

Abstract
Thiamin diphosphate (ThDP) is a key coenzyme in sugar metabolism. The 4′-aminopyrimidine ring of ThDP cycles through several ionization and tautomeric states during enzyme catalysis, but it is not fully understood which states are adopted during the individual steps of the catalytic cycle. Thiamin has been synthesized with labels selectively inserted into the C2 and C6′ positions, as well as into the amino group, creating [C2, C6′-13C2] thiamin and [N4′-15N] thiamin. Magic angle spinning (MAS) NMR spectroscopy has been employed to record the 13C and 15N chemical shift anisotropy (CSA) tensors for C2, C6′ and N4′ atoms. Our results indicate that the isotropic chemical shifts as well as the principal components of the 13C and 15N CSA tensors are very sensitive to the protonation states in these compounds and therefore permit to differentiate between the two ionization states, 4-aminopyrimidine (AP) and 4-aminopyrimidinium (APH+). Using Density Functional Theory (DFT), we have calculated the magnetic shielding anisotropy tensors of C2, C6′ and N4′, and found excellent agreement between the computed and the experimental tensors. Our findings indicate that MAS NMR spectroscopy in conjunction with DFT calculations is a sensitive probe of ionization states in the thiamin cofactor. The results of this study will serve as a guide for characterization of ionization and tautomeric states of thiamin in complexes with thiamin-dependent enzymes.
Keywords: solid-state NMR, magic angle spinning, MAS, thiamin, ROCSA, protonation states, ionization, protonation, chemical shift anisotropy, CSA, Density Functional Theory, DFT
INTRODUCTION
Thiamin diphosphate (ThDP) is an important coenzyme in a variety of enzyme systems that catalyze the transfer of aldehyde or acyl groups and the decarboxylation of α-keto acids. It is comprised of thiazolium and 4′-aminopyrimidine aromatic rings. Only recently has the participation of the 4′-aminopyrimidine group in enzyme catalysis gained wider acceptance.1,2 During catalysis by the thiamin diphosphate (ThDP) superfamily of enzymes, the 4′-aminopyrimidine moiety of ThDP can interconvert among four ionization/tautomeric states: the positively charged N1′-protonated 4′-aminopyrimidinium (APH+), and the three neutral forms, 4′-aminopyrimidine (AP), 1′, 4′-iminopyrimidine (IP) and the ylide (Yl) (Scheme 1). However, not all of these ionization/tautomeric forms have been clearly characterized yet. In particular, previous research has shown that APH+ and the ylide forms are not directly detectable by other spectroscopic methods, such as circular dichroism3 or solution NMR.7 Therefore, central questions concerning the molecular basis for activation of C2-H bond of ThDP and how the enzymes favorably stabilize the IP form during various steps of the catalytic cycle still remain unanswered.3–10 Solid-state NMR spectroscopy, which could provide atomic-level information in these large enzymes, would go the farthest in resolving such long-standing questions, due to the exquisite sensitivity of NMR chemical shifts to both the ionization and tautomeric states.11–13
Scheme 1.

Various ionization and tautomerization states of ThDP bound to enzymes
While isotropic chemical shifts available from solution NMR spectra are in many cases sufficient to infer the ionization and tautomeric states,14,15 chemical shift anisotropy (CSA) tensors recorded in the solid state provide much more detailed insight.16–20 Furthermore, Density Functional Theory (DFT) calculations of the magnetic shielding anisotropy tensors and their analysis in light of the experimental NMR results yield comprehensive understanding of the electronic structure of a molecule.21–23 Solid-state NMR measurements are often the only way to record chemical shifts and CSA tensors, for example, in large proteins, where solution lines are broadened beyond detection. The large molecular weight of the ThDP-dependent enzymes (varying between 160,000 and 250,000) has so far limited their solution NMR studies, while solid-state NMR spectroscopy presents a promising alternative for investigation of their catalytic mechanisms.
Despite this enormous potential of solid-state NMR methods, there have been no reports in the literature so far on the use of the CSA tensors as probes for various ionization/tautomeric forms of thiamin-dependent enzymes. Therefore, the focus of our work presented in the current report has been the investigation of 13C and 15N chemical shift anisotropy tensors for different ionization states of free thiamin in the solid state as a starting point for subsequent studies of 13C and 15N chemical shifts of several ionization and tautomeric states in a series of thiamin-enzyme complexes. This work represents the first step toward understanding the tautomeric and ionization states among which the protein-bound ThDP cofactor undergoes interconversion during the enzymatic cycle. We have synthesized thiamin molecules containing selective 13C and 15N isotopic labels at key positions of the 4′-aminopyrimidine ring (C6′, N4′) and of the thiazolium ring (C2). Protonated thiamin is in the 4′-aminopyrimidinium form (APH+) and unprotonated is in the 4′-aminopyrimidine form (AP), both on and off the enzymes. We have recorded the 13C and 15N CSA tensors for each ionization form by magic angle spinning (MAS) NMR spectroscopy and performed DFT calculations of the corresponding magnetic shielding anisotropy tensors. Our results indicate that the isotropic chemical shifts as well as the principal components of the 13C and 15N CSA tensors are very sensitive to the protonation states in thiamin and therefore permit to differentiate between the two forms (AP and APH+). The calculated magnetic shielding anisotropy tensors of C2, C6′ and N4′ exhibit excellent agreement with the experimental CSA tensors. Our findings indicate that MAS NMR spectroscopy in conjunction with DFT calculations is a sensitive probe of ionization states in the ThDP cofactor, and this approach can be applied to studies of the ionization and tautomeric forms in ThDP-dependent enzymes.
