

Abstract
Whole mount in situ hybridization is a very informative approach for defining gene expression patterns in embryos. The in situ hybridization procedures are lengthy and technically demanding with multiple important steps that collectively contribute to the quality of the final result. This protocol describes in detail several key quality control steps for optimizing probe labeling and performance. Overall, our protocol provides a detailed description of the critical steps necessary to reproducibly obtain high quality results. First, we describe the generation of digoxygenin (DIG) labeled RNA probes via in vitro transcription of DNA templates generated by PCR. We describe three critical quality control assays to determine the amount, integrity and specific activity of the DIG-labeled probes. These steps are important for generating a probe of sufficient sensitivity to detect endogenous mRNAs in a whole mouse embryo. In addition, we describe methods for the fixation and storage of E8.5-E11.5 day old mouse embryos for in situ hybridization. Then, we describe detailed methods for limited proteinase K digestion of the rehydrated embryos followed by the details of the hybridization conditions, post-hybridization washes and RNase treatment to remove non-specific probe hybridization. An AP-conjugated antibody is used to visualize the labeled probe and reveal the expression pattern of the endogenous transcript. Representative results are shown from successful experiments and typical suboptimal experiments.
Protocol
1. Riboprobe generation via in vitro transcription
- Preparing PCR products for in vitro transcription templates.
- Designing PCR primers with phage transcription promoter sequences in their 5' ends. Note: The promoter sequence added to 5'end of the sense-strand PCR primer will be used for transcribing the sense probe, and the promoter sequence added to 5'end of the antisense PCR primer will be used for synthesizing the anti-sense probe1-3. We have not detected any difference in transcription efficiency between templates that contain only the core promoter sequences vs. templates that contain the core promoter plus additional 5' sequences. This method has been used with the T3, T7 and SP6 phage promoters. Sequences that are highly conserved among gene family members or highly repetitive sequences should be avoided since they can result in non-specific hybridization and thereby increase background staining. Probes with high GC content will have limited digoxigenin-rUTP incorporation and DNA template sequences with runs of T residues will limit the incorporation of dig-UTP due to steric interference between the digoxigenin molecules. The probe length can range from 300bp to 1kb. But as long as the above criteria are met, usually longer probes have higher specific activities.
- The template for the PCR can be a plasmid clone, genomic clone or genomic DNA. Typically we purchase full-length EST clones (e.g. from ATCC or Open Biosystems) to use as PCR templates. Amplify the template DNA using standard PCR procedures to produce a PCR product containing the promoter sequences. To make enough probe templates to support several probe synthesis reactions we typically set up 8 x 50μl PCRs. The reactions are pooled for the next step. In general a few large volume PCRs or more small reactions (such as the 50μl reactions we use) are sufficient to generate enough template for several probe synthesis reactions.
- To remove contaminating proteins, especially traces of RNase in plasmid preparations, digest each 100μl of PCR reaction with 1μl of 20mg/ml Proteinase K at 55° C for at least 30 minutes. All reagents and other items used after this Proteinase K digestion should be RNase free and specifically dedicated for RNA work.
- During the Proteinase K digestion, run a sample of the PCR reaction on a gel to check integrity of the PCR product. We use acrylamide mini gels to resolve smaller PCR products (less than 600-700 bp) and agarose gels to resolve larger PCR products (>600-700 bp) for quality control analysis. If you see multiple bands on the gel, we recommend optimizing the PCR reaction so that you obtain a single PCR product of the correct size.
- Thoroughly extract the Proteinase K digested PCR reaction with one equal volume of a phenol/chloroform (1:1) mixture, followed by an extraction with one equal volume of chloroform. Mix at each step by vortexing the tube for at least 30 seconds. These steps must be performed in a chemical fume hood.
- Precipitate the purified PCR product by adding 0.1 volume of 3M sodium acetate and 2 volumes of 100% ethanol. Leave in -20° C for at least 30 minutes.
- Spin for 5 minutes at 13,000 rpm in a microcentrifuge, remove supernatant, and wash with 70% Ethanol. Allow the DNA pellet to dry completely.
- Resuspend DNA in suitable volume (e.g. 50μl) of 1xTE. Measure the DNA concentration using a fluorometer.
- Determine the nucleotide sequence of the each PCR template prep using primers that correspond to the phage promoter sequences or to an internal sequence within the probe. This is a critical quality control step that ensures that the probe template is of the correct sequence.
