

Abstract
Epigenetics describes the heritable changes in gene function that occur independently to the DNA sequence. The molecular basis of epigenetic gene regulation is complex, but essentially involves modifications to the DNA itself or the proteins with which DNA associates. The predominant epigenetic modification of DNA in mammalian genomes is methylation of cytosine nucleotides (5-MeC). DNA methylation provides instruction to gene expression machinery as to where and when the gene should be expressed. The primary target sequence for DNA methylation in mammals is 5'-CpG-3' dinucleotides (Figure 1). CpG dinucleotides are not uniformly distributed throughout the genome, but are concentrated in regions of repetitive genomic sequences and CpG "islands" commonly associated with gene promoters (Figure 1). DNA methylation patterns are established early in development, modulated during tissue specific differentiation and disrupted in many disease states including cancer. To understand the biological role of DNA methylation and its role in human disease, precise, efficient and reproducible methods are required to detect and quantify individual 5-MeCs.
This protocol for bisulphite conversion is the "gold standard" for DNA methylation analysis and facilitates identification and quantification of DNA methylation at single nucleotide resolution. The chemistry of cytosine deamination by sodium bisulphite involves three steps (Figure 2). (1) Sulphonation: The addition of bisulphite to the 5-6 double bond of cytosine (2) Hydrolic Deamination: hydrolytic deamination of the resulting cytosine-bisulphite derivative to give a uracil-bisulphite derivative (3) Alkali Desulphonation: Removal of the sulphonate group by an alkali treatment, to give uracil. Bisulphite preferentially deaminates cytosine to uracil in single stranded DNA, whereas 5-MeC, is refractory to bisulphite-mediated deamination. Upon PCR amplification, uracil is amplified as thymine while 5-MeC residues remain as cytosines, allowing methylated CpGs to be distinguished from unmethylated CpGs by presence of a cytosine "C" versus thymine "T" residue during sequencing.
DNA modification by bisulphite conversion is a well-established protocol that can be exploited for many methods of DNA methylation analysis. Since the detection of 5-MeC by bisulphite conversion was first demonstrated by Frommer et al.1 and Clark et al.2, methods based around bisulphite conversion of genomic DNA account for the majority of new data on DNA methylation. Different methods of post PCR analysis may be utilized, depending on the degree of specificity and resolution of methylation required. Cloning and sequencing is still the most readily available method that can give single nucleotide resolution for methylation across the DNA molecule.
Keywords: Genetics, Issue 56, epigenetics, DNA methylation, Bisulphite, 5-methylcytosine (5-MeC), PCR
Protocol
Bisulphite conversion protocol
A standard protocol for the conversion of 2 μg of genomic DNA is described below. Smaller or larger amounts of genomic DNA (100 pg-200 μg) can also be bisulphite treated successfully. These reaction conditions result in complete conversion (99.5-99.7%) of almost every target DNA sequence3.
1. DNA Preparation
Prepare samples by incubating genomic DNA with bisulphite DNA Lysis Buffer (2 μg tRNA, 280 ng/μl Proteinase K, 1% SDS) in a total volume of 18 μl for 1 hr at 37°C. Note: This is important to achieve maximal bisulphite conversion, especially with DNA isolated from clinical samples where there may still be protein associated with the DNA.
2. DNA Denaturation
Denature 2 μg DNA in a volume of 20 μl by adding 2 μl of freshly prepared 3M NaOH to a final concentration of 0.3M.
Incubate the samples at 37°C for 15 min in a water bath, followed by incubation at 90°C for 2 min in a heat block. Immediately place the tubes on ice for 5 min.
Centrifuge the tubes at 4°C for 10 s at 10,000 g, to ensure the DNA is at the bottom of the tube.
3. Bisulphite Deamination
Sulphonation & Hydrolytic Deamination
Prepare fresh solutions of 10 mM Quinol and saturated sodium metabisulphite pH 5.0 (7.6 g Na2S2O5 with 464 μl of 10 M NaOH, made up to 15 ml with water). The solution of saturated sodium bisulphite is achieved by gently inverting the reagent/H2O mixture, with minimum mixing and aeration. If necessary, adjust the pH with 10 M NaOH, for full dissolution of the metabisulphite. Note: As it is a saturated solution, small lumps may still remain undissolved.
Add 208 μl of the saturated metabisulphite and 12 μl of 10mM Quinol to the denatured DNA (20 μl), in a final volume of 240 μl to a final concentration of 2.31M bisulphite/0.5mM Quinol, pH 5.0. Gently mix all reagents and centrifuge for 10 sec to ensure all of the droplets are at the bottom of the tube.