RESULTS AND DISCUSSION
Selective labeling
Thiamin (Th) and thiamin hydrochloride (Th•HCl) correspond to the AP and APH+ form of ThDP, respectively, in enzyme catalysis (Scheme 1). Since only the 4′-aminopyrimidinium ring undergoes change in ionization, we anticipated that between the two selectively labeled carbon atoms, C2 and C6′, the 13C CSA tensor of C6′ would be the prime reporter of the protonation state of the 4′-aminopyrimidine ring, along with the 15N CSA tensor of the N4′ atom. The chemical structures of Th and Th•HCl with selective 13C labels introduced at positions 2 and 6′, and with 15N label incorporated at N4′ are depicted in Figure 1.
Figure 1.

Chemical structures of Th•HCl and Th with 13C/15N enrichment at positions C2, C6′ and N4′.
Assignments of 13C Resonances in Different Ionization States of Thiamin
Since there are two 13C isotopically labeled sites (C2 and C6′) in both Th and Th•HCl, ambiguity arises in the resonance assignment of the 13C spectra of both samples in the absence of a priori knowledge about the two carbon chemical shifts. In order to resolve this ambiguity, we have measured the 1J13C-1H coupling constant for these two carbons by solution NMR, and compared the experimental results with the DFT computed J-couplings in Th•HCl and Th. The results are summarized in Table 1. Based on the DFT calculations, JC2-H is larger than JC6′-H by more than 22 Hz in Th•HCl and by 35 Hz in Th. The experimental J-couplings obtained from the proton-coupled 13C solution NMR spectra of Th•HCl and Th are in excellent agreement with the theoretical predictions, and immediately permit us to assign the two carbons in both ionization states; the corresponding isotropic solution chemical shifts are also indicated in Table 1.
Table 1.
Experimental and Computed JCH Couplings for 2,6′-13C/4′-15N Th•HCl and Th.
| Sample | JCH (Hz) | δiso (ppm) | ||
|---|---|---|---|---|
| Solution NMR | DFT | Solution NMR | ||
| Th•HCl | C2 | 214.1 | 208.8 | 154.6 |
| C6′ | 185.6 | 186.7 | 145.1 | |
|
| ||||
| Th | C2 | 213.6 | 210.0 | 157.8 |
| C6′ | 177.5 | 175.3 | 161.0 | |
13C and 15N Chemical Shift Anisotropies in Different Ionization States of Thiamin: Solid-State NMR
The 13C CSA tensors were recorded by two methods: i) slow-MAS spectra at three different spinning frequencies with the subsequent Herzfeld-Berger analysis to extract the CSA tensor components;24 ii) symmetry-based Recoupling Of Chemical Shift Anisotropy (ROCSA) method suitable for high MAS frequencies.25 The 15N CSA parameters were determined using ROCSA because of the much smaller 15N CSA tensor resulting in rather weak sideband intensities even at slow-MAS frequencies and thus prohibiting their accurate measurement.
The 13C spectra, slow-MAS and ROCSA, are illustrated in Figure 2. From Table 2, it is obvious that the two methods give consistent results for 13C CSA tensors in both ionization states. The C2 CSA tensor is not very different in the two ionization forms. The three principal components (δ11, δ22 and δ33) display a small shift of ca. 3 ppm in the upfield direction upon protonation, from Th to Th•HCl. As a result, the net change is only in the isotropic chemical shift, from 156.5 ppm to 153.4 ppm, while the reduced anisotropy δσ and the asymmetry parameter ησ remain nearly the same in the two states. As expected, the CSA parameters of C6′ are significantly different in the two protonation states. The C6′ isotropic chemical shift changes by about 12.5 ppm upfield upon protonation, from Th to Th•HCl. The principal components of the CSA tensor, δ11 and δ33 also move upfield by 17 and 15 ppm, respectively. The δ22 component displays only a small shift of 6 ppm. The resulting change in the reduced anisotropy δσ and the asymmetry parameter ησ is negligible- these remain the same within the error bars in the two protonation states, as the difference between the relative shifts of δiso and δ33 is small.
Figure 2.

13C ROCSA and slow-MAS spectra of Th•HCl and Th: experimental (black) and simulated (red). a) ROCSA spectrum of C2 of Th•HCl; b) ROCSA spectrum C6′ of Th•HCl; c) slow-MAS spectrum C2 and C6′ of Th•HCl at 2900 Hz rotor frequency; d) ROCSA spectrum of C2 of Th; e) ROCSA spectrum of C6′ of Th; and f) slow-MAS spectrum of C2 and C6′ of Th at 2075 Hz rotor frequency.
Table 2.