- The DNA is ready to serve as a template for in vitro transcription. Store the DNA template at 4° C.
- in vitro transcription using the PCR product as a template.
- Set up the an RNA transcription reaction in a final volume of 50μl. The transcription reaction includes 500 - 1000 ng of template DNA, 5μl of 10X Transcription Buffer (with 100mM DTT), 10μl 2.5mM dig-NTP mix, 3μl (50˜90 units) of RNA Polymerase, 1μl Alpha-32P CTP (up to 6 months old) and the remainder of the volume is made up by adding diethylpyrocarbonate treated water. The 2.5mM dig-NTP mix is typically made up as a 40μl working stock containing 10μl 10mM CTP, 10μl 10mM GTP, 10μl 10mM ATP, 6.5μl 10mM UTP, 3.5μl 10mM digoxigenin-11 UTP. In assembling the transcription reaction, be sure that all of the reaction components (except for the enzyme) are warmed to room temperature. Mix the DNA and water first, then add the reaction buffer, mix and then add the other components otherwise the spermidine in the transcription buffer will precipitate the DNA. Incubate for 2 hrs at the temperature recommended for the RNA polymerase used. Note: the α-32P is added to the reaction to allow you to determine the proportion of the starting nucleotide pool that is incorporated into the RNA probe. This is an important measure of the efficiency of the reaction. All of the following steps follow procedures for handling 32P. An alternative method for measuring the amount of RNA probe synthesized is to use a small volume spectrophotometer. This avoids the use of 32P labeling (see notes in step 1.3.1)
- During last few minutes of incubation, prepare the Quick Spin Column (Roche) according the manufacturer's instructions.
- After incubation, add 1μl of RNase free DNase I and incubate at 37° C for 10 min.
- Dilute to 100μl with 50μl of Reaction Dilution buffer (20mM Tris pH7.5, 1% SDS, 20mM EDTA, 100mM NaCl (RNase free, make a 50ml stock and store at room temperature)). Take 1μl (usually into 4μl of 1x TE) for scintillation counting of the total starting counts and for gel analysis.
- Apply the rest of the reaction onto the center of the column bed and spin at 1100 X g for 4 min in a table top clinical centrifuge. After the spin your RNA probe is in the collection tube.
- Add 2 volumes of 100% ethanol into the eluted reaction. Mix well and keep on ice for 5min (or in -20° C or 30 min).
- Spin the tube in a microcentrifuge at maximum rpm for 5 minutes to pellet the RNA probe.
- Completely remove the supernatant by pipette and let the pellet air dry. Do not let the pellet over-dry because it will be very difficult to dissolve it.
- Dissolve the RNA probe with 50 μl - 100 μl 1xTE or DEPC-H2O. Take a 1 μl sample for determining percent incorporation and for analysis on a denaturing acrylamide gel. Adjust your probe concentration to 100 ng/μl according to the estimated yield.
- Probes that have passed all three quality controls can be then stored in -20° C for 6 to12 months. We usually exhaust the probe stock within 12 months. It is very likely that probes can be stored for very extended periods at -20C.
- Quality assessment of the riboprobe - measuring probe yield.
- To estimate the probe yield, divide the counts recovered after the spin column by the counts in the reaction before the spin column (measured with the samples taken in the probe synthesis protocol). For this reaction, 100% incorporation = 33 μg. A successful reaction usually gives a 15-50% yield. Probes with less than 15% incorporation (<5 μg yield) should not be used. Note: An alternative method for measuring probe yield is to use a small volume spectrophotometer or fluorometer to directly measure the amount of RNA present after spin column chromatography. This alternative approach avoids the use of 32P trace labeling. We have measured riboprobe concentration using a BioTek Epoch Microplate Spectrophotometer. A comparison of the values obtained from the spectrophotometer to the estimations of riboprobe mass from the 32P incorporation showed that the two methods give comparable results using a similar small volume (2 μl) sample from the final probe preparation.Nearly always a very low or no yield is due to RNase contamination originating from the plasmid preparation used as template for the PCR. Be sure to treat your plasmid DNA and PCR product with proteinase K as described in step 1.1.3.
- Quality assessment of the riboprobe - measuring specific activity.
