E and F). Although the origin and fate of these proliferative cells is still under investigation, they likely contribute to mIGF-1–mediated cardiac recovery in response to tissue damage, contrarily to the BrdU-positive cells in wild-type hearts, which are probably unable to elicit tissue restoration in the unfavorable infarcted environment. Notably, cardiomyocytes isolated from injured wild-type and mIGF-1 transgenic hearts did not show signs of evident hypertrophy or polyploidy (Figure 6D), indicating that neither CTX injection neither mIGF-1 overexpression elicited a hypertrophic response to the viable myocardium (Figure 6D).
Figure 6.
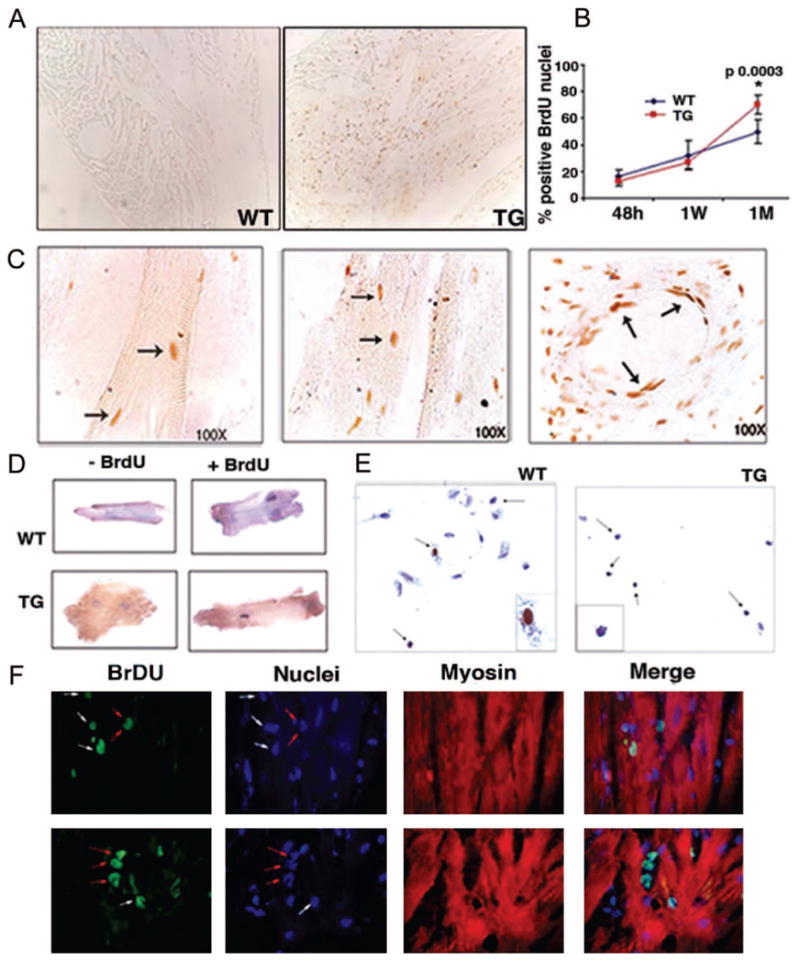
Late cell proliferation in mIGF-1 transgenic hearts. A, Positive nuclei in paraffin sections visualized with biotinylated anti-BrdU antibody in the hearts of WT and TG mice. B, Statistical analysis of BrdU-positive nuclei in paraffin sections stained with biotinylated anti-BrdU antibody and counted at different time points after CTX injection. Asterisk indicates significant relative values in the BrdU-positive hearts at 1 month (P<0.05). Values are the average of 3 independent experiments. C, Characteristics of BrdU-positive cells in mIGF-1 hearts 1 month after CTX injection. BrdU-positive cells were photographed at 100× magnification. Arrows indicate BrdU-positive cardiomyocytes (left and middle panels) and cells lining blood vessel (right panel). Cardiac myocytes (D) and nonmuscle cells (E) were isolated from BrdU-labeled WT and TG hearts 1 month after CTX injection. Dissociated cell cultures were analyzed for BrdU and hematoxylin to visualize proliferating nuclei. The experiment was performed on 3 hearts each from WT and TG mice. F, Confocal microscopic analysis of BrdU-positive cells in TG heart tissue at 100× magnification. Cardiomyocytes were visualized by an anti-myosin antibody. White arrows indicate BrdU-positive cardiac myocytes; red arrows indicate noncardiomyocyte cells.
