

Abstract
Aim
To characterize the intracellular signaling mechanisms mediating the synergistic anticancer effects of combined γ-tocotrienol and celecoxib treatment in neoplastic +SA mouse mammary epithelial cells in vitro.
Methods
+SA mammary tumor cells in different treatment groups were maintained in serum-free defined media containing 10ng/ml EGF as a mitogen and exposed to various doses of γ-tocotrienol and celecoxib alone or in combination. After a 96 hr culture period, cells were collected and whole cell lysates were subjected to Western blot analysis to determine treatment effects on intracellular signaling proteins associated with EGF-dependent mitogenesis and survival, and prostaglandin synthesis and responsiveness.
Results
Treatment with high doses of γ-tocotrienol or celecoxib alone inhibited Akt activation and downstream signaling and NFκB activation. Similar treatment with γ-tocotrienol also decreased concentration and activation of ErbB2-4 receptors, whereas celecoxib only inhibited ErbB2-4 receptor activation. In contrast, combined treatment with subeffective doses of γ-tocotrienol and celecoxib resulted in a large decrease ErbB2-4 receptor levels and activation, a decrease in PGE2 levels, and a corresponding increase in prostaglandin EP2 and EP4 receptor levels. Combined treatment also induced an increase in the prostaglandin catabolizing enzyme, PGDH.
Conclusion
The synergistic anticancer effects of combined low dose γ-tocotrienol and celecoxib treatment in +SA mammary tumor cells are mediated by COX-2-dependent mechanisms associated with a suppression in PGE2 levels, as well as, COX-2-independent mechanisms associated with a reduction in ErbB2-4 receptor levels, activation, and subsequent reduction in downstream Akt and NFκB mitogenic signaling.
Keywords: γ-Tocotrienol, Celecoxib, ErbB, Prostaglandin Receptors, PGDH, PGE2, NFκB
Introduction
γ-Tocotrienol represents one of the eight natural isoforms that make up the family of vitamin E compounds and displays potent antiproliferative and apoptotic activity against neoplastic mammary epithelial cells at treatment doses that have little or no effect on normal cell growth and function [1,2]. Celecoxib is a specific COX-2 inhibitor that has also been shown to inhibit mammary tumor cell growth and viability [3,4]. However, clinical use of these agents in the treatment of cancer has been limited due to COX-2 inhibitor-induced high dose toxicity [5,6] and poor bioavailability of γ-tocotrienol following oral administration [7-10]. However, recent studies have shown that combined treatment with low doses of γ-tocotrienol and celecoxib resulted in a synergistic inhibition in mammary tumor cell growth, and this synergistic antiproliferative effect was associated with a relatively large suppression in COX-2 expression and PGE2 production [11]. These findings suggested that combination therapy with these agents may provide enhanced therapeutic response in breast cancer patients, while avoiding the toxicity associated with high-dose COX-2 inhibitor monotherapy and the need for high dose γ-tocotrienol levels in the blood.
Experimental evidence also suggests that the growth inhibitory effects of γ-tocotrienol and celecoxib might also be mediated through COX-2-independent mechanisms. Studies have shown that γ-tocotrienol treatment inhibits EGF-dependent activation of multiple ErbB receptor family members and subsequent downstream mitogenic signaling in mouse +SA mammary tumor cells [12,13] and celecoxib inhibits Akt [14-16] and NFκB [17] activity in breast cancer cells. Furthermore, COX-2 activation and prostaglandin synthesis has been shown to modulate EGF receptor tyrosine kinase activation and mitogenic signaling [18-20]. PGE2 is a direct indicator of COX-2 activation that acts to modulate ErbB receptor activation through an intracellular signaling mechanism that involves Src-dependent events [21]. The biological actions of prostaglandins are mediated through the activation of G-protein coupled receptors called EP1-4 [22] and studies have shown that PGE2 acts primarily through EP2 and EP4 receptor activation [23]. PGE2 biological activity is regulated by the catabolizing enzyme prostaglandin dehydrogenase (PGDH) [24].
Since both γ-tocotrienol and celecoxib inhibit COX-2-dependent prostaglandin synthesis, EGF-dependent ErbB receptor activation, and mitogenic signaling, it was hypothesized that the synergistic antiproliferative effects resulting from combined low dose treatment of these compounds might also involve suppression of PGE2-dependent modulation of ErbB receptor activation and mitogenic signaling. Therefore, studies were conducted to determine if growth inhibitory effects resulting from combined low dose treatment with γ-tocotrienol and celecoxib are mediated through suppression in EGF-dependent ErbB receptor activation in +SA mammary tumor cells.
Materials and Methods
Reagent and chemicals
All reagents were purchased from Sigma (St. Louis, MO) unless otherwise stated. Isolated γ-tocotrienol (>98% pure) was provided by First Tech International Ltd. (Hong Kong). Celecoxib was purchased from LC Laboratories (Woburn, MA). Antibodies for Akt, phospho-Akt (Ser 473), PTEN, phospho-PTEN (Ser 380), phospho-GSK-3β(Ser 9), phospho-NFκB p65 (Ser 536), phospho-IKKα/β (Ser 176/180), phospho-IκB-α (Ser 32), ErbB1, phospho-ErB1 (Tyr 1173), ErbB2, phospho-ErbB2 (Tyr 877), ErbB3, phospho-ErbB3 (Tyr 1289), phospho-ErbB4 (Tyr 1284) were purchased from Cell Signaling Technology (Beverly, MA). PGDH (FL-266) primary antibody was purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). EP2 and EP4 primary antibodies were purchased from Cayman Chemical Co. (Ann Arbor, MI). Goat anti-rabbit secondary antibody was purchased from PerkinElmer Biosciences (Boston, MA). Mouse anti-actin and peroxidase goat anti-mouse antibody were purchased from Calbiochem (San Diego, CA). The PGE2 EIA-Monoclonal assay kit was purchased from Cayman Chemical Co. (Ann Arbor, MI).