- To measure the extent of digoxigenin - UTP incorporation perform a spot test. This is a crucial quality control test. Adjust the probe concentration to 100 ng/μl. Spot 1 μl of serial dilutions (10-2 to 10-5) of the probe onto an 82mm diameter of Hybond-N+ (GE Healthcare) or other equivalent nylon filter. Include an unlabeled DNA sample as a negative control. Crosslink the damp filter immediately in a UV crosslinker at 125mJoules or according to the manufacturer's instructions for the nylon filter you are using. All of the following steps (1.4.2 - 1.4.6) are performed in a petri dish on a rocking platform at room temperature. You do not have to worry about RNase after spotting the probe dilutions on the filter
- Block the filter for 30 minutes at room temperature in 10ml of 1% Blocking reagent (Roche) in 1X TN (10X TN buffer = 1M Tris pH7.5, 1.5M NaCl)
- Wash 1 X 15 min in 10ml 1X TN buffer.
- Incubate with anti digoxigenin Fab fragment at 1/5000 dilution in 1X TN buffer for 30 minutes.
- Wash 2 X 15 min in 10ml 1X TN buffer.
- The color reaction is performed with BM purple substrate (need about 5ml in the dish). Color should be visible on the spot from the 10-4 dilution in 30 minutes. At that point, stop the color reaction by washing 2 times for 5 minutes each with 5X TE. If the signal is not visible in the spot that corresponds to the 10-4 dilution then the probe should be discarded. Note: In our experience, inadequate specific activity is only found with short probes. Increasing the size of the probe template (up to 1kb) while adhering to the other design criteria specified in this protocol should increase the specific activity of the probe. Low specific activity leads to low or undetectable levels of signal.
- Test the integrity of the probe by electrophoresis through a 5% denaturing acrylamide gel. This is another important quality control assay. This protocol assumes that the probe has been labeled with 32P during the synthesis reaction (see step 1.2.1). We use denaturing acrylamide gels to provide very high resolution, allowing us to easily detect degraded or incomplete probes. Typically a 5% denaturing acrylamide minigel (7.5g Urea, 1.9ml 40% acrylamide, 1.9ml 2% bis-acrylamide (or use a 20:1 acrylamide: bis-acrylamide solutions), 1.5ml 10X MOPS gel buffer (0.2 M morpholinopropanesulfonic acid, pH7.0, 50mM sodium acetate 5mM EDTA) and H2O to a final volume of 15 ml) works very well for resolving probes in the size ranges used in this protocol. Note: As an alternative, TBE-urea precast gels can be purchased from various suppliers. Follow the manufacturer's instructions for running precast gels.
- Mix the sample with an equal volume of Gel Loading Buffer (Ambion) and mix well.
- Heat to 95° C for 5 minutes to denature any secondary structure.
- Immediately cool on ice before loading on the gel to prevent re-annealing.
- Run the before and after the column samples together at 200V until the blue dye runs to the end of the gel. The gel is run in 1X MOPS gel buffer.
- Wrap the gel in plastic wrap and expose it to X-ray film or phosphor imager screen to detect radiolabeled RNA. If 32P labeling is not used you can stain the gel with a conventional fluorescent dye to detect the RNA
- A successful probe synthesis will result in an RNA probes that migrates as a sharp band very close to the top of the gel.
2. Collection and storage of mouse embryos
Mouse embryos should be collected in ice cold PBST (PBS + 0.1% Tween 20, (diethylpyrocarbonate (DEPC) treated)) and kept on ice during the dissection.
The embryos are processed in 4 ml screw capped glass vials. Multiple embryos can be fixed in one vial. As many as 10 to 15 E9.5 or 5 E10.5 embryos can be fixed in a single vial. Due to the increasing size of the embryo, E11.5 embryos should be hemisected with a razor blade prior to fixation to aid in probe access during the hybridization. Up to 3 hemisected E11.5 embryos can be placed in a single vial.
To maximize the effectiveness of the steps in this protocol and to reduce damage to the embryos, embryo fixation and all washes are performed in sample vials with solutions filled to the lower level of the vial neck so that only a small air bubble remains, unless otherwise specified. For all of the wash steps vials are laid down on their side on a rocker platform.
Fix for 6 hours to overnight at 4° C on a rocking table with 4% paraformaldehyde (PFA) in PBST. Vials should be placed horizontally and rocked on a rocker platform at 4° C.
After fixation, embryos are washed twice for 5 minutes each in PBST on ice and then dehydrated stepwise through a methanol/PBST series (25% methanol in PBST, 50% methanol in PBST and 75% methanol in PBST) into 100% methanol by replacing solutions in the vial using glass Pasteur pipettes. Embryos can be stored at -20C in 100% methanol for 8 months and maybe longer.