Discussion
In this study we have intervened in the normal signaling mechanisms at work in the cardiac repair process to increase the efficiency of mammalian morphological and functional tissue restoration. The beneficial effects of the mIGF-1 isoform have been previously documented in mammalian skeletal muscle,10 a tissue with considerable endogenous regenerative capacity. The present work extends these observations to the mammalian heart, and elucidates the molecular mechanisms whereby the mIGF-1 isoform improve cardiac function on tissue damage.
Despite shifts in signaling pathways accompanied by modest effects on morphology and hemodynamic parameters, continuous expression of mIGF-1 throughout postnatal life did not produce significant perturbations in normal heart physiology, and in contrast to previous studies with other IGF-1 transgenes7 did not progress to a pathological phenotype. In response to injury, however, the pathways activated by mIGF-1 were sufficient to restore form and function of damaged cardiac tissue. The program induced by mIGF-1 followed a sequential course (Figure 7), involving early resolution of inflammation at the site of injury to prevent scar formation and to make way for the subsequent tissue replacement. Interestingly, the mIGF-1 transgene activates p21, which has been reported to be important for IGF-1–mediated cell survival on UV irradiation26 and to suppress production of IL6 in rheumatoid arthritis.27 Notably, prolonging the initial induction of p21 in damaged cardiac tissue enhances DNA repair and genome stability, without precluding cell replacement.28 Modulating expression of these downstream effectors of inflammation is important for providing a conducive environment for cell replacement and tissue restoration.
Figure 7.

Mechanisms of mIGF-1 induce recovery in injured hearts. A, Transgenic mIGF-1 induces protein synthetic and cell survival pathways in cardiac tissue through a PDK1/SGK1 phosphorylation cascade, bypassing canonical Akt, mTOR, and p70S6K intermediates. B, On myocardial infarction (MI), pathways induced by mIGF-1 result in rapid repression of proinflammatory cytokines such as IL6 that promote fibrosis and cardiac decompensation while activating cytokines such as IL4 and IL10 that resolve inflammation. C, This permissive tissue environment enables efficient cardiac wall replacement, as shown by increased proliferation of cells at 1 month after injury, and functional repair in mIGF-1 transgenic hearts.
The increased expression of antioxidant and survival transcripts, such as UCP1,25 adiponectin,24 and methallothionein 2 (Figure 5C and 5D and supplemental Figure V) during the first hours after CTX injection indicates that mIGF-1 over-expression protects the heart from toxins, generally produced in pathophysiological conditions. Interestingly, several studies have shown that UCP family members (UCP1, -2, and -3) are involved in decreasing ROS formation when overexpressed in pathological conditions.23,29 Our analysis for the first time showed that UCP1 is expressed in the infarcted hearts of mIGF-1 mice, most likely playing an important role in oxidant detoxification.
The novel intracellular signaling cascades set in motion by mIGF-1 isoform (Figure 7) presumably underlie the improved healing capacity conferred on the transgenic hearts. Under both physiological and pathological conditions, transgenic mIGF-1 upregulated S6 ribosomal protein activity through PDK1, an important downstream mediator of IGF1 signaling,30 rather than through Akt as previously reported for other IGF-1 isoforms.31 Thus, enhanced protein synthesis is likely to play a critical role during the regenerative process. Because this pathway does not involve the canonical activation of p70S6 kinase in the mIGF-1 transgenic hearts, other as yet unidentified mediators may come into play during cardiac growth and regenerative processes.
One promising candidate is SGK, which is implicated in different cell signaling leading to cell survival, apoptotic response, and osmoregulation.32 In C elegans, SGK functions in an insulin/IGF-I receptor–mediated signaling pathway to regulate metabolism, development, and longevity.33 In the heart, SGK1 is dynamically regulated and promotes cardiomyocyte survival.21 Decreased SGK1 phosphorylation in transgenic hearts expressing chronically active Akt provides further evidence for its independent signaling capacity.19 The fact that PDK1 interacts with SGK1 but with none of the Akt isoforms in mIGF-1 transgenic hearts suggests that it may be directly implicated in S6 ribosomal protein phosphorylation during physiological growth and in enhancing repair signaling. Alternatively, downstream mediators of PDK1/SGK1 interaction, such as the forkhead family member FKHRL1, the B-Raf kinase, and the sodium channels,32 may mediate more complex pathways in mIGF-1 response.