Cell line and culture conditions
Experiments conducted in the present study represent a logical continuation of previous studies that have extensively characterized the antiproliferative and apoptotic effects of γ-tocotrienol in the highly malignant +SA mammary epithelial cell line [25,26]. The highly malignant +SA mammary epithelial cell line was derived from an adenocarcinoma that developed spontaneously in a BALB/c female mouse [27-29]. The +SA cell line is characterized as being highly malignant, estrogen-independent, and displays anchorage-independent growth when cultured in soft agarose gels. When +SA cells are injected back into the mammary gland fat pad of syngeneic female mice, they form rapidly growing anaplastic adenocarcinomas that are highly invasive and metastasize to the lung [27-29]. Cell culture and the experimental procedures used in this present study have been previously described in detail [1,11]. Briefly, cells were grown and maintained in serum-free Dulbecco's modified Eagle's medium (DMEM)/F12 control media containing 5 mg/ml bovine serum albumin (BSA), 10 μg/ml transferrin, 100 μg/ml soybean trypsin inhibitor, and 100 U/ml penicillin and 100 μg/ml streptomycin, 10μg/ml insulin, and 10 ng/ml EGF as a mitogen. Cells were maintained at 37°C in a humidified atmosphere of 95.0% air and 5.0% CO2.
Experimental treatments
For all experiments, an aqueous stock solution of highly lipophilic γ-tocotrienol was prepared as previously described [1,11]. Briefly, an appropriate amount of γ-tocotrienol was first dissolved in 100 μL of 100% ethanol, then added to a small volume of sterile 10% BSA in water and incubated overnight at 37°C with continuous shaking. This stock solution was then used to prepare various concentrations of 0-3.5 μM γ-tocotrienol-supplemented treatment media. A stock solution of celecoxib was prepared by dissolving a known amount in sterile dimethyl sulfoxide (DMSO) at the start of each experiment. This stock solution was then used to prepare various concentrations of 0-20 μM celecoxib-supplemented treatment media. Final concentration of DMSO and/or ethanol was maintained as the same in all treatments groups within a given experiment and never exceeded 0.1%. Treatment doses used for experimentation were based on previous dose-response studies characterizing treatment effects in +SA mammary tumor cells [11].
Measurement of PGE2
+SA cells were initially plated at a density of 5 × 104 cells/well (6 wells/group) in serum-free defined control media in 24-well culture plates and allowed to adhere overnight. The following day, cells were divided into different treatment groups and media was removed and replaced with fresh control or treatment media, and then returned to the incubator for a 72 h culture period. Cells were treated alone or in combination with vehicle, γ-tocotrienol (0.25 μM) or celecoxib (2.5 μM). In these particular experiments, media was not replaced at any time after the start of treatment exposure. At the end of the 72 h treatment period, media was collected and assayed for PGE2 according to the methods described in the EIA kit provided by the manufacturer (Cayman Chemical Co. Ann Arbor, MI). Optical density was measured at 420 nm on a Synergy-2 Multi Mode Microplate Reader (BioTek Instruments Inc., Winooski, VT). Differences among the treatment groups were determined by analysis of variance (ANOVA) followed by Dunnett's t-test using the SAS software package (SAS Institute Inc., NC). A difference of P < 0.05 was considered to be significant as compared to vehicle-treated controls.
Electrophoresis and western blot analysis
+SA cells were plated at a density of 1 × 106 cells/100 mm culture plates and grown in serum-free defined control or treatment media. At the end of 96 h treatment period, cells were isolated with trypsin, washed, and then whole cell lysates were prepared for subsequent electrophoresis, as previously described in detail [48]. Briefly, protein concentration in each sample was determined using the BioRad protein assay kit (BioRad, Hercules, CA). Equal amounts of protein (30 μg) from each sample were loaded on 7.5–15 % SDS-polyacrylamide minigels and electrophoresed. Proteins were then transblotted (30 V for 12-16 h at 4oC) to polyvinylidene difluoride (PVDF) membranes (Dupont, Boston, MA) and then blocked with 2% bovine serum albumin (BSA) in 0.1% Tween Tris buffered saline (TBST) for 2 h. The PVDF membranes were probed with specific primary antibodies against Akt (1:5000), phospho-Akt (1:2000), PTEN (1:5000), phospho-PTEN (1:5000), phospho-GSK-3β (1:5000), phospho-NFκB p65 (1:2000), phospho-IKK-α/β (1:2000), phospho-IκB-α (1:2000), PGDH (1:5000), EP2 (1:10000) and EP4 (1:10000) receptors, total and phosphorylated ErbB1-4 receptors (1:2000) in 2% BSA/TBST for 2 h at room temperature. Membranes were washed five times with TBST and then incubated with horseradish peroxidase-conjugated goat anti-rabbit secondary antibody diluted 1:4000 in 2% BSA/TBST for 1 h followed by washing. Antibody bound proteins were visualized with the SuperSignal enhanced chemiluminescence kit (Pierce, Rockford, IL). The Kodak Gel Logic-1500 imaging system (Carestream Molecular Imaging, New Haven, CT) was used to visualize the luminescent proteins. All experiments were repeated at least three times. A representative Western blot image from each experiment is shown in each Figure. The visualization of β-actin was used to ensure equal sample loading in each lane. Densitometric analysis was performed using Kodak Molecular Imaging Software 4.5 (Carestream Health Inc, New Haven, CT). For quantification, the values obtained from densitometry of Western blot images for the various treatment groups were normalized to their respective β-actin and control densitometric values to clearly visualize the differences between treatment groups.