3. Hybridization of the digoxigenin labeled probe to whole mouse embryos
Puncture hearts and heads of E10.5 and older embryos with a microdissection knife while the embryos are still in methanol (if this wasn't done during the dissection). This simple step allows the wash solutions to enter the brain vesicles and the heart chambers thereby greatly reducing background staining.
Rehydrate the embryos by successive washes in a methanol/PBST series (75% methanol in PBST, 50% methanol in PBST and then 25% methanol in PBST) for 5-10 minutes at each methanol concentration. Wash twice for 5 minutes each wash in 100% PBST.
Incubate in 4:1 PBST: 30% H2O2 for 1 hour on ice. Then wash with 3 changes 5 min each in 1X PBST.
Remove the last PBST wash and replace with 1ml 10 μg/ml proteinase K in PBST. During the proteinase K digestion place the tubes vertically in a rack in a 25°C water bath. Note: Solutions of proteinase K lose activity over time due to self-digestion. To ensure maximal and consistent activity the proteinase K solution must be made fresh immediately before each use. For each use make a small amount of a 1mg/ml stock then dilute it. The incubation time should be determined for each lot of proteinase K powder and for each embryo age. For example, for E9.5 embryos a good time is usually 10-15 min at 25° C while for E10.5 it is usually 20-30 min at 25°C. The proteinase K incubation time should also be measured precisely during experiments to allow for valid comparisons between vials and between experiments. The temperature should be carefully maintained during the digestion to increase reproducibility. Differences as small as 1°C can alter the amount of digestion between experiments. It is strongly recommended that the proteinase K digestions be done in an incubator or a waterbath for accurate temperature control. This step is critical for allowing the probe to penetrate the tissue thereby generating strong signal. However, over-digestion will damage the embryos and they will gradually disintegrate over the course of the remaining steps.
Stop the Proteinase K digestion by washing twice for 5 minutes each wash at room temperature with freshly made 2 mg/ml glycine in PBST. Then wash twice for 5 minutes each in PBST.
Fix the digested embryos for 20 minutes at room temperature in 4% PFA/PBST, 0.2% gluteraldehyde (Polysciences). Do not over- or underfix. Wash 3 times for 5 minutes each in PBST.
At this point, embryos should be divided into groups for hybridization. Remove the PBST. Add 1 ml hybridization buffer (50% ultrapure formamide (Invitrogen), 5x SSC, pH 5, 1% SDS, 50 μg/ml heparin (Acros), 50 μg/ml Torula RNA (Sigma)) that has been warmed to 65°C (without probe). Allow embryos to settle, then remove all of the hybrization buffer and replace with 1 ml fresh warmed hybridization buffer without probe.
Prehybridize for 1 hour at 65° C in a shaking water bath or in an oven on a rocking platform (for example the Boekle Shake 'n' Bake Oven, see Table 1). If you incubate the vials in a water bath be sure water comes up to, but not over, the tops of the vials. The hybridization temperature can be increased up to 70° C. Hybridization temperatures above 65° C do not affect signal intensity or background with most probes we have tested. The embryos become translucent, sticky, and fragile in formamide buffers (e.g. hybridization buffer) be careful when handling them.
Replace the prehybridization buffer with 0.4ml hybridization buffer containing probe at a concentration of 0.25-1 μg/ml. Use 0.5 μg/ml of probe for embryos up to about E8.5. For older embryos, probes should be titrated from 1-0.1 μg/ml to determine the optimal concentration for a good signal with low background (usually 0.25-0.5 μg/ml or so). Note that larger hybridization volumes may be necessary for the larger embryos. Hybridize overnight at 65° C.
If the embryos have been over treated with proteinase K. they will appear extremely transparent. They may also stick to the sides of the vial, or fall apart. Embryos that appear this way should be discarded at this point in the protocol. Processing them further will not result in interpretable data.
Replace the hybridization buffer with warmed Solution I (50% formamide, 5x SSC pH 5, 1% SDS) at 70°C. Wash twice for 30 minutes each wash for E8.5 embryos, three times for 30 minutes per wash for E9.5 and older.
Wash once with pre-warmed 50% Solution I: 50% Solution II (0.5M NaCl, 10mM Tris HCl pH 7.5, 0.1% Tween 20) at 70°C for 10 minutes.
Wash three times for 5 minutes per wash with Solution II at room temperature.