Overlay the samples with 200 μl of mineral oil. Incubate the samples at 55°C in a water bath, for 4-16 hrs. The length of bisulphite treatment is dependant on the quantity and quality of the DNA to be converted. For DNA of poor quality or degraded DNA, limit the incubation time to 4 hrs. Note: it is important that that the bisulphite conversion takes place in the dark to avoid oxidation, so if that is not possible wrap the tubes in foil prior to incubation.
At the end of the appropriate incubation time, spin the tubes briefly in a microcentrifuge to ensure all the liquid is at the bottom of the tube.
Recover the bisulphite treated DNA from under the oil layer by carefully pipetting out the DNA solution from the bottom of the tube, without taking up any of the mineral oil.
Desalting
Remove any free bisulphite ions by passing through a desalting column and eluting in 50 μl of milli-Q water (MQH2O). Depending on the quantity and quality of the genomic DNA and how it is prepared, different desalting columns can be used. We routinely use Promega Wizard Clean up columns, as these columns are suitable to remove the SDS used in the preparation of the DNA prior to conversion.
Alkali Desulphonation
The bisulphite adduct is removed from the uracil ring by desulphonation. Desulphonate by adding 5.5 μl of freshly prepared 3M NaOH to a final concentration of 0.3M, then incubate the samples at 37°C for 15 min.
Centrifuge briefly and add 1 μl of tRNA (10 mg/ml).
Neutralize the solution by the addition of 33 μl of ammonium acetate (NH4O Ac), pH 7.0 to a final concentration of 3M.
Ethanol-precipitate the DNA by adding 330 μl of ice cold 100% ethanol, mix well by inversion. Leave at -20μC for 1 hr to overnight. Centrifuge at 14,000 g for 15 min at 4°C. Remove all traces of supernatant and air dry the precipitated DNA for ~20 min.
Re-suspend the DNA pellet in 50 μl/μg of 0.1 TE (10mM Tris-HCL, 0.1mM EDTA, pH 8.0) or H2O. Leave at room temperature for approximately 2hrs. Occasionally vortex the tubes to ensure the DNA is dissolved, followed by a quick centrifuge.
Use immediately for PCR amplification, or store at -20°C for 1-10 years depending on the quantity and quality of DNA.
4. PCR Amplification
Primer Design
Effective design of PCR bisulphite conversion-specific primers is crucial for ensuring that efficient, unbiased amplification of completely converted DNA occurs. The following guidelines are used to aid primer design.
Primer Design Guidelines

Thermocyling
To test for proportional PCR amplification of methylated and unmethylated DNA, use a 50:50 Methylated/Unmethylated fully bisulphite converted control sample and amplify with the bisulphite conversion-specific primers under the optimized PCR reaction conditions (Figure 3). For the 50:50 Methylated:Unmethylated control, use in vitro SssI methylated DNA and either whole genome amplified (WGA) DNA or whole blood DNA. Please be aware that some genes will be naturally methylated in blood and therefore in these cases it is best to only use WGA DNA as the "Unmethylated" control.
Prepare PCR amplification reaction mixtures in 100 μl aliquots containing 2 μl of bisulphite converted genomic DNA, 200 μM dNTP's, 1 μM primers, 1.5 mM MgCl2, 50 mM KCl, 10mM Tris-HCL, pH 8.3 and 0.15 μl Taq polymerase.
In a temperature gradient thermocycler, set the run reaction in a gradient +/- 3°C from the predicted Tm of the primer across 10 tubes

To test that the methylated and unmethylated amplicons have been amplified in proportion, the amplicon can be digested with an informative restriction enzyme, such as Taq 1 (TCGA), that will digest methylated DNA but will not digest unmethylated DNA when the restriction enzyme site is altered after bisulphite conversion to TTGA. The extent of digestion can be visualized by agarose gel electrophoresis (Figure 3A). Alternatively, SYBRGreen (0.2 μl of 1:1000 dilution) can be added to the PCR reaction and the extent of methylation can be assessed by performing heat dissociation plots in a real-time PCR thermocycler (Figure 3B).
PCR Reaction mix including MgCl2 concentration and thermocycling conditions may need to be adjusted to increase PCR efficiency or to ensure proportional PCR amplification of methylated and unmethylated PCR fragments.
Once the PCR conditions are optimized, PCR amplification can be performed on bisulphite converted samples. Note: Once the Tm is optimized a temperature gradient PCR need not be used.