Results
Effects of γ-tocotrienol and celecoxib on PGDH and PGE2 levels
Treatment with 3.5 μM γ-tocotrienol, 2.5 μM or 20 μM celecoxib alone or the combination of 0.25 μM γ-tocotrienol and 2.5 μM celecoxib induced a moderate increase, whereas treatment with 0.25 μM γ-tocotrienol alone had little or no effect on intracellular levels of PGDH as compared to +SA cells in the vehicle-treated control group (Figure 1A). Treatment with 0.25 μM γ-tocotrienol or 2.5 μM celecoxib alone induced a slight decrease in PGE2 levels as compared to vehicle-treated control, but this decrease was not found to be significant (Figure 1B). However, combined treatment with the same subeffective doses of γ-tocotrienol and celecoxib resulted in a significant decrease in PGE2 levels as compared to vehicle-treated controls (Figure 1B).
Figure 1.
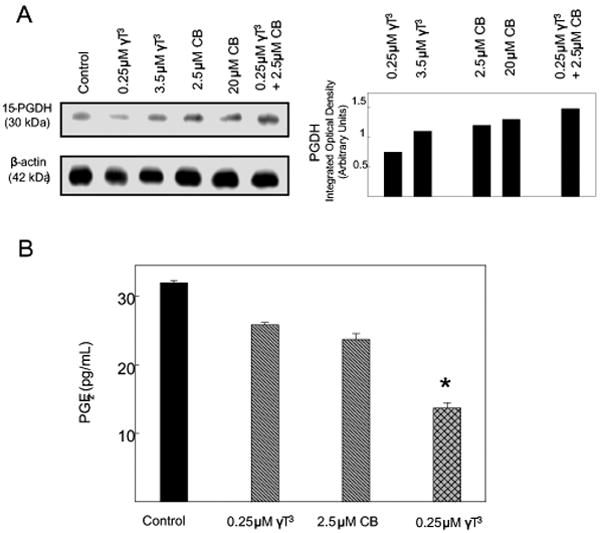
(A) Western blot analysis of γ-tocotrienol and celecoxib treatment effects on PGDH in neoplastic +SA mammary epithelial cells. Cells were initially plated at a density of 1×106 cells/100 mm culture dishes, divided into different treatment groups, and then maintained on their respective control or treatment media for a 4-day treatment period. Afterwards, cells were isolated and prepared for Western blot analysis. Samples were analyzed for relative levels of the PGDH. Scanning densitometric analysis was performed for each blot to visualize the relative levels of proteins. Integrated optical density of each band was normalized with their corresponding β-actin and control treatment bands and then shown in bar graphs. Vertical bars indicate the fold-change in protein levels in various treatment groups as compared with their respective controls. (B) Effects of celecoxib and γ-tocotrienol treatment alone and in combination on PGE2 synthesis in neoplastic +SA mammary epithelial cells. Cells were initially plated at a density of 5×104 cells/well in 24-well culture plates, divided into the different treatment groups, and then maintained on their respective control or treatment media for a 72 hr treatment period. Afterward, media was collected from the different treatment groups and prepared for use in the EIA assay for PGE2. Vertical bars indicate the mean cell count ± SEM in each treatment group. *P<0.05 as compared with the vehicle-treated control group.
Effects of γ-tocotrienol and celecoxib on EP2 and EP4 prostaglandin receptor levels
Treatment with subeffective doses of γ-tocotrienol (0.25 μM) and celecoxib (2.5 μM) alone had no effect, whereas treatment with growth inhibitory doses of γ-tocotrienol (3.5 μM) or celecoxib (20 μM) alone caused a slight increase in EP2 and EP4 prostaglandin receptor levels as compared to +SA cells in the vehicle-treated control group (Figure 2). Combined treatment with subeffective doses of γ-tocotrienol (0.25 μM) and celecoxib (2.5 μM) was also found to result in a slight increase in EP2 and EP4 prostaglandin receptor levels as compared to vehicle-treated controls (Figure 2).
Figure 2.
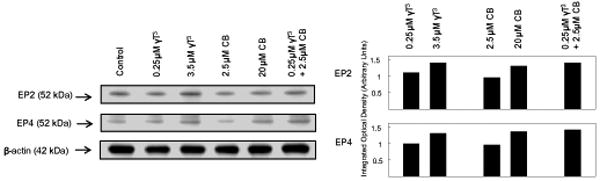
Western blot analysis of γ-tocotrienol and celecoxib treatment on EP2 and EP4 receptors in neoplastic +SA mammary epithelial cells. Cells were initially plated at a density of 1×106 cells/100 mm culture dishes, divided into different treatment groups, and then maintained on their respective control or treatment media for a 4-day treatment period. Afterwards, cells were isolated and prepared for Western blot analysis. Samples were analyzed for relative levels of the proteins. Scanning densitometric analysis was performed for each blot to visualize the relative levels of proteins. Integrated optical density of each band was normalized with their corresponding β-actin and control treatment bands and then shown in bar graphs. Vertical bars indicate the fold-change in protein levels in various treatment groups as compared with their respective controls.
Effects of γ-tocotrienol and celecoxib on EGF-Dependent Akt Mitogenic Signaling
The effects of IC50 growth inhibitory doses of γ-tocotrienol (3.5 μM) and celecoxib (20 μM) given alone where compared to subeffective doses γ-tocotrienol (0.25 μM) and celecoxib (2.5 μM) given alone or in combination on EGF-dependent Akt activation and downstream signaling in +SA mammary tumor cells. Treatment doses were selected based on results obtained from previous dose-response studies with these cells [11]. Western blot (Figure 3A) and scanning densitometric analysis (Figure 3B) showed that treatment with 3.5 μM γ-tocotrienol or 20 μM celecoxib alone caused a relatively large decrease, whereas treatment with 0.25 μM γ-tocotrienol or 2.5 μM celecoxib alone had no effect on phospho-Akt (activated) levels as compared to cells in the vehicle-treated control group. In contrast, combined treatment with 0.25 μM γ-tocotrienol and 2.5 μM celecoxib resulted in a relatively large decrease in phosphorylated-Akt levels (Figure 3A and 3B). Similar treatment effects were observed on phospho-GSK-3β levels, a downstream target for activated Akt phosphorylation (Figure 3A and 3B). Total Akt levels were not found to differ among any of the treatment groups (Figure 3A and 3B). Since Akt inactivation is regulated by the phosphatase, PTEN, it was of interest to determine treatment effects on total PTEN and phospho-PTEN (activated) levels. Results showed that total PTEN and phospho-PTEN levels were similar in all treatment groups (Figure 3A and 3B).