Replace buffer with Solution II containing RNase A 100 μg/ml, RNase T1 100 units/ml. Incubate on a rocker in the warm room at 37C twice for 30 minutes per wash. The RNase wash is essential for reducing background resulted from nonspecifically hybridized probes.
Wash in Solution III (50% formamide, 2x SSC pH5) at 65°C twice for 30 minutes each wash for E8.5, 3 times for 30 minutes per wash for E9.5 and older.
Wash 3 times for 5 minutes per wash with 1X TBST (100 ml of 10X TBS contains 8g NaCl, 0.2g KCl, 12.5ml 2M Tris HCl pH 7.6 in water. To make 1X TBST dilute the 10X TBS in an appropriate volume of water and add Tween 20 to 0.1% final concentration)
Unless specified, all washes are performed in the sample vials with solutions filled to the lower level of the vial neck. For all wash steps, vials are laid down on their side on a rocker platform.
4. Antibody detection of the digoxygenin labeled probe
Embryos are pre-blocked in 0.5 ml 1X TBST + 10% heat treated sheep serum. Then rock the vials vertically at room temperature (˜ 20°C) for at least 2.5 hr.
Replace the blocking solution with fresh 1X TBST + 10% serum containing a 1:2000 - 1:5000 dilution of alkaline phosphatase-conjugated sheep anti-digoxigenin Fab fragments (subtracted by incubating with embryo acetone powder, see below). Use higher dilutions for E9.5 and older embryos, up to 1:5000 to minimize background. Incubate vertically on rocker at 4°C overnight. Note: We have found that incubation of the anti-digoxigenin Fab fragments with an embryo acetone powder reduces levels of background staining in this protocol. To prepare the embryo acetone powder homogenize pooled E12.5-14.5 mouse embryos in a minimum volume of PBS. Add 4 volumes of ice cold acetone, mix and incubate on ice for 30 minutes. Remove supernatant by spinning at 10,000xg for 10 minutes. Wash the pellet with ice cold acetone and spin again. Spread the pellet out and grind into powder on a sheet of filter paper and allow it to air dry. The powder can then be stored in an air-tight tube at 4°C for years. For the antibody subtraction, the antibody should be pre-absorbed to the embryo acetone powder to reduce nonspecific binding during or before pre-blocking. Incubate antibody at 1/100 with a small amount of embryo powder (the amount is not critical) in 1xTBST/10% serum for at least 1hr at 4°C with rocking. Remove powder by spinning in a microcentrifuge for 10 minutes at 4°C. Subtracted antibody can be stored at 4°C for at least 6 months.
Wash three times 5 minutes each with 1X TBST, then 5-6 times for 1hr each at room temperature to remove excess antibody. If preferred, do an extended overnight wash at 4°C to reduce background. The overnight wash can dramatically reduce background levels thereby increasing the signal to noise ratio.
Wash twice for 20 minutes per wash with freshly made alkaline phosphatase buffer (100mM Tris, pH 9.5, 50mM MgCl2, 100mM NaCl, 0.1% Tween 20. Add 0.5mg/ml levamisole, an endogenous AP inhibitor, for E9.5 and older embryos).
Replace the last wash with BM Purple AP substrate (Roche) prewarmed to room temperature.
Allow the color reaction to incubate at room temperature, and watch for signal periodically via a dissecting microscope. For extended color reactions (several hours to overnight) the vials should be shielded from light. Signal for abundant mRNAs should be visible within 15 minutes. Reactions should be stopped when you are satisfied with the signal, or when the background begins to be a problem. If the solution itself starts to turn dark you can extend the color reaction by replacing the substrate solution.
Stop reaction by washing twice for 5 minutes per wash in 1X PBST/5mM EDTA.
Transfer to 4% paraformaldehyde/PBST to re-fix, from several hours to overnight. Or embryos can be stored in 4% paraformaldehyde/1X PBST for years or can be immediately processed for sectioning. This final fixation step is crucial for long term preservation of the staining pattern.
5. Paraffin embedding and sectioning of stained embryos
Before the embryos are fixed after the color reaction (step 4.8) pictures of the whole mount stained embryos are taken when the staining level is at the desired intensity. After documenting the expression patterns in the whole embryos they are incubated in the BM purple substrate overnight to 24 hours at room temperature until the whole embryo becomes dark blue. This step is to ensure that the staining will be sufficiently dark to show up in the tissue sections. Please note that the blue color of the embryo is due to the deposition of a very thin layer of pigment all over the embryo surface. This background staining will not detectable within the tissue of interest after sectioning.