The delayed cell proliferative response seen in mIGF-1 transgenic hearts stands in contrast to the effects of direct myocardial injection of fully processed IGF-1 protein, which rapidly induces the appearance of small new myocytes within the infarct at 1 to 2 days after coronary ligation.34 In that system the maximum benefit required a dual stimulation by IGF-1 injection together with the chemotactic effects of coinjected hepatocyte growth factor. In the present study, full cardiac restoration without additional hepatocyte growth factor points either to a different mode of action used by an expressed IGF-1 transgene product or to a qualitative difference in the action of the mIGF-1 isoform itself. Interestingly, a recent study suggests that induction of IGF-1 may account for the favorable effect of Sonic Hedgehog gene therapy on recovery of myocar-dial ischemia.35 Elucidation of the roles played by native signal and E peptides in enhancing the beneficial response in mIGF-1 transgenic hearts, without the requirement for additional growth factors, will inform the design of clinically feasible therapeutic strategies to counteract the normal fibrotic tissue formation and consequent cardiac functional impairment in heart disease.
Supplementary Material
Acknowledgments
We thank David Sancho, Cancer Research Institute, London, for IL10 and IL4 primers; and Philip Cohen and James Murray for SGK1 antibody. We are grateful to Rosenthal group members for discussion. We thank Dr Melissa Little for reading the manuscript.
Sources of Funding
This work was supported by grants to N.R. from the Muscular Dystrophy Association (USA), the European Union (QLRT-2001-00930), the Foundation Leducq, a CNIC fellowship to E.L.P, and Ricerca Finalizzata (Italian Ministry of Health).
Footnotes
Disclosure
Catherine Theodoropoulos manages scientific Applications at Visualsonics. In this article she served as both a consultant and collaborator.
References
- 1.Longo VD, Finch CE. Evolutionary medicine: from dwarf model systems to healthy centenarians? Science. 2003;299:1342–1346. doi: 10.1126/science.1077991. [DOI] [PubMed] [Google Scholar]
- 2.Adamo ML, Neuenschwander S, LeRoith D, Roberts CT., Jr Structure, expression, and regulation of the IGF-I gene. Adv Exp Med Biol. 1993;343:1–11. doi: 10.1007/978-1-4615-2988-0_1. [DOI] [PubMed] [Google Scholar]
- 3.Goldspink G. Gene expression in skeletal muscle. Biochem Soc Trans. 2002;30:285–290. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 4.Chan JM, Stampfer MJ, Giovannucci E, Gann PH, Ma J, Wilkinson P, Hennekens CH, Pollak M. Plasma insulin-like growth factor-I and prostate cancer risk: a prospective study. Science. 1998;279:563–566. doi: 10.1126/science.279.5350.563. [DOI] [PubMed] [Google Scholar]
- 5.Reiss K, Cheng W, Ferber A, Kajstura J, Li P, Li B, Olivetti G, Homcy CJ, Baserga R, Anversa P. Overexpression of insulin-like growth factor-1 in the heart is coupled with myocyte proliferation in transgenic mice. Proc Natl Acad Sci U S A. 1996;93:8630–8635. doi: 10.1073/pnas.93.16.8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Q, Li B, Wang X, Leri A, Jana KP, Liu Y, Kajstura J, Baserga R, Anversa P. Overexpression of insulin-like growth factor-1 in mice protects from myocyte death after infarction, attenuating ventricular dilation, wall stress, and cardiac hypertrophy. J Clin Invest. 1997;100:1991–1999. doi: 10.1172/JCI119730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delaughter MC, Taffet GE, Fiorotto ML, Entman ML, Schwartz RJ. Local insulin-like growth factor I expression induces physiologic, then pathologic, cardiac hypertrophy in transgenic mice. Faseb J. 1999;13:1923–1929. doi: 10.1096/fasebj.13.14.1923. [DOI] [PubMed] [Google Scholar]