5. Representative Results:
An example of bisulphite PCR amplification optimization is shown in Figure 3. Optimal PCR amplification conditions should amplify methylated and unmethylated amplicons in proportion and without bias. Figure 3A shows an agarose gel with a temperature gradient PCR amplification profile from a mixture of 50% methylated and unmethylated DNA. The PCR amplicons have been treated with a restriction enzyme, Taq 1 (TCGA), that will digest methylated (M) DNA but will not digest unmethylated (U) DNA as the restriction enzyme site is altered by bisulphite conversion to TTGA. An equal amount of cut and uncut PCR product is expected if methylated and unmethylated DNA is amplified in proportion. It can be seen on this gel that the optimal thermocyling conditions for non-bias amplification is at 56.1°C (T). Complete conversion of the bisulphite DNA can also be analyzed by digestion with cytosine-site specific enzyme such as HpaIII (CCGG). HpaIII will only digest if the conversion has failed, as the restriction site will be maintained. If the conversion is complete the restriction site will be converted to TCGG or TTGG depending on the methylation state of the DNA. Complete bisulphite DNA conversion can be seen in Figure 3A (H).
Figure 3B shows a real time dissociation plot that can also be used as a tool to determine whether there is any amplification bias based on the temperature at which the different molecules will dissociate. In this figure it can be seen that the PCR has amplified methylated (M) and unmethylated (U) DNA in proportion from a mix of 50% methylated and unmethylated DNA compared to the control amplification of fully methylated and unmethylated DNA which dissociate at 82.1μC and 78.9μC respectively.
After optimization of thermocyling and reaction conditions Bisulphite treated samples can be amplified with strand specific and bisulphite specific primers in either a single or semi nested PCR reaction. The resulting PCR fragments can be visualized by agarose gel electrophoresis and sequenced either directly (Figure 4A) or by cloning and sequencing. After cloning and sequencing the methylation state of the individual molecules can be tabulated, in a bisulphite map (Figure 4B), to visualize the heterogeneity of methylation.
High throughput quantitative methylation analysis can be preformed using technology such as that utilized by the Sequenom EpiTYPER method. Using this method, bisulphite converted DNA is amplified with bisulphite specific PCR primers and followed by a proprietry cleavage process. The resulting fragments are quantitated by MALDI-TOF mass spectrometry with the specific spectrum dependent on the presence of methylated cytosines (Figure 5A). A summary of methylation ratios in the sample can then be extrapolated in the form of an Epigram (Figure 5B) or methylation plot (Figure 5C).
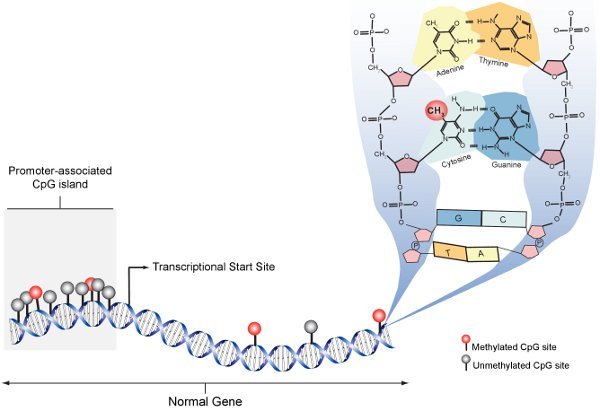 Figure 1. Methylated CpG schematic. In the normal cell, promoter-associated CpG islands are predominantly unmethylated (grey) whereas CpG sites within gene bodies are sparse and generally methylated (red). The panel on the right expands the molecular structure of DNA at an individual CpG site and shows methylation with a CH3 molecule at carbon 5 of cytosine.
Figure 1. Methylated CpG schematic. In the normal cell, promoter-associated CpG islands are predominantly unmethylated (grey) whereas CpG sites within gene bodies are sparse and generally methylated (red). The panel on the right expands the molecular structure of DNA at an individual CpG site and shows methylation with a CH3 molecule at carbon 5 of cytosine.