Experimental and Computed 13C and 15N CSA Parameters for 2,6′-13C/4′-15N Th•HCl and Th.
| Sample | Atom | Method | δiso (ppm) | δσ (ppm) | ησ | δ11 (ppm) | δ22 (ppm) | δ33 (ppm) |
|---|---|---|---|---|---|---|---|---|
| Th•HCl | C2 | Slow-MAS | 153.4 | −98.5 ± 1.6 | 0.59 ± 0.03 | 231.7 | 173.6 | 54.9 |
| ROCSA | 153.4 | −103.8 ± 5.0 | 0.61 ± 0.06 | 237.0 | 173.6 | 49.6 | ||
| DFT/with Cl− | 154.9 | −105.9 | 0.80 | 250.2 | 165.5 | 49.0 | ||
| DFT/without Cl− | 158.7 | 110.3 | 1.00 | 269.0 | 158.7 | 48.4 | ||
|
| ||||||||
| C6′ | Slow-MAS | 147.8 | −109.6 ± 4.5 | 0.78 ± 0.03 | 245.3 | 159.9 | 38.2 | |
| ROCSA | 147.8 | −111.3 ± 7.8 | 0.81 ± 0.08 | 248.5 | 158.4 | 36.5 | ||
| DFT/with Cl− | 153.7 | −114.0 | 0.87 | 260.3 | 161.1 | 39.7 | ||
| DFT/without Cl− | 149.7 | −110.2 | 0.88 | 253.3 | 156.3 | 39.5 | ||
|
| ||||||||
| N4′ | ROCSA | 116.5 | 74.9 ± 3.5 | 0.27 ± 0.09 | 191.4 | 89.2 | 68.9 | |
| DFT/with Cl− | 113.1 | 70.8 | 0.11 | 183.9 | 81.6 | 73.8 | ||
| DFT/without Cl− | 103.8 | 78.4 | 0.25 | 182.2 | 74.4 | 54.8 | ||
|
| ||||||||
| Th | C2 | Slow-MAS | 156.5 | −100.8 ± 1.2 | 0.66 ± 0.05 | 240.2 | 173.6 | 55.7 |
| ROCSA | 156.5 | −103.1 ± 6.4 | 0.62 ± 0.08 | 240.0 | 176.1 | 53.4 | ||
| DFT/with Cl− | 155.6 | −107.0 | 0.75 | 249.2 | 169.0 | 48.6 | ||
| DFT/without Cl− | 162.8 | −114.9 | 0.80 | 266.2 | 174.3 | 47.9 | ||
|
| ||||||||
| C6′ | Slow-MAS | 160.4 | −107.8 ± 2.2 | 0.92 ± 0.03 | 263.9 | 164.7 | 52.6 | |
| ROCSA | 160.4 | −108.7 ± 5.6 | 0.93 ± 0.07 | 265.3 | 164.2 | 51.7 | ||
| DFT/with Cl− | 162.2 | 109.2 | 0.90 | 271.4 | 156.7 | 58.5 | ||
| DFT/without Cl− | 163.4 | −108.6 | 0.99 | 271.5 | 163.9 | 54.8 | ||
|
| ||||||||
| N4′ | ROCSA | 91.2 | 59.6 ± 2.6 | 0.28 ± 0.06 | 150.8 | 69.7 | 53.1 | |
| DFT/with Cl− | 98.2 | 59.5 | 0.51 | 157.7 | 83.6 | 53.3 | ||
| DFT/without Cl− | 70.2 | −56.2 | 0.90 | 123.5 | 73.0 | 14.0 | ||
The 15N ROCSA spectra are presented in Figure 3. The N4′ CSA parameters are found to be even more sensitive to the protonation state than those for C6′. The isotropic chemical shift, δiso, differs by more than 25 ppm in the two forms. Of the three principal components of the CSA tensor, the δ11 component of N4′ CSA tensor is observed to be the most sensitive to the protonation, moving downfield by ca. 40 ppm from Th to Th•HCl. As a result, the reduced anisotropy, δσ, increases by 15 ppm in Th•HCl. The δ22 and δ33 principal components also display considerable downfield shift of 20 and 16 ppm, respectively. We note that, it is not possible to accurately measure ησ in our experiments, because this parameter has a strong correlation with the line-broadening parameter used in fitting the experimental 15N ROCSA lineshapes with nearly axial symmetry. As a consequence, the position of both δ22 and δ33 is not as well defined as that of δ11 especially when ησ is in the range of 0.1 – 0.4.26
Figure 3.
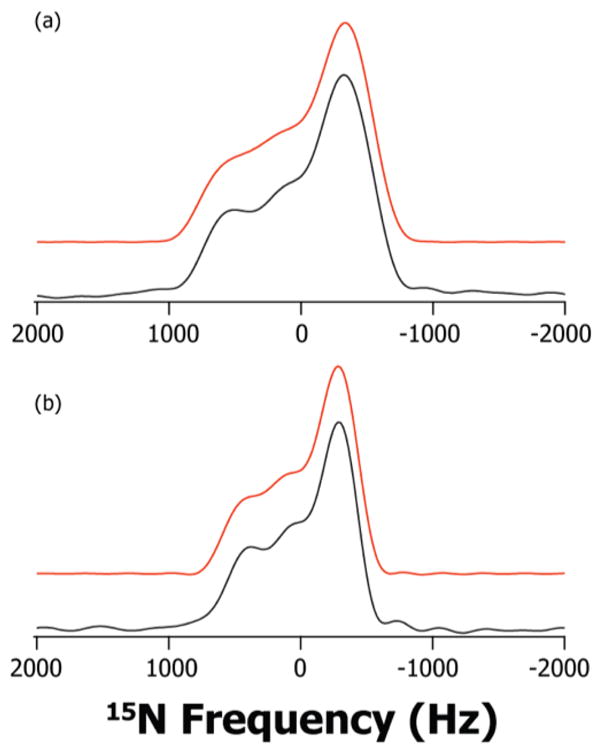
15N ROCSA spectra of Th•HCl and Th: experimental (black) and simulated (red). a) N4′ of Th•HCl; b) N4′ of Th.