Figure 3.
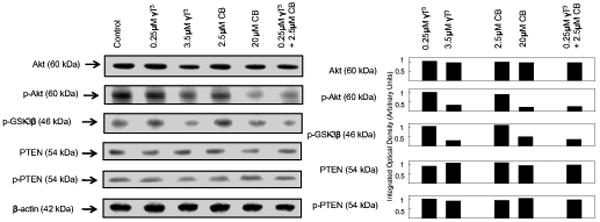
Western blot analysis of γ-tocotrienol and celecoxib on Akt, p-Akt (Ser 473), PTEN, p-PTEN and p-GSK-3β in neoplastic +SA mammary epithelial cells. Cells were initially plated at a density of 1×106 cells/100 mm culture dishes, divided into different treatment groups, and then maintained on their respective control or treatment media for a 4-day treatment period. Afterwards, cells were isolated and prepared for Western blot analysis. Samples were analyzed for relative levels of the proteins. Scanning densitometric analysis was performed for each blot to visualize the relative levels of proteins. Integrated optical density of each band was normalized with their corresponding β-actin and control treatment bands and then shown in bar graphs. Vertical bars indicate the fold-change in protein levels in various treatment groups as compared with their respective controls.
Effects of γ-tocotrienol and celecoxib on EGF-dependent NFκB activation
Western blot (Figure 4A) and scanning densitometric analysis (Figure 4B) showed that treatment with higher doses of γ-tocotrienol (3.5 μM) or celecoxib (20 μM) alone caused a large decrease in phospho-NFκB p65 (Ser 536), phospho-IKK-α/β (Ser 176/180) and phospho-IκB-α (Ser 32) in +SA cells as compared to vehicle-treated controls. Treatment with subeffective doses of γ-tocotrienol (0.25 μM) and celecoxib (2.5 μM) alone had no effect, whereas combined treatment with 0.25 μM γ-tocotrienol and 2.5 μM celecoxib induced a relatively large decrease in phospho-NFκB p65 (Ser 536), phospho-IKK-α/β (Ser 176/180) and phospho-IκB-α (Ser 32) levels in +SA cells as compared to vehicle-treated controls (Figure 4A and 4B).
Figure 4.
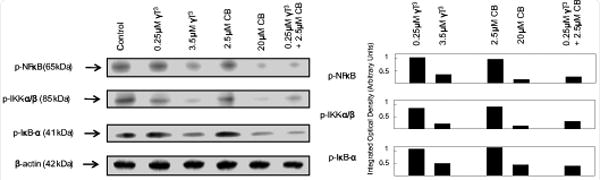
Western blot analysis of γ-tocotrienol and celecoxib treatment alone and in combination on p-NFκB p65 (Ser 536), p-IKK-α/β (Ser 176/180) and p-IκB-α (Ser 32) in neoplastic +SA mammary epithelial cells. Cells were initially plated at a density of 1×106 cells/100 mm culture dishes, divided into different treatment groups, and then maintained on their respective control or treatment media for a 4-day treatment period. Afterwards, cells were isolated and prepared for Western blot analysis. Samples were analyzed for relative levels of the proteins. Scanning densitometric analysis was performed for each blot to visualize the relative levels of proteins. Integrated optical density of each band was normalized with their corresponding β-actin and control treatment bands and then shown in bar graphs. Vertical bars indicate the fold-change in protein levels in various treatment groups as compared with their respective controls.
Effects of γ-tocotrienol and celecoxib on ErbB receptor levels and activation
Treatment with 0.25 μM γ-tocotrienol, 2.5 μM celecoxib or 20 μM celecoxib alone had no effect on total ErbB2-4 levels in +SA mammary tumor cells (Figure 5). However, treatment with 3.5 μM γ-tocotrienol alone or combined treatment with subeffective doses of γ-tocotrienol (0.25 μM) and celecoxib (2.5 μM) was found to cause a relatively large decrease in total levels ErbB2-4 as compared to vehicle-treated controls (Figure 5). ErbB1 levels were not found to be altered in any of the treatment groups (Figure 5). Treatment with 0.25 μM γ-tocotrienol or 2.5 μM celecoxib alone had no effect on the phosphorylated (activated) ErbB1-4 levels (Figure 5). However treatment with growth inhibitory doses of γ-tocotrienol (3.5 μM) or celecoxib (20 μM) alone, or combined treatment with subeffective doses of γ-tocotrienol (0.25 μM) and celecoxib (2.5 μM) caused relatively large decrease in phosphorylated (activated) ErbB2-4 levels as compared to vehicle-treated controls (Figure 5). Phosphorylated ErbB1 levels were not found to differ among any of the different treatment groups (Figure 6).
Figure 5.