Wash the stained embryos with PBS three times to remove the excess substrate precipitates and fix overnight in 4% PFA at 4°C.
Wash with PBS three times, and then dehydrate the embryos into 100% methanol (as described in 2.4).
Replace the last methanol with xylene. Wash in xylene 2 to 3 times for 2 min each wash until the embryos become clear. Do not over wash in xylene since it will make the tissue very brittle after it is embedded making it very difficult to recover intact sections. Visually monitor each xylene incubation to make sure embryos are just to the point of being clear and be very careful to not allow the xylene incubation to extend beyond this point.
Transfer embryos (E10.5 and older) into Biopsy Sure-Tek cassettes (Fisherbrand), and incubate the whole cassette in wax (Fisherbrand) for 15˜30 min at 65°C. Change into fresh wax for another 15˜30min and then change again. Smaller embryos/tissue trunks require shorter wax incubation time. E9.5 embryos can be placed in the tissue embedding molds for all wax changes. In those cases you can remove the wax from the mold instead of transferring the embryos.
After three wax changes, transfer embryos into the tissue embedding molds (Polysciences) and embed with fresh wax. Carefully position the embryos for the desired plane of sectioning. Let the wax to cool until solidified.
Section the wax blocks into 8˜14μm slices using standard microtome (e.g. Leica RM2155). Collect sections onto microscope slides (Fisherbrand).
Let the slides lay flat and dry on a slidewarmer. The slides can be stored at 4°C for months.
Dried sections are dewaxed by two 5 min xylene washes and rehydrated into water by serial methanol/water washes (100% methanol 2 min, 100% methanol 2 min, 90% methanol/water 2min, 70% methanol/water 2min, deionized water 5 min).
Stain the sections with nuclear fast red (Sigma) staining solution for 1 minute. To generate the nuclear fast red staining solution, dilute a concentrated stock of nuclear fast red (0.1% nuclear fast red in 5% aluminum sulfate in water, dissolve with heat and filter through Whatman filter paper) 1:5 in an aqueous solution of 5% aluminum sulfate . After staining wash the slides with running water until the water becomes clear. The nuclear fast red acts as a counterstain and reveals overall tissue structure in the sections. Its color contrasts well with the purple-blue staining that marks the localization of the RNA probe. Nuclear fast red is a saturating stain and the staining intensity is controlled by the concentration of the staining solution. If the intensity of the nuclear fast red staining is inadequate when using this staining solution you can repeat this step with a more concentrated nuclear fast red solution.
Prepare the slides for mounting coverslips by washes in each of the solutions in the following series: 90% methanol/water followed by 2 washes in 100% methanol followed by 2 washes in xylenes. Incubate the slide for 2 minutes in each of the washes.
Mount the slides with Cytoseal 60 mounting medium (Richard-Allan Scientific). Please note that aqueous mounting media will dissolve the nuclear fast red stain. Note: we have also used a plastic embedding resin (Immuno-Bed, Polysciences Inc) to generate sections of stained embryos with excellent results 4.
6. Representative Results:
Figure 1 illustrates representative results obtained using this protocol. Figure 1A shows the expression pattern of Gata3 in E10.5 mouse embryos. This panel illustrates a good result with a high signal to background ratio. Figure 1C, in contrast, shows a failed in situ hybridization for Gata3 mRNA that used a partially degraded RNA probe. A low specific activity probe will result in similar results with low signal to background contrast. A comparison of panels A and C illustrates the importance of performing careful quality control assessments of probe quality to ensure high quality results. The result obtained with the Foxg1 probe (Figure 1B) also illustrates a good result with specific staining in the telencephalon with low background in other parts of the brain. The brain and the heart are common sites of background staining caused by probe trapping. Puncturing the brain and heart with forceps allows wash solutions into the embryo and minimizes background staining (see step 3.1). Figure 1D illustrates the typical light blue background found when the brain is not punctured. This shows a suboptimal result obtained using a Pax1 probe. Pax1 is not expressed in any part of the brain and all of the staining seen in the brain of this embryo is due to non-specific background.