- 8.Winn N, Paul A, Musaro A, Rosenthal N. Insulin-like growth factor isoforms in skeletal muscle aging, regeneration, and disease. Cold Spring Harb Symp Quant Biol. 2002;67:507–518. doi: 10.1101/sqb.2002.67.507. [DOI] [PubMed] [Google Scholar]
- 9.Hill M, Goldspink G. Expression and splicing of the insulin-like growth factor gene in rodent muscle is associated with muscle satellite (stem) cell activation following local tissue damage. J Physiol. 2003;549:409–418. doi: 10.1113/jphysiol.2002.035832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Musaro A, Giacinti C, Borsellino G, Dobrowolny G, Pelosi L, Cairns L, Ottolenghi S, Cossu G, Bernardi G, Battistini L, Molinaro M, Rosenthal N. Stem cell-mediated muscle regeneration is enhanced by local isoform of insulin-like growth factor 1. Proc Natl Acad Sci U S A. 2004;101:1206–1210. doi: 10.1073/pnas.0303792101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajnoch C, Chachques JC, Berrebi A, Bruneval P, Benoit MO, Carpentier A. Cellular therapy reverses myocardial dysfunction. J Thorac Cardiovasc Surg. 2001;121:871–878. doi: 10.1067/mtc.2001.112937. [DOI] [PubMed] [Google Scholar]
- 12.Hamer PW, McGeachie JM, Davies MJ, Grounds MD. Evans Blue Dye as an in vivo marker of myofibre damage: optimising parameters for detecting initial myofibre membrane permeability. J Anat. 2002;200:69–79. doi: 10.1046/j.0021-8782.2001.00008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ren G, Dewald O, Frangogiannis NG. Inflammatory mechanisms in myocardial infarction. Curr Drug Targets Inflamm Allergy. 2003;2:242–256. doi: 10.2174/1568010033484098. [DOI] [PubMed] [Google Scholar]
- 14.Liechty KW, Kim HB, Adzick NS, Crombleholme TM. Fetal wound repair results in scar formation in interleukin-10-deficient mice in a syngeneic murine model of scarless fetal wound repair. J Pediatr Surg. 2000;35:866–872. doi: 10.1053/jpsu.2000.6868. discussion 872–3. [DOI] [PubMed] [Google Scholar]
- 15.Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001;3:1014–1019. doi: 10.1038/ncb1101-1014. [DOI] [PubMed] [Google Scholar]
- 16.Matsui T, Tao J, del Monte F, Lee KH, Li L, Picard M, Force TL, Franke TF, Hajjar RJ, Rosenzweig A. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation. 2001;104:330–335. doi: 10.1161/01.cir.104.3.330. [DOI] [PubMed] [Google Scholar]
- 17.Bock-Marquette I, Saxena A, White MD, Dimaio JM, Srivastava D. Thymosin beta4 activates integrin-linked kinase and promotes cardiac cell migration, survival and cardiac repair. Nature. 2004;432:466–472. doi: 10.1038/nature03000. [DOI] [PubMed] [Google Scholar]
- 18.Chao W, Matsui T, Novikov MS, Tao J, Li L, Liu H, Ahn Y, Rosenzweig A. Strategic advantages of insulin-like growth factor-I expression for cardioprotection. J Gene Med. 2003;5:277–286. doi: 10.1002/jgm.347. [DOI] [PubMed] [Google Scholar]
- 19.Nagoshi T, Matsui T, Aoyama T, Leri A, Anversa P, Li L, Ogawa W, del Monte F, Gwathmey JK, Grazette L, Hemmings B, Kass DA, Champion HC, Rosenzweig A. PI3K rescues the detrimental effects of chronic Akt activation in the heart during ischemia/reperfusion injury. J Clin Invest. 2005;115:2128–2138. doi: 10.1172/JCI23073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pullen N, Dennis PB, Andjelkovic M, Dufner A, Kozma SC, Hemmings BA, Thomas G. Phosphorylation and activation of p70s6k by PDK1. Science. 1998;279:707–710. doi: 10.1126/science.279.5351.707. [DOI] [PubMed] [Google Scholar]
- 21.Aoyama T, Matsui T, Novikov M, Park J, Hemmings B, Rosenzweig A. Serum and glucocorticoid-responsive kinase-1 regulates cardiomyocyte survival and hypertrophic response. Circulation. 2005;111:1652–1659. doi: 10.1161/01.CIR.0000160352.58142.06. [DOI] [PubMed] [Google Scholar]
- 22.Mora A, Sakamoto K, McManus EJ, Alessi DR. Role of the PDK1-PKB-GSK3 pathway in regulating glycogen synthase and glucose uptake in the heart. FEBS Lett. 2005;579:3632–3638. doi: 10.1016/j.febslet.2005.05.040. [DOI] [PubMed] [Google Scholar]