13C and 15N Chemical Shift Anisotropies in Thiamin Hydrochloride: Density Functional Theory
Experimental results and DFT calculations are summarized in Table 2. DFT calculations performed on the geometry-optimized structure of Th•HCl predict the experimental trend very well in the following respects: i) C6′ is more shielded than C2; and ii) δσ of C6′ is larger than that of C2.
The calculations predict the principal components of the C2 CSA tensor to within 9 ppm. The calculated isotropic chemical shift of C2, 154.9 ppm, agrees well with the experimental value of 153.4 ppm. Maximum deviation is observed for δ11 (13 ppm) and δ22 (−8 ppm) with δ33 showing no deviation at all. For C6′, the computed principal components of the magnetic shielding anisotropy tensor are in even better agreement with experiment, with the difference being only of the order of 7 ppm. Since for C6′ the three computed principal components deviate in the same direction (downfield) with respect to the experimental values, the isotropic chemical shift also differs by 6 ppm.
For N4′ CSA, the agreement between experiment and theory is excellent, and even tighter agreement is observed than for the 13C tensors. The average deviation of the three principal components is less than 7 ppm. The computed δ11 and δ22 are 7.5 ppm upfield, while the calculated δ33 is downfield by 5 ppm compared with experiment. The calculated isotropic chemical shift is therefore only 3.4 ppm different from the experimental value.
We also addressed the effect of the presence of two chloride anions in the DFT calculations. We have performed an additional round of calculations with geometry optimization on the twice positively charged Th•HCl without the two chloride anions. The following trends were observed:
The predicted C6′ CSA tensor is in much better agreement with experiment without the two chloride anions, with the average deviations between experiment and theory of less than 4 ppm for the three principal components. The predicted isotropic chemical shift of 149.7 ppm is in much better agreement with the experimental value of 147.8 ppm.
At the same time, the computed principal components of the C2 CSA tensor show greater deviations from the experimental values (by more than 20 ppm on average). As observed in the calculations with chloride anions, this deviation can be largely attributed to changes in δ11 (30 ppm) and to a lesser extent to δ22 (−15 ppm) with δ33 showing no deviation at all.
The computed N4′ CSA tensor deviates more from the experimental values without the chloride anions with an average difference of 13 ppm for the individual principal components. Both δ22 and δ33 display the maximum deviation from experiments as well as from their computed values in the presence of chloride ions. The δ11 component of N4′ remains relatively unchanged upon the removal of the chloride ions.
13C and 15N Chemical Shift Anisotropies in Thiamin: Density Functional Theory
For the DFT calculations on thiamin, we used a starting model that was derived from the Th•HCl X-ray structure by removing the HCl, and performed further geometry optimization to obtain the final structure for magnetic shielding anisotropy tensor calculations.
As in the case of Th•HCl, DFT calculations on the geometry-optimized Th structure correctly predict that C6′ is more deshielded than C2, which is consistent with the experimental results. The calculated reduced anisotropy, δσ, of C6′ is higher than that of C2, as observed in experiments.
The calculated principal components of the C2 CSA tensor agree with their experimental values to within 7 ppm. As in the Th•HCl calculation, δ11 of C2 shows greater deviation from experiment (9 ppm downfield), while δ22 and δ33 are upfield compared with experiment. As a result, the calculated isotropic chemical shift is different by only 1 ppm from experiment. For C6′, the average deviation of the calculated principal components is also less than 7 ppm. δ11 and δ33 are downfield compared with experiment (by 6.2 and 6.8 ppm, respectively), while δ22 is upfield by 7.5 ppm. This results in the calculated isotropic chemical shift (162.2 ppm) in close agreement with the experimental value (160.4 ppm).
The calculated N4′ CSA components are also in close agreement with experiment, to within 9 ppm. As observed in the Th•HCl calculations, both δ11 and δ22 contribute to the difference between calculations and experiment with δ33 showing no deviation.
The perturbation of the carbon CSA tensors due to the removal of the chloride anions in Th is nearly the same as in Th•HCl. The C6′ CSA tensor is observed to agree more closely with the experimental values, while C2 CSA tends to deviate to a greater extent from the experiment; nevertheless, the agreement between theory and experiment is still very good.
Conversely, the absence of the two chloride anions has a great effect on the N4′ CSA tensor in Th. The δ11 and δ33 components differ from their experimental values by −27 and −39 ppm, respectively. As a result, the computed isotropic chemical shift is off by more than 20 ppm upfield. Moreover, the calculations predict an almost rhombic tensor (ηδ = 0.90), whereas in experiments the tensor is close to axial (ηδ = 0.28).
The sensitivity of the C2 and N4′ CSA tensors to the absence of the chloride ions in both AP and APH+ forms can be attributed to the chloride ion located close to the sulfur atom in the thiazolium ring (see Figure S1 of the Supporting Information). The interatomic C2-Cl and N4′-Cl distances are ca. 3.7 Å and 3.2 Å, respectively, in the X-ray structure as well as in the geometry-optimized structures used in our shielding tensor calculations.