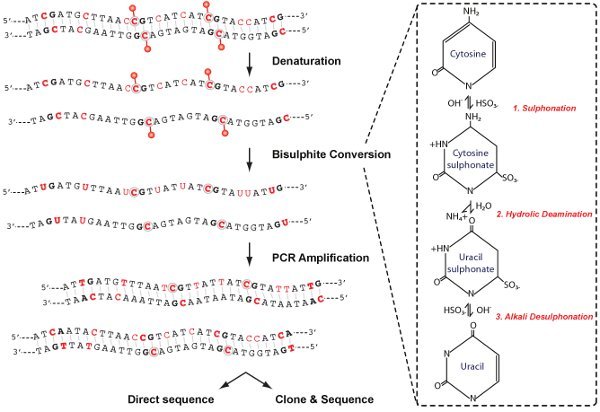 Figure 2. Chemical conversion schematic. Analysis of DNA methylation includes four main stages as shown; denaturation, bisulphite conversion, PCR amplification and analysis. In the right panel, modifications to the cytosine molecule that occur during bisulphite conversion are depicted.
Figure 2. Chemical conversion schematic. Analysis of DNA methylation includes four main stages as shown; denaturation, bisulphite conversion, PCR amplification and analysis. In the right panel, modifications to the cytosine molecule that occur during bisulphite conversion are depicted.
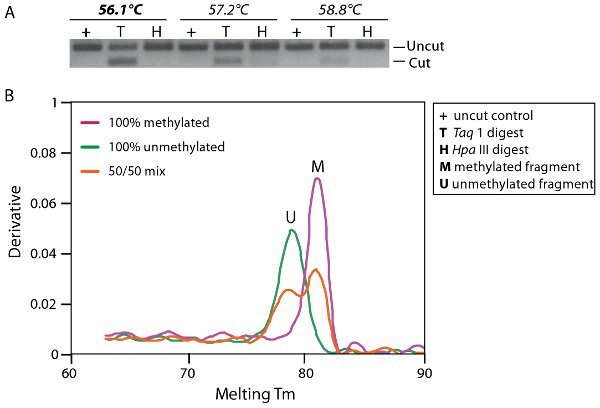 Figure 3. Optimisation of PCR amplification. A. Agarose gel electrophoresis with a temperature gradient PCR amplification profile from a mixture of 50% methylated and unmethylated DNA. Fragments have been digested with either Taq1 (T) or HpaIII (H) which digest methylated DNA and non bisulphite converted DNA respectively. B. Heat dissociation curve analysis shows an equal proportion of methylated and unmethylated DNA (red line 50/50 mix) compared to the control amplification of fully methylated (pink line, M) and unmethylated DNA (green line, U) which dissociate at 82.1°C and 78.9°C respectively.
Figure 3. Optimisation of PCR amplification. A. Agarose gel electrophoresis with a temperature gradient PCR amplification profile from a mixture of 50% methylated and unmethylated DNA. Fragments have been digested with either Taq1 (T) or HpaIII (H) which digest methylated DNA and non bisulphite converted DNA respectively. B. Heat dissociation curve analysis shows an equal proportion of methylated and unmethylated DNA (red line 50/50 mix) compared to the control amplification of fully methylated (pink line, M) and unmethylated DNA (green line, U) which dissociate at 82.1°C and 78.9°C respectively.
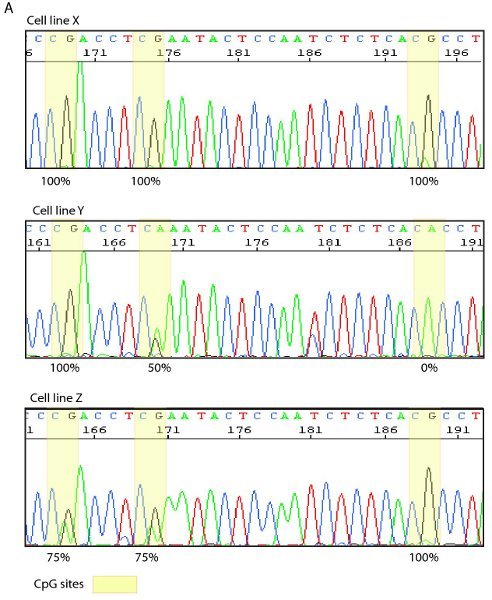
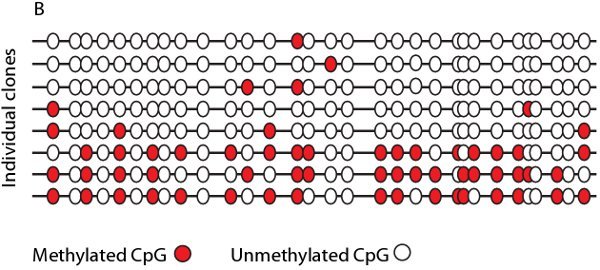 Figure 4. Example of direct sequencing and a bisulphite map. A. Sequence trace from three different cell lines with CpG sites highlighted in yellow. Cell line X displays 100% methylation at all three CpG sites whereas cell lines Y and Z show varying degrees of methylation as seen by overlapping G/A signals. B. Representative bisulphite map showing the density of methylation (red dots) at individual CpG sites, as determined by direct sequencing of individual clones.