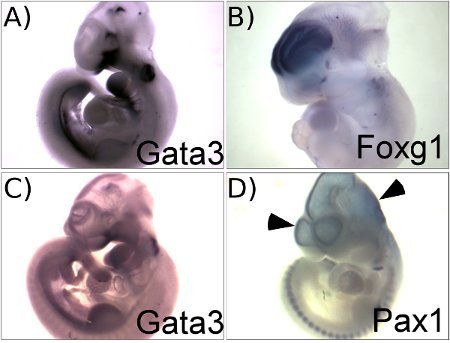 Figure 1. Representative results showing examples of successful (A, B) and of suboptimal (C, D) outcomes. A. E10.5 embryo hybridized with Gata3 probe. B. E11.5 embryo hybridized with Foxg1 probe. C. E10.5 embryo hybridized with Gata3 probe that is partially degraded, showing very weak signal but high background through out the whole embryo. D. E10.5 embryo hybridized with Pax1 probe without puncturing the head, showing background staining in the ventricles (arrow heads).
Figure 1. Representative results showing examples of successful (A, B) and of suboptimal (C, D) outcomes. A. E10.5 embryo hybridized with Gata3 probe. B. E11.5 embryo hybridized with Foxg1 probe. C. E10.5 embryo hybridized with Gata3 probe that is partially degraded, showing very weak signal but high background through out the whole embryo. D. E10.5 embryo hybridized with Pax1 probe without puncturing the head, showing background staining in the ventricles (arrow heads).
Discussion
The methods described in this protocol have been adapted from a number of different sources and optimized for whole mount E8.5-E11.5 day old mouse embryos. Methods for whole mount in situ hybridization of vertebrate embryos first appeared in the early 1990s 2,5-12. This protocol was adapted primarily from methods developed for Xenopus embryos 7,8 as well as mouse 2,11. In our protocol a great deal of emphasis is placed on careful quality assessment of the probe. Careful attention to generating a high quality and high specific activity RNA probe as determined by the earliest steps in this protocol will greatly increase the quality of the data and save considerable amounts of time and effort in the long run. Our protocol also includes the use of PCR generated DNA templates for phage RNA polymerase transcription. This approach is rapid and very scalable. It can enable the generation of a large number of RNA probes for large scale in situ hybridization screens 13-15.
Several steps in this protocol should be considered when the background is high but signal is low. One consideration is the probe. We have never had high background with RNA probes that met the design criteria specified in this protocol and that had passed all three quality control tests (yield , specific activity and probe length). Another factor that can affect background levels is the ability of the probe to penetrate into the embryonic tissues. Care must be taken during the dissection to completely remove the extraembryonic membranes that cover the embryo. In addition, the proteinase K digestion step is absolutely essential for embryos older than E9.5. Longer proteinase K digestion times give lower background and stronger signal, however, excessive digestion times can lead to increased tissue damage and may result in the disintegration of the embryo during the procedure. Optimization of the digestion time for each lot of proteinase K is required to obtain the best results. Another step that can be optimized to obtain high signal to background ratios is the RNase incubation (step 3.14). Our protocol uses a mixture of RNase A and RNase T1. Both enzymes digest single stranded RNA but each enzyme has a different base specificity. The combination of the two enzymes results in extensive digestion of single stranded RNA to small oligoribonucleotides. This enables most of the non-specifically hybridized probe to be removed by the post-hybridization washes resulting in a large decrease in background staining. Use of RNase A or T1 alone results in increased background and should be avoided. We have also found that the quality of the final color reaction is improved by using fresh AP buffer, made immediately prior to use.
Our protocol can be adapted for older embryos or adult tissues with additional processing. For example, we have also used this protocol to perform in situ hybridizations on thick (100 μm) vibratome sections of adult mouse brain (data not shown). We have also used this protocol to study gene expression in older embryos. In some cases have used this protocol to visualize gene expression on the surface of E13.5 and E14.5 embryos, such as Gad1 expression in the follicles of the vibrissae 4. In other cases we have used a razor blade or scalpel to cut E12.5-E14.5 embryos to provide probe access to specific tissues within the embryo. Embryo fragments prepared in this manner can be processed using this protocol with some modifications. In these cases the length of all treatments and washing steps needs to be adjusted according to tissue size and optimized empirically.
Our protocol also includes methods for post-hybridization sectioning and analysis of expression patterns. Combined with the whole mount visualization of gene expression this additional step can add a great deal of additional information regarding the expression pattern of a gene. We have embedded embryos after undergoing in situ hybridization in paraffin as well as in plastic resin 4,16. Sections of stained embryos can be used to generate a three dimensional reconstruction of the expression pattern revealed by the whole mount in situ hybridization procedure. We have used reconstruction software from SURFdriver software for this purpose.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was supported by NIH grants R21MH082360 (BGC) and R01HD056315 (NRM) as well as the University of Georgia.