In summary, the calculations predict accurately the C2 and N4′ CSA tensors in the presence of the chloride anions, and the C6′ CSA tensor in the absence of chloride anions. This trend is observed to be the same for both Th as well as Th•HCl. The correlation between the computed and the experimental CSA components is shown in Figure 4 for both 13C and 15N atoms. As discussed, the correlation between the 15N CSA predictions and the experimental values is much better in the presence of the chloride anions than without chloride anions (see Figure 4b and 4c, respectively).
Figure 4.

Correlation between the principal components δii of the calculated (DFT) and experimental (NMR) chemical shift anisotropy tensors of Th•HCl and Th: a) 13C of C2 and C6′; b) 15N of N4′ with chloride anions; and c) 15N of N4′ without chloride anions. The solid lines in both figures indicate perfect agreement (δcal = δexp), and the dotted lines are the least-square fits: (a) δcal = 1.05 * δexp − 4.88, R2 = 0.99; (b) δcal = 0.94 * δexp + 7.86, R2 = 0.98; and (c) δcal = 1.03 * δexp − 20.28, R2 = 0.94.
In an effort to determine whether there is any marked difference in the orientation of CSA tensors of the key atoms in the two ionization states, we calculated the C6′ and N4′ tensor orientations with respect to the molecular frame. We observed that the N4′ and C6′ CSA orientations are relatively unchanged in both AP and APH+ forms in the presence of the chloride anions. However, in the absence of the chloride ions, the N4′ CSA orientation changes significantly. Further details are presented in the Supporting Information.
CONCLUSIONS
Our work demonstrates that the 13C and the 15N CSA tensors of C2, C6′ and N4′ atoms are sensitive probes of ionization states of thiamin. Both the principal components of the CSA tensors and the isotropic chemical shifts exhibit significant changes upon protonation/deprotonation. The Density Functional Theory calculations are in excellent agreement with the experimental findings. The predicted C6′ and N4′ isotropic chemical shifts are within 6.0 ppm and 7.0 ppm of the experimental values, respectively. Considering that the isotropic chemical shift difference of these atoms in the two ionization states far exceeds this range of deviation, we conclude that the DFT calculation of CSA parameters can be used in conjunction with MAS NMR measurements for characterization of the different ionization states of thiamin. Our conclusions can be extended to thiamin diphosphate (ThDP), the biologically active form of thiamin since the β-diphosphate moiety in ThDP is located too far from the heterocyclic rings to make any contribution to the chemical shifts, nor does the diphosphate come near to the key atoms under consideration in the enzyme-bound forms. The results of this work are expected to guide future investigations of thiamin ionization and tautomeric states in thiamin-dependent enzymes, which will address some of the remaining open questions about their enzymatic mechanisms.
EXPERIMENTS AND METHODS
Synthesis of [C2, C6′ – 13C2] thiamin chloride hydrochloride is reported elsewhere.28
Synthesis of [N4′ – 15N] thiamin chloride hydrochloride
Compounds I, II & III (ref. Scheme S1) were synthesized with some minor modifications to the previously described methods.29 Compound IV was synthesized according to a method described by Uray et al.30; and compounds V, VI & VII were synthesized with minor modifications to the method described by Contant et al.31 and Nicewonger et a.l32 The details are described in Supporting Information.
[N4′– 15N] Thiamin and [C2, C6′-13C2] Thiamin were prepared by neutralization of [N4′ – 15N] thiamin chloride hydrochloride and [C2, C6′-13C2] thiamin chloride hydrochloride respectively under anhydrous conditions (See Supporting Information section).
[N4′– 15N] Thiamin was also prepared using a chemo-enzymatic approach by the Begley group.55
Preparation of solid-state NMR samples
Crystalline [C2, C6′-13C2] thiamin chloride hydrochloride (10.4 mg, 0.031 mmol) or [N4′-15N] thiamin chloride hydrochloride (9.0 mg, 0.027 mmol) were packed into 3.2 mm MAS rotors and used in the subsequent MAS NMR experiments as models for the N1′ protonated form (Figure 1a, 1b).
[C2, C6′-13C2] thiamin (8.0 mg, 0.027 mmol) or [N4′-15N] thiamin (6.7 mg, 0.022 mmol) were packed into 3.2 mm MAS rotors and used in the subsequent MAS NMR experiments as models for the N1′ unprotonated form (Figure 1c, 1d).
Solid-state NMR spectroscopy
All 13C and 15N MAS spectra presented in this work were acquired at 9.4 T (400.17 MHz 1H Larmor frequency) on a Tecmag Discovery spectrometer outfitted with a 3.2 mm wide bore Varian HXY T3 probe. The actual sample temperature includes a correction because of sample spinning which is about +5° C for 10 kHz spinning frequency. The temperature was calibrated for this probe at different MAS frequencies using a PbNO3 temperature sensor33, and the actual temperature at the sample was 18° C maintained to within ±1 °C throughout the experiments using the Tecmag temperature controller. 13C chemical shifts were referenced to the downfield peak of adamantane (38.56 ppm with respect TMS). 15N chemical shifts were referenced to ammonium chloride (39.2 ppm with respect to liquid NH3).