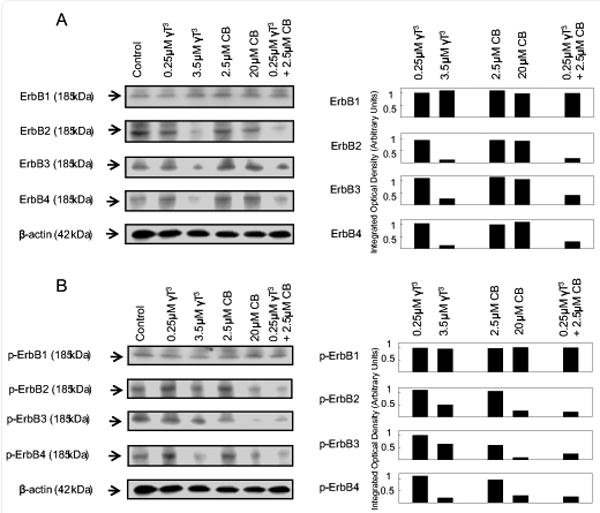
Western blot analysis of γ-tocotrienol and celecoxib treatment alone and in combination on (A) ErbB1, ErbB2, ErbB3, and ErbB4, and (B) p-ErbB1 (Tyr 1173), p-ErbB2 (Tyr 877), p-ErbB3 (Tyr 1289) and p-ErbB4 (Tyr 1284) in neoplastic +SA mammary epithelial cells. Cells were initially plated at a density of 1×106 cells/100 mm culture dishes, divided into different treatment groups, and then maintained on their respective control or treatment media for a 4-day treatment period. Afterwards, cells were isolated and prepared for Western blot analysis. Samples were analyzed for relative levels of the proteins. Scanning densitometric analysis was performed for each blot to visualize the relative levels of proteins. Integrated optical density of each band was normalized with their corresponding β-actin and control treatment bands and then shown in bar graphs. Vertical bars indicate the fold-change in protein levels in various treatment groups as compared with their respective controls.
Figure 6.
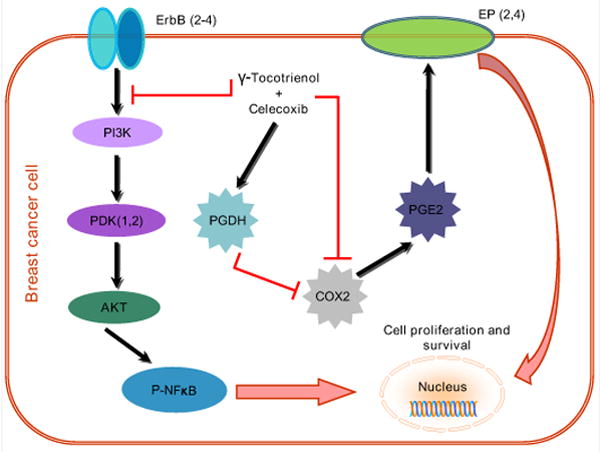
Schematic illustration of the COX-2 dependent and independent mechanisms involved in mediating the synergistic antiproliferative effects of combined γ-tocotrienol and celecoxib treatment. The COX-2-dependent mechanism involve a suppression in COX-2 and PGE2 levels and a corresponding increase in PGDH levels, while the COX-2-independent mechanisms involve a reduction in the levels and EGF-dependent activation ErbB2-4 receptor levels and subsequent reductions in downstream Akt and NFκB mitogenic signaling.
Discussion
Previous studies demonstrated that the synergistic antiproliferative effects of combined treatment with low doses of γ-tocotrienol with celecoxib in +SA mammary tumor cells were associated with a large reduction in intracellular levels of COX-2 and a corresponding significant decrease in PGE2 production [11]. Results in the present study further extend these previous findings and demonstrate that combination treatment with these agents also attenuates EGF-dependent ErbB receptor activation and downstream mitogenic signaling. Specifcally, combined treatment with subeffective doses γ-tocotrienol and celecoxib reduced EGF-dependent ErbB2-4 phosphorylation. Since ErbB2-4 receptor phosphorylation is required for substrate interaction and activation through tyrosine phosphorylation [30,31], a reduction in ErbB receptor activation also resulted in a corresponding decrease in EGF-dependent phosphorylation (activation) of Akt, NFκB, and GSK-3β. These findings indicate that the synergistic anticancer effects of combined γ-tocotrienol and celecoxib treatment in +SA mammary tumor cells are mediated by both COX-2-dependent and -independent mechanisms.
Previous studies showed that treatment with moderate doses of celecoxib or γ-tocotrienol alone resulted in a significant decrease, whereas treatment with subeffective of these agents alone had no significant effect on +SA mammary tumor cell growth or PGE2 synthesis [11]. However, combined treatment with subeffective doses of γ-tocotrienol and celecoxib were found to cause a significant reduction in COX-2, but not COX-1 levels, and a corresponding significant decrease in PGE2 levels [11]. The present study further demonstrates that combination treatment with these agents also results with a modest increase in PGDH levels. Since this enzyme inactivates prostaglandins, particularly PGE2, it is possible that treatment-induced elevations in PGDH levels may also play a role in reducing PGE2 levels in these mammary tumor cells. Interestingly, combined treatment with subeffective doses of γ-tocotrienol and celecoxib was also found to cause increase in prostaglandin receptors EP2 and EP4 levels. However, this elevation in prostaglandin receptor levels as a counter-regulatory response to decreased PGE2 levels was not enough to reverse the growth inhibitory effects of combined low dose γ-tocotrienol and celecoxib treatment.
The intracellular mechanisms involved in mediating the antiproliferative effects of γ-tocotrienol in mammary tumor cells have been previously reviewed in detail [11,16,26]. Studies have shown that treatment with growth inhibitory doses of γ-tocotrienol inhibits EGF-dependent growth and is associated with a reduction in Akt and NFκB activation in +SA mammary tumor cells [11,16,32,33]. Similarly, treatment with high doses of celecoxib has also been shown to suppress Akt and NFκB activation and mitogenesis in various tumor cell types [14,17]. However, the exact intracellular mechanisms involved in mediating the antiproliferative effects of combined treatment with subeffective doses of celecoxib and γ-tocotrienol in +SA mammary tumor cells had not previously been determined.