References
- Divjak M, Glare EM, Walters EH. Improvement of non-radioactive in situ hybridization in human airway tissues: use of PCR-generated templates for synthesis of probes and an antibody sandwich technique for detection of hybridization. J. Histochem. Cytochem. 2002;50:541–548. doi: 10.1177/002215540205000411. [DOI] [PubMed] [Google Scholar]
- Wilkinson DG, Nieto MA. Detection of messenger RNA by in situ hybridization to tissue sections and whole mounts. Methods Enzymol. 1993;225:361–373. doi: 10.1016/0076-6879(93)25025-w. [DOI] [PubMed] [Google Scholar]
- Logel J, Dill D, Leonard S. Synthesis of cRNA probes from PCR-generated DNA. Biotechniques. 1992;13:604–610. [PubMed] [Google Scholar]
- Maddox DM, Condie BG. Dynamic expression of a glutamate decarboxylase gene in multiple non-neural tissues during mouse development. BMC Dev Biol. 2001;1:1–1. doi: 10.1186/1471-213X-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon RA, Rossant J. Exogenous retinoic acid rapidly induces anterior ectopic expression of murine Hox-2 genes in vivo. Development. 1992;116:357–368. doi: 10.1242/dev.116.2.357. [DOI] [PubMed] [Google Scholar]
- Krauss S. Zebrafish pax[zf-a]: a paired box-containing gene expressed in the neural tube. EMBO J. 1991;10:3609–3619. doi: 10.1002/j.1460-2075.1991.tb04927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmati-Brivanlou A. Localization of specific mRNAs in Xenopus embryos by whole-mount in situ hybridization. Development. 1990;110:325–330. doi: 10.1242/dev.110.2.325. [DOI] [PubMed] [Google Scholar]
- Harland RM. In situ hybridization: an improved whole-mount method for Xenopus embryos. Methods Cell Biol. 1991;36:685–695. doi: 10.1016/s0091-679x(08)60307-6. [DOI] [PubMed] [Google Scholar]
- Herrmann BG. Expression pattern of the Brachyury gene in whole-mount TWis/TWis mutant embryos. Development. 1991;113:913–917. doi: 10.1242/dev.113.3.913. [DOI] [PubMed] [Google Scholar]
- Conlon RA, Herrmann BG. Detection of messenger RNA by in situ hybridization to postimplantation embryo whole mounts. Methods Enzymol. 1993;225:373–383. doi: 10.1016/0076-6879(93)25026-x. [DOI] [PubMed] [Google Scholar]
- Parr BA, Shea MJ, Vassileva G, McMahon AP. Mouse Wnt genes exhibit discrete domains of expression in the early embryonic CNS and limb buds. Development. 1993;119:247–261. doi: 10.1242/dev.119.1.247. [DOI] [PubMed] [Google Scholar]
- Rosen B, Beddington RS. Whole-mount in situ hybridization in the mouse embryo: gene expression in three dimensions. Trends Genet. 1993;9:162–167. doi: 10.1016/0168-9525(93)90162-b. [DOI] [PubMed] [Google Scholar]
- Quiring R. Large-scale expression screening by automated whole-mount in situ hybridization. Mech. Dev. 2004;121:971–976. doi: 10.1016/j.mod.2004.03.031. [DOI] [PubMed] [Google Scholar]
- Thut CJ, Rountree RB, Hwa M, Kingsley DM. A large-scale in situ screen provides molecular evidence for the induction of eye anterior segment structures by the developing lens. Dev. Biol. 2001;231:63–76. doi: 10.1006/dbio.2000.0140. [DOI] [PubMed] [Google Scholar]
- Neidhardt L. Large-scale screen for genes controlling mammalian embryogenesis, using high-throughput gene expression analysis in mouse embryos. Mech. Dev. 2000;98:77–94. doi: 10.1016/s0925-4773(00)00453-6. [DOI] [PubMed] [Google Scholar]
- Gordon J, Bennett AR, Blackburn CC, Manley NR. Gcm2 and Foxn1 mark early parathyroid- and thymus-specific domains in the developing third pharyngeal. 2001;103:141–143. doi: 10.1016/s0925-4773(01)00333-1. [DOI] [PubMed] [Google Scholar]