The low spinning-frequency 13C CPMAS spectra were acquired on Th•HCl at ωr/2π = 2900, 4070 and 5980 Hz. The MAS frequency was controlled with Tecmag MAS controller: within ±5 Hz at 2900 kHz, while at 4070 and 5980 Hz the variation was up to ±100 Hz. The standard CP sequence was used with 1 ms contact time; the 1H radio frequency field strength was 60 kHz, the 13C field was linearly ramped 80–100% with the center of the ramp being 50 kHz. The 1H 90° pulse width was 2.5 μs. The same experimental parameters were used at every spinning frequency. The 1H decoupling was performed using TPPM34 with 100 kHz 1H radio frequency field strength maintained for the duration of the acquisition period, 25.6 ms. The CPMAS spectra were acquired with either 32 scans (at ωr/2π = 2900 Hz) or with 4 scans (ωr/2π = 4070 and 5080 Hz); the recycle delay was 10 s. The spectra were processed without any line broadening, and the intensities of each sideband were extracted for the two carbons in the 2, 6′-13C Th•HCl sample.
The low spinning-frequency 13C CP-MAS spectra of 2, 6′-13C Th were acquired at ωr/2π = 2075, 3810, and 5973 Hz. The rest of the experimental conditions were the same as for the thiamin hydrochloride sample. At all MAS frequencies, 16 scans were used with 10 sec pulse delay.
The Herzfeld-Berger analysis24 was performed to calculate the CSA parameters from the intensities of the spinning side-band envelopes for the two carbon atoms in both forms of thiamin.
The 15N CP-MAS NMR spectra of 4′-15N Th•HCl and Th were acquired at the spinning frequency of 10 kHz. The standard CP sequence was used with 1 ms contact; the 1H radio frequency field strength was 60 kHz, the 15N field was linearly ramped 80–100% with the center of the ramp being 50 kHz.100 kHz. TPPM decoupling was applied during the acquisition time (30 ms); 1H radio frequency field strength was 100 kHz. 32 scans were acquired with a recycle delay of 10 s.
ROCSA (Recoupling Of Chemical Shift Anisotropy) pulse sequence25 was used to recouple the 13C and 15N CSA interaction at MAS frequencies above 10 kHz. The 13C ROCSA spectra were collected at the MAS frequency of 12 kHz. The 13C radio frequency field strength during the ROCSA period was 51.3 kHz, and that of the 1H decoupling was 122 kHz. The 15N ROCSA spectra were collected at the MAS frequency of 10 kHz. The 15N rf field strength during the ROCSA period was 42.8 kHz and that of the 1H decoupling was 100 kHz. 16 transients were added up with a recycle delay of 10 sec for each t1 point. 32 and 24 t1 points were acquired using the States method35 for 15N and 13C ROCSA spectra, respectively. The CSA dimension was zero-filled to 512 points; no apodization was applied prior to the Fourier transformation.
CSA tensor convention
In this work, we use two conventions for reporting CSA tensors. 1) Standard convention with the three principal components defined as follows: δ11 ≥ δ22 ≥ δ33. 2) Haeberlen-Mehring-Spiess convention where the CSA tensor is characterized by three parameters: i) isotropic chemical shift (δiso) ii) reduced anisotropy (δσ) iii) asymmetry parameter (ησ). The relation between the two conventions is as follows: The isotropic chemical shift is defined as δiso = (δ11 + δ22 + δ33)/3. The reduced anisotropy (δσ) and the asymmetry parameter (ησ) are defined as: δσ = δ11 − δiso and ησ = (δ22 − δ33)/δσ if |δ11 − δiso| ≥ |δ33 − δiso| ≥ |δ22 − δiso| (positive anisotropy), or δσ = δ33 − δiso and ησ = (δ22 − δ11)/δσ if |δ33 − δiso| ≥ |δ11 − δiso| ≥ |δ22 − δiso| (negative anisotropy). The asymmetry parameter is always in the range between 0 and 1.
NMR spectra simulations
The slow spinning frequency 13C MAS spectra were simulated in SIMPSON36 using the CSA parameters obtained from HB analysis. An exponential line broadening of 88 Hz was applied to the simulated spectra to match the natural line width. SIMPSON was also used to simulate 13C and 15N ROCSA lineshapes. The MINUIT procedure embedded in SIMPSON was applied for fitting the experimental ROCSA spectra using three variable parameters: δσ, ησ and the line-broadening parameter. The uncertainties for the fitting parameters δσ and ησ were determined from the corresponding values of δσ and ησ giving .
Density Functional Theory calculations
Quantum chemistry calculations were carried out using the DFT method in Gaussian03.37 Geometry optimization38 was employed using the B3LYP functional39–41 and tzvp bais set.42,43 13C/15N shielding tensor calculations were performed using the same functional and the basis set and GIAO approximation.44–47 The starting structure for Th•HCl geometry optimization was obtained from the X-ray structure of Th•HCl.48 The starting structure for the unprotonated thiamin used in the subsequent geometry optimization was derived from the crystal structure of Th•HCl by removing the HCl. Two sets of calculations were performed for both Th•HCl and Th: one with chloride anions, the other one- without the anions (charge +1 for Th and charge +2 for Th•HCl). The J-coupling constant calculations were performed using B3LYP and IGLO-III basis set49 with the polarizeable continuum model (PCM).50–52 Most of the calculations were performed under GridChem computational resources and services, Computational Chemistry Grid53 (www.gridchem.org)
The 13C chemical shifts were referenced with respect to adamantane (the downfield resonance at 38.56 ppm) whose absolute magnetic shielding tensor was calculated at the same level of theory as those for the thiamin models. The 15N chemical shift was referenced with respect to 244.6 ppm according to the previously described procedure.54
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIH Grants P20RR017716 to TP, DK44083 to TB and GM050380 to FJ). GridChem is acknowledged for computational resources (www.gridchem.com). The authors thank Lex Vega for helpful discussions.