Results in the present study show that the growth inhibitory effects of combined treatment with subeffective doses of γ-tocotrienol and celecoxib are associated with a decrease in phospho-Akt (active form) and phospho-GSK-3β a downstream substrate of activated Akt [34-37]. Results also showed that this decrease in Akt activity did not result from an increase in PTEN phosphatase levels, an enzyme involved in reducing phospho-Akt levels [35]. In addition, combined γ-tocotrienol and celecoxib treatment was also found to inhibit NFκB phosphorylation (active form). NFκB exist primarily in the cytoplasm in an inactive state bound to the inhibitory protein, IκB [38-40]. Several mitogen-dependent kinases such as Akt can activate IκB-kinase, which will then phosphorylate IκB and promotes its degradation and leads to the release and enhanced phosphorylation and activation of NFκB [38-40]. Results showed that the inhibitory effects of combined γ-tocotrienol and celecoxib treatment on NFκB phosphorylation appear to result from a decrease in IκB-kinase phosphorylation and subsequent decrease in IκB phosphorylation. Since γ-tocotrienol and celecoxib inhibits mitogen-induced Akt or NFκB activation, but are not found to directly inhibit Akt or NFκB activity [15,36], the inhibitory effects of these agents must occur upstream of these signaling proteins, possible at the level of the EGF receptor.
Characterization of ErbB receptors in +SA mammary tumor cells has shown that ErbB1-4 receptors are expressed in these cells and display tyrosine phosphorylation following EGF exposure [12,13]. EGF-induced activation of ErbB1 receptors leads to the formation of receptor homo- and heterodimers [31,41]. Furthermore, although the ErbB2 receptor lacks a ligand binding site and the ErbB3 receptor has no tyrosine kinase activity, these receptors can initiate mitogenic signaling by forming heterodimers [30,42] that are particularly potent in activating Akt [19,43]. Furthermore, elevated Akt signaling is associated with advanced breast cancer progression and a poor prognosis [44].
In the present study, combined treatment with subeffective doses γ-tocotrienol and celecoxib had no effect on ErbB1 receptor levels or EGF-dependent phosphorylation (activation). However, combined treatment was found to induce a large reduction in total ErbB3, and to a lesser extent ErbB2 and ErbB4 receptor levels, and corresponding decrease in EGF-dependent phosphorylation in ErbB2-4 receptors in +SA mammary tumor cells. Since activation of ErbB2 and ErbB3 heterodimers is associated with enhanced Akt mitogenic signaling, these results indicate that the antiproliferative effects induced by combined treatment with subeffective doses of γ-tocotrienol and celecoxib are mediated, at least in part, through COX-2-independent mechanisms that involve a reduction in levels and EGF-dependent activation of multiple ErbB receptor family members. At present, it is not known if combined treatment-induced reductions in ErbB2-4 levels result from a decrease in receptor synthesis or increase in receptor degradation. Further studies are required to determine if one or both of these possibilities are correct.
Overexpression of COX-2 plays a major role in nearly all stages of tumor development [45,46], and COX-2 inhibitors have been shown to be potent anticancer agents. Nevertheless, the use of COX-2 inhibitors in cancer trials has been greatly limited by their high-dose toxicity that is characterized by severe gastrointestinal and cardiovascular toxicities [5,6]. Similarly, it is now well established that γ-tocotrienols display potent antiproliferative and apoptotic activity against mammary tumor cells at treatment doses that have little or no effect on normal cell growth and function. Although experimental evidence has been very promising, oral supplementation of tocotrienols in clinical studies has produced inconsistent results. It is now evident that the part of the reason for the discrepancies in anticancer effectiveness between in vitro and in vivo studies is due to poor γ-tocotrienol bioavailability. Recent studies have shown that it is very difficult to obtain and/or sustain therapeutic levels of γ-tocotrienol in the blood and target tissues following oral administration because of inefficient tocotrienol intestinal absorption and delivery to target tissues in the body [7-10]. Results in this study indicate that combined low doses γ-tocotrienol and celecoxib treatment may have potential value as a potent therapeutic regiment in the treatment of breast cancer, while at the same time reducing or eliminating the problems associated with celecoxib high dose toxicity and γ-tocotrienol poor bioavailability when high doses of these agents are as monotherapy.
In summary, these studies have shown that the synergistic antiproliferative effects of combined low dose γ-tocotrienol and celecoxib treatment in +SA mammary tumor cells are mediated by both COX-2-dependent and COX-2-independent mechanisms. The COX-2-dependent mechanism involve a suppression in COX-2 and PGE2 levels, while the COX-2-independent mechanisms involve a reduction in the levels and EGF-dependent activation ErbB2-4 receptor levels and subsequent reductions in downstream Akt and NFκB mitogenic signaling.
Acknowledgments
This work was supported in part by grants from NIH (grant # CA86833) and First Tech International Ltd. The authors would like to thank First Tech International Ltd. For generously providing γ-tocotrienol for use in these experiments.