- 23.Cannon B, Shabalina IG, Kramarova TV, Petrovic N, Nedergaard J. Uncoupling proteins: A role in protection against reactive oxygen species-or not. Biochim Biophys Acta. 2006;1757:449–458. doi: 10.1016/j.bbabio.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 24.Efstathiou SP, Tsioulos DI, Tsiakou AG, Gratsias YE, Pefanis AV, Mountokalakis TD. Plasma adiponectin levels and five-year survival after first-ever ischemic stroke. Stroke. 2005;36:1915–1919. doi: 10.1161/01.STR.0000177874.29849.f0. [DOI] [PubMed] [Google Scholar]
- 25.Hoerter J, Gonzalez-Barroso MD, Couplan E, Mateo P, Gelly C, Cassard-Doulcier AM, Diolez P, Bouillaud F. Mitochondrial uncoupling protein 1 expressed in the heart of transgenic mice protects against ischemic-reperfusion damage. Circulation. 2004;110:528–533. doi: 10.1161/01.CIR.0000137824.30476.0E. [DOI] [PubMed] [Google Scholar]
- 26.Murray SA, Zheng H, Gu L, Jim Xiao ZX. IGF-1 activates p21 to inhibit UV-induced cell death. Oncogene. 2003;22:1703–1711. doi: 10.1038/sj.onc.1206327. [DOI] [PubMed] [Google Scholar]
- 27.Perlman H, Bradley K, Liu H, Cole S, Shamiyeh E, Smith RC, Walsh K, Fiore S, Koch AE, Firestein GS, Haines GK, 3rd, Pope RM. IL-6 and matrix metalloproteinase-1 are regulated by the cyclin-dependent kinase inhibitor p21 in synovial fibroblasts. J Immunol. 2003;170:838–845. doi: 10.4049/jimmunol.170.2.838. [DOI] [PubMed] [Google Scholar]
- 28.Rossig L, Badorff C, Holzmann Y, Zeiher AM, Dimmeler S. Glycogen synthase kinase-3 couples AKT-dependent signaling to the regulation of p21Cip1 degradation. J Biol Chem. 2002;277:9684–9689. doi: 10.1074/jbc.M106157200. [DOI] [PubMed] [Google Scholar]
- 29.Negre-Salvayre A, Hirtz C, Carrera G, Cazenave R, Troly M, Salvayre R, Penicaud L, Casteilla L. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. Faseb J. 1997;11:809–815. [PubMed] [Google Scholar]
- 30.Mora A, Komander D, van Aalten DM, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol. 2004;15:161–170. doi: 10.1016/j.semcdb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 31.Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, Yancopoulos GD, Glass DJ. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol. 2001;3:1009–1013. doi: 10.1038/ncb1101-1009. [DOI] [PubMed] [Google Scholar]
- 32.Firestone GL, Giampaolo JR, O’Keeffe BA. Stimulus-dependent regulation of serum and glucocorticoid inducible protein kinase (SGK) transcription, subcellular localization and enzymatic activity. Cell Physiol Biochem. 2003;13:1–12. doi: 10.1159/000070244. [DOI] [PubMed] [Google Scholar]
- 33.Hertweck M, Gobel C, Baumeister R. C. elegans SGK-1 is the critical component in the Akt/PKB kinase complex to control stress response and life span. Dev Cell. 2004;6:577–588. doi: 10.1016/s1534-5807(04)00095-4. [DOI] [PubMed] [Google Scholar]
- 34.Urbanek K, Rota M, Cascapera S, Bearzi C, Nascimbene A, De Angelis A, Hosoda T, Chimenti S, Baker M, Limana F, Nurzynska D, Torella D, Rotatori F, Rastaldo R, Musso E, Quaini F, Leri A, Kajstura J, Anversa P. Cardiac stem cells possess growth factor-receptor systems that after activation regenerate the infarcted myocardium, improving ventricular function and long-term survival. Circ Res. 2005;97:663–673. doi: 10.1161/01.RES.0000183733.53101.11. [DOI] [PubMed] [Google Scholar]
- 35.Kusano KF, Pola R, Murayama T, Curry C, Kawamoto A, Iwakura A, Shintani S, Ii M, Asai J, Tkebuchava T, Thorne T, Takenaka H, Aikawa R, Goukassian D, von Samson P, Hamada H, Yoon YS, Silver M, Eaton E, Ma H, Heyd L, Kearney M, Munger W, Porter JA, Kishore R, Losordo DW. Sonic hedgehog myocardial gene therapy: tissue repair through transient reconstitution of embryonic signaling. Nat Med. 2005;11:1197–1204. doi: 10.1038/nm1313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.