Footnotes
Protocols for synthesis of isotopically labeled thiamin and thiamin chloride hydrochloride, discussion and illustration of relative orientation of C6′ and N4′ CSA tensors, and full citation of reference 37. This material is available free of charge via the Internet at http://pubs.acs.org
Contributor Information
Sivakumar Paramasivam, Email: siva@udel.edu.
Anand Balakrishnan, Email: anandbk@andromeda.rutgers.edu.
Olga Dmitrenko, Email: odmitr@udel.edu.
Amy Godert, Email: agodert@wells.edu.
Tadhg P. Begley, Email: begley@chem.tamu.edu.
Frank Jordan, Email: frjordan@andromeda.rutgers.edu.
Tatyana Polenova, Email: tpolenov@udel.edu.
References
- 1.Jordan F. Nat Prod Rep. 2003;20(2):184–201. doi: 10.1039/b111348h. [DOI] [PubMed] [Google Scholar]
- 2.Schellenberger A. Biochim Biophys Acta. 1998;1385(2):177–86. doi: 10.1016/s0167-4838(98)00067-3. [DOI] [PubMed] [Google Scholar]
- 3.Nemeria N, Chakraborty S, Baykal A, Korotchkina LG, Patel MS, Jordan F. PNAS. 2007;104(1):78–82. doi: 10.1073/pnas.0609973104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nemeria N, Korotchkina L, McLeish MJ, Kenyon GL, Patel MS, Jordan F. Biochemistry. 2007;46(37):10739–44. doi: 10.1021/bi700838q. [DOI] [PubMed] [Google Scholar]
- 5.Nemeria N, Baykal A, Joseph E, Zhang S, Yan Y, Furey W, Jordan F. Biochemistry. 2004;43(21):6565–75. doi: 10.1021/bi049549r. [DOI] [PubMed] [Google Scholar]
- 6.Jordan F, Nemeria NS, Zhang S, Yan Y, Arjunan P, Furey W. J Am Chem Soc. 2003;125(42):12732–8. doi: 10.1021/ja0346126. [DOI] [PubMed] [Google Scholar]
- 7.Kern D, Kern G, Neef H, Tittmann K, Killenberg-Jabs M, Wikner C, Schneider G, Hubner G. Science. 1997;275(5296):67–70. doi: 10.1126/science.275.5296.67. [DOI] [PubMed] [Google Scholar]
- 8.Kaplun A, Binshtein E, Vyazmensky M, Steinmetz A, Barak Z, Chipman DM, Tittmann K, Shaanan B. Nat Chem Biol. 2008;4(2):113–8. doi: 10.1038/nchembio.62. [DOI] [PubMed] [Google Scholar]
- 9.Meshalkina LE, Kochetov GA, Brauer J, Hubner G, Tittmann K, Golbik R. Biochem Biophys Res Commun. 2008;366(3):692–7. doi: 10.1016/j.bbrc.2007.11.180. [DOI] [PubMed] [Google Scholar]
- 10.Tittmann K, Neef H, Golbik R, Hubner G, Kern D. Biochemistry. 2005;44(24):8697–700. doi: 10.1021/bi050522x. [DOI] [PubMed] [Google Scholar]
- 11.Reynolds WF, Peat IR, Freedman MH, Lyerla JR. J Am Chem Soc. 1973;95(2):328–331. doi: 10.1021/ja00783a006. [DOI] [PubMed] [Google Scholar]
- 12.Rabenstein DL, Sayer TL. J Magn Reson. 1976;24(1):27–39. [Google Scholar]
- 13.Surprenant HL, Sarneski JE, Key RR, Byrd JT, Reilley CN. J Magn Reson. 1980;40(2):231–243. [Google Scholar]
- 14.Bachovchin WW, Roberts JD. J Am Chem Soc. 1978;100(26):8041–8047. [Google Scholar]
- 15.Sudmeier JL, Bradshaw EM, Haddad KEC, Day RM, Thalhauser CJ, Bullock PA, Bachovchin WW. J Am Chem Soc. 2003;125(28):8430–8431. doi: 10.1021/ja034072c. [DOI] [PubMed] [Google Scholar]
- 16.Naito A, Ganapathy S, Akasaka K, McDowell CA. J Chem Phys. 1981;74(6):3190–3197. [Google Scholar]
- 17.Haberkorn RA, Stark RE, Van Willigen H, Griffin RG. J Am Chem Soc. 1981;103(10):2534–2539. [Google Scholar]
- 18.Smith SO, Farr-Jones S, Griffin RG, Bachovchin WW. Science. 1989;244(4907):961–964. doi: 10.1126/science.2499045. [DOI] [PubMed] [Google Scholar]
- 19.Gu Z, McDermott A. J Am Chem Soc. 1993;115(10):4282–4285. [Google Scholar]
- 20.Gu Z, Zambrano R, McDermott A. J Am Chem Soc. 1994;116(14):6368–6372. [Google Scholar]
- 21.Bagno A, Scorrano G. J Phys Chem. 1996;100(5):1545–1553. [Google Scholar]