Abbreviations
- COX
Cyclooxygenase
- PGE2
Prostaglandin E2
- PGDH
Prostaglandin Dehydrogenase
- TBS
Tris Buffered Saline
Footnotes
Publisher's Disclaimer: This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.McIntyre BS, Briski KP, Gapor A, Sylvester PW. Antiproliferative and apoptotic effects of tocopherols and tocotrienols on preneoplastic and neoplastic mouse mammary epithelial cells. Proc Soc Exp Biol Med. 2000;224:292–301. doi: 10.1046/j.1525-1373.2000.22434.x. [DOI] [PubMed] [Google Scholar]
- 2.McIntyre BS, Briski KP, Tirmenstein MA, Fariss MW, Gapor A, et al. Antiproliferative and apoptotic effects of tocopherols and tocotrienols on normal mouse mammary epithelial cells. Lipids. 2000;35:171–180. doi: 10.1007/BF02664767. [DOI] [PubMed] [Google Scholar]
- 3.Howe LR, Subbaramaiah K, Patel J, Masferrer JL, Deora A, et al. Celecoxib, a selective cyclooxygenase 2 inhibitor, protects against human epidermal growth factor receptor 2 (HER-2)/neu-induced breast cancer. Cancer Res. 2002;62:5405–5407. [PubMed] [Google Scholar]
- 4.Lanza-Jacoby S, Miller S, Flynn J, Gallatig K, Daskalakis C, et al. The cyclooxygenase-2 inhibitor, celecoxib, prevents the development of mammary tumors in Her-2/neu mice. Cancer Epidemiol Biomarkers Prev. 2003;12:1486–1491. [PubMed] [Google Scholar]
- 5.Laible B. COX-2 inhibitors and cardiovascular toxicity: a class effect? S D J Med. 2005;58:93–94. [PubMed] [Google Scholar]
- 6.Shafiq N, Malhotra S, Pandhi P, Nada R. Comparative gastrointestinal toxicity of selective cyclooxygenase (COX-2) inhibitors. Indian J Exp Biol. 2005;43:614–619. [PubMed] [Google Scholar]
- 7.Hayes KC, Pronczuk A, Liang JS. Differences in the plasma transport and tissue concentrations of tocopherols and tocotrienols: observations in humans and hamsters. Proc Soc Exp Biol Med. 1993;202:353–359. doi: 10.3181/00379727-202-43546. [DOI] [PubMed] [Google Scholar]
- 8.Ikeda I, Imasato Y, Sasaki E, Sugano M. Lymphatic transport of alpha-, gamma- and delta-tocotrienols and alpha- tocopherol in rats. Int J Vitam Nutr Res. 1996;66:217–221. [PubMed] [Google Scholar]
- 9.Yap SP, Yuen KH, Lim AB. Influence of route of administration on the absorption and disposition of alpha-, gamma- and delta-tocotrienols in rats. J Pharm Pharmacol. 2003;55:53–58. doi: 10.1111/j.2042-7158.2003.tb02433.x. [DOI] [PubMed] [Google Scholar]
- 10.Yap SP, Yuen KH, Wong JW. Pharmacokinetics and bioavailablity of alpha-, gamma-, and delta-tocotrienols under different food status. J Pharmaceut Pharmacol. 2001;56:67–71. doi: 10.1211/0022357011775208. [DOI] [PubMed] [Google Scholar]
- 11.Shirode AB, Sylvester PW. Synergistic anticancer effects of combined gamma-tocotrienol and celecoxib treatment are associated with suppression in Akt and NFkappaB signaling. Biomed Pharmacother. 2010;64:327–332. doi: 10.1016/j.biopha.2009.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bachawal SV, Wali VB, Sylvester PW. Combined gamma-tocotrienol and erlotinib/gefitinib treatment suppresses Stat and Akt signaling in murine mammary tumor cells. Anticancer Res. 2010a;30:429–437. [PubMed] [Google Scholar]
- 13.Bachawal SV, Wali VB, Sylvester PW. Enhanced antiproliferative and apoptotic response to combined treatment of gamma-tocotrienol with erlotinib or gefitinib in mammary tumor cells. BMC Cancer. 2010b;10:84. doi: 10.1186/1471-2407-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kucab JE, Lee C, Chen CS, Zhu J, Gilks CB, et al. Celecoxib analogues disrupt Akt signaling, which is commonly activated in primary breast tumours. Breast Cancer Res. 2005;7:R796–807. doi: 10.1186/bcr1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samant GV, Sylvester PW. gamma-Tocotrienol inhibits ErbB3-dependent PI3K/Akt mitogenic signalling in neoplastic mammary epithelial cells. Cell Prolif. 2006;39:563–574. doi: 10.1111/j.1365-2184.2006.00412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shah SJ, Sylvester PW. Gamma-tocotrienol inhibits neoplastic mammary epithelial cell proliferation by decreasing Akt and nuclear factor kappaB activity. Exp Biol Med (Maywood) 2005b;230:235–241. doi: 10.1177/153537020523000402. [DOI] [PubMed] [Google Scholar]
- 17.van Wijngaarden J, van Beek E, van Rossum G, van der Bent C, Hoekman K, et al. Celecoxib enhances doxorubicin-induced cytotoxicity in MDA-MB231 cells by NF-kappaB-mediated increase of intracellular doxorubicin accumulation. Eur J Cancer. 2007;43:433–442. doi: 10.1016/j.ejca.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 18.Al-Salihi MA, Ulmer SC, Doan T, Nelson CD, Crotty T, et al. Cyclooxygenase-2 transactivates the epidermal growth factor receptor through specific E-prostanoid receptors and tumor necrosis factor-alpha converting enzyme. Cell Signal. 2007;19:1956–1963. doi: 10.1016/j.cellsig.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim HH, Vijapurkar U, Hellyer NJ, Bravo D, Koland JG. Signal transduction by epidermal growth factor and heregulin via the kinase-deficient ErbB3 protein. Biochem J. 1998;334(Pt 1):189–195. doi: 10.1042/bj3340189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu R, Abramson AL, Shikowitz MJ, Dannenberg AJ, Steinberg BM. Epidermal growth factor-induced cyclooxygenase-2 expression is mediated through phosphatidylinositol-3 kinase, not mitogen-activated protein/extracellular signal-regulated kinase kinase, in recurrent respiratory papillomas. Clin Cancer Res. 2005;11:6155–6161. doi: 10.1158/1078-0432.CCR-04-2664. [DOI] [PubMed] [Google Scholar]
- 21.Buchanan FG, Wang D, Bargiacchi F, DuBois RN. Prostaglandin E2 regulates cell migration via the intracellular activation of the epidermal growth factor receptor. J Biol Chem. 2003;278:35451–35457. doi: 10.1074/jbc.M302474200. [DOI] [PubMed] [Google Scholar]
- 22.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 23.Chang SH, Liu CH, Conway R, Han DK, Nithipatikom K, et al. Role of prostaglandin E2-dependent angiogenic switch in cyclooxygenase 2-induced breast cancer progression. Proc Natl Acad Sci U S A. 2004;101:591–596. doi: 10.1073/pnas.2535911100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tai HH, Ensor CM, Tong M, Zhou H, Yan F. Prostaglandin catabolizing enzymes. Prostaglandins Other Lipid Mediat. 2002;68-69:483–493. doi: 10.1016/s0090-6980(02)00050-3. [DOI] [PubMed] [Google Scholar]
- 25.Sylvester PW, Shah SJ. Mechanisms mediating the antiproliferative and apoptotic effects of vitamin E in mammary cancer cells. Front Biosci. 2005;10:699–709. doi: 10.2741/1565. [DOI] [PubMed] [Google Scholar]
- 26.Sylvester PW, Theriault A. Role of Tocotrienols in the Prevention of Cardiovascular Disease and Breast Cancer. Current Topics in Nutraceutical Research. 2003;1:121–135. [Google Scholar]
- 27.Anderson LW, Danielson KG, Hosick HL. New cell line. Epithelial cell line and subline established from premalignant mouse mammary tissue. In Vitro. 1979;15:841–843. doi: 10.1007/BF02618037. [DOI] [PubMed] [Google Scholar]
- 28.Anderson LW, Danielson KG, Hosick HL. Metastatic potential of hyperplastic alveolar nodule derived mouse mammary tumor cells following intravenous inoculation. Eur J Cancer Clin Oncol. 1981;17:1001–1008. doi: 10.1016/s0277-5379(81)80005-3. [DOI] [PubMed] [Google Scholar]
- 29.Danielson KG, Anderson LW, Hosick HL. Selection and characterization in culture of mammary tumor cells with distinctive growth properties in vivo. Cancer Res. 1980;40:1812–1819. [PubMed] [Google Scholar]
- 30.Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. Embo J. 2000;19:3159–3167. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 32.Shah SJ, Sylvester PW. Tocotrienol-induced cytotoxicity is unrelated to mitochondrial stress apoptotic signaling in neoplastic mammary epithelial cells. Biochem Cell Biol. 2005c;83:86–95. doi: 10.1139/o04-127. [DOI] [PubMed] [Google Scholar]
- 33.Sylvester PW. Vitamin E and apoptosis. Vitam Horm. 2007;76:329–356. doi: 10.1016/S0083-6729(07)76012-0. [DOI] [PubMed] [Google Scholar]
- 34.Downward J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr Opin Cell Biol. 1998;10:262–267. doi: 10.1016/s0955-0674(98)80149-x. [DOI] [PubMed] [Google Scholar]
- 35.Kandel ES, Hay N. The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp Cell Res. 1999;253:210–229. doi: 10.1006/excr.1999.4690. [DOI] [PubMed] [Google Scholar]
- 36.Shah SJ, Sylvester PW. Gamma-tocotrienol inhibits neoplastic mammary epithelial cell proliferation by decreasing Akt and nuclear factor kappaB activity. Exp Biol Med. 2005a;230:235–241. doi: 10.1177/153537020523000402. [DOI] [PubMed] [Google Scholar]
- 37.Toker A. Protein kinases as mediators of phosphoinositide 3-kinase signaling. Mol Pharmacol. 2000;57:652–658. [PubMed] [Google Scholar]
- 38.Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 39.Khwaja A. Akt is more than just a Bad kinase. Nature. 1999;401:33–34. doi: 10.1038/43354. [DOI] [PubMed] [Google Scholar]
- 40.Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W. IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J Biol Chem. 1999;274:30353–30356. doi: 10.1074/jbc.274.43.30353. [DOI] [PubMed] [Google Scholar]
- 41.Normanno N, Bianco C, Strizzi L, Mancino M, Maiello MR, et al. The ErbB receptors and their ligands in cancer: an overview. Curr Drug Targets. 2005;6:243–257. doi: 10.2174/1389450053765879. [DOI] [PubMed] [Google Scholar]
- 42.Sliwkowski MX, Schaefer G, Akita RW, Lofgren JA, Fitzpatrick VD, et al. Coexpression of erbB2 and erbB3 proteins reconstitutes a high affinity receptor for heregulin. J Biol Chem. 1994;269:14661–14665. [PubMed] [Google Scholar]
- 43.Karunagaran D, Tzahar E, Beerli RR, Chen X, Graus-Porta D, et al. ErbB-2 is a common auxiliary subunit of NDF and EGF receptors: implications for breast cancer. Embo J. 1996;15:254–264. [PMC free article] [PubMed] [Google Scholar]
- 44.Witton CJ, Reeves JR, Going JJ, Cooke TG, Bartlett JM. Expression of the HER1-4 family of receptor tyrosine kinases in breast cancer. J Pathol. 2003;200:290–297. doi: 10.1002/path.1370. [DOI] [PubMed] [Google Scholar]
- 45.Soslow RA, Dannenberg AJ, Rush D, Woerner BM, Khan KN, et al. COX-2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer. 2000;89:2637–2645. doi: 10.1002/1097-0142(20001215)89:12<2637::aid-cncr17>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 46.Wang D, Dubois RN. Prostaglandins and cancer. Gut. 2006;55:115–122. doi: 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim YM, Park SY, Pyo H. Cyclooxygenase-2 (COX-2) negatively regulates expression of epidermal growth factor receptor and causes resistance to gefitinib in COX-2-overexpressing cancer cells. Mol Cancer Res. 2009;7:1367–1377. doi: 10.1158/1541-7786.MCR-09-0004. [DOI] [PubMed] [Google Scholar]
- 48.Shah S, Gapor A, Sylvester PW. Role of caspase-8 activation in mediating vitamin E-induced apoptosis in murine mammary cancer cells. Nutr Cancer. 2003;45:236–246. doi: 10.1207/S15327914NC4502_14. [DOI] [PubMed] [Google Scholar]
- 49.Yarden Y. Biology of HER2 and its importance in breast cancer. Oncology. 2001;61:1–13. doi: 10.1159/000055396. [DOI] [PubMed] [Google Scholar]