- 22.Ooms KJ, Bolte SE, Smee JJ, Baruah B, Crans DC, Polenova T. Inorg Chem. 2007;46(22):9285–9293. doi: 10.1021/ic7012667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ooms K, Polenova T, Shough AM, Doren DJ, Nash MJ, Lobo RF. J Phys Chem C. 2009;113(24):10477–10484. [Google Scholar]
- 24.Herzfeld J, Berger AE. J Chem Phys. 1980;73(12):6021–6030. [Google Scholar]
- 25.Chan JCC, Tycko R. J Chem Phys. 2003;118(18):8378–8389. [Google Scholar]
- 26.Wylie BJ, Franks WT, Rienstra CM. J Phys Chem B. 2006;110(22):10926–10936. doi: 10.1021/jp060507h. [DOI] [PubMed] [Google Scholar]
- 27.Perrin DD, Armarego WLF, Perrin DR. Purification of Laboratory Chemicals. 2. Pergamon Press Inc; Elmsford, N.Y: 1980. [Google Scholar]
- 28.Chakraborty S. PhD Thesis. Rutgers, the State University of New Jersey; Newark: 2006. [Google Scholar]
- 29.Todd AR, Bergel F. J Chem Soc (Resumed) 1937:364–367. [Google Scholar]
- 30.Uray G, Celotto C, Ibovnik A, Zoltewicz JA. J Org Chem. 1989;54(16):3941–3945. [Google Scholar]
- 31.Contant P, Forzy L, Hengartner U, Moine G. Helv Chim Acta. 1990;73(5):1300–1305. [Google Scholar]
- 32.Nicewonger R, Costello CA, Begley TP. J Org Chem. 1996;61(12):4172–4174. doi: 10.1021/jo960234u. [DOI] [PubMed] [Google Scholar]
- 33.Neue G, Dybowski C. Solid State Nucl Magn Reson. 1997;7(4):333–336. doi: 10.1016/s0926-2040(96)01291-x. [DOI] [PubMed] [Google Scholar]
- 34.Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG. J Chem Phys. 1995;103(16):6951–6958. [Google Scholar]
- 35.States DJ, Haberkorn RA, Ruben DJ. J Magn Reson. 1982;48(2):286–292. [Google Scholar]
- 36.Bak M, Rasmussen JT, Nielsen NC. J Magn Reson. 2000;147(2):296–330. doi: 10.1006/jmre.2000.2179. [DOI] [PubMed] [Google Scholar]
- 37.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA. Gaussian, Inc. Wallingford, CT: 2004. [Google Scholar]
- 38.Schlegel HB. J Comput Chem. 1982;3(2):214–218. [Google Scholar]
- 39.Becke AD. Phys Rev A. 1988;38(6):3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 40.Becke AD. J Chem Phys. 1993;98(7):5648–5652. [Google Scholar]
- 41.Lee CT, Yang WT, Parr RG. Phys Rev B. 1988;37(2):785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 42.Schafer A, Horn H, Ahlrichs R. J Chem Phys. 1992;97(4):2571–2577. [Google Scholar]
- 43.Schafer A, Huber C, Ahlrichs R. J Chem Phys. 1994;100(8):5829–5835. [Google Scholar]
- 44.Wolinski K, Hinton JF, Pulay P. J Am Chem Soc. 1990;112(23):8251–8260. [Google Scholar]
- 45.McWeeny R. Phys Rev. 1962;126(3):1028. [Google Scholar]
- 46.Wolinski K, Sadlej AJ. Mol Phys. 1980;41(6):1419– 1430. [Google Scholar]
- 47.Ditchfield R. Mol Phys. 1974;27(4):789– 807. [Google Scholar]
- 48.Te RL, Griesser UJ, Morris KR, Byrn SR, Stowell JG. Cryst Growth Des. 2003;3(6):997–1004. [Google Scholar]
- 49.Kutzelnigg W, Fleischer U, Schindler M. The IGLO-Method: Ab Initio Calculation and Interpretation of NMR Chemical Shifts and Magnetic Susceptibilities. Vol. 23 Springer Verlag; 1990. [Google Scholar]
- 50.Cossi M, Barone V, Cammi R, Tomasi J. Chem Phys Lett. 1996;255(4–6):327–335. [Google Scholar]
- 51.Miertus S, Tomasi J. Chem Phys. 1982;65(2):239–245. [Google Scholar]
- 52.Miertus S, Scrocco E, Tomasi J. Chem Phys. 1981;55(1):117–129. [Google Scholar]
- 53.Dooley R, Milfeld K, Guiang C, Pamidighantam S, Allen G. J Grid Comput. 2006;4(2):195–208. [Google Scholar]
- 54.Brender JR, Taylor DM, Ramamoorthy A. J Am Chem Soc. 2001;123(5):914–922. doi: 10.1021/ja001980q. [DOI] [PubMed] [Google Scholar]
- 55.Melnick JS, Sprinz KI, Reddick JJ, Kinsland C, Begley TP. Bioorganic & Medicinal Chemistry Letters. 2003;13(22):4139–4141. doi: 10.1016/j.bmcl.2003.07.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.