Figure 4. Example of direct sequencing and a bisulphite map. A. Sequence trace from three different cell lines with CpG sites highlighted in yellow. Cell line X displays 100% methylation at all three CpG sites whereas cell lines Y and Z show varying degrees of methylation as seen by overlapping G/A signals. B. Representative bisulphite map showing the density of methylation (red dots) at individual CpG sites, as determined by direct sequencing of individual clones.
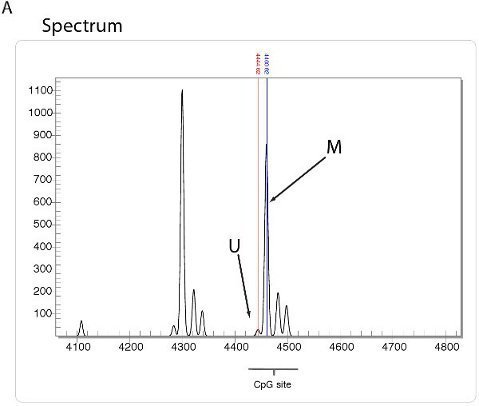
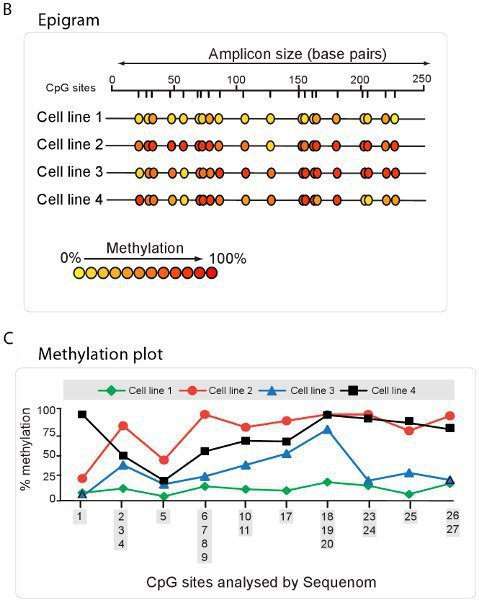 Figure 5. High throughput DNA methylation analysis using Sequenom. A. Spectrum view using Sequenom epiTYPER technology. DNA fragments display specific spectra, depending on amount of DNA methylation present. B. Epigram summary of the percentage of DNA methylation at each CpG site for four different cell lines. C. Methylation plot summary derived from the Sequenom Epigram.
Figure 5. High throughput DNA methylation analysis using Sequenom. A. Spectrum view using Sequenom epiTYPER technology. DNA fragments display specific spectra, depending on amount of DNA methylation present. B. Epigram summary of the percentage of DNA methylation at each CpG site for four different cell lines. C. Methylation plot summary derived from the Sequenom Epigram.
Discussion
DNA methylation analysis by genomic sequencing of bisulphite converted DNA is a well established and versatile method that facilitates the identification and quantification of DNA methylation at single nucleotide resolution. However, bisulphite conversion and subsequent analysis relies on the premise that the DNA has been fully converted with every unmethylated cytosine being deaminated to uracil and only methylated cytosines remaining un-reactive. If conversion is incomplete, problems can arise with analysis since unconverted unmethylated cytosines may be incorrectly interpreted as methylated cytosines. Bisulphite conversion can be optimized at various stages to maximize the percentage of conversion. For DNA to be fully converted it first must be single stranded so that cytosine residues are exposed to the bisulphite ions. The first step, DNA denaturation, is critical and can be the source of incomplete conversion if the DNA is not fully denatured. To ensure that the DNA is fully denatured it is important that the reaction parameters including removal of all associated protein, appropriate salt concentration, incubation temperature and time are suitable to maintain DNA in single stranded conformation. It is important to use fresh 3M NaOH and to allow adequate incubation time. If the DNA is resisting denaturation, some modifications to the protocol including extending the incubation time to 30 minutes or fragmenting the DNA prior to denaturation may facilitate DNA denaturation. In addition, single stranded DNA (ssDNA) may re-anneal to double stranded DNA (dsDNA) during the bisulphite conversion reaction. Carrying out the reaction at a higher temperature (90-95°C) either periodically, or continuously during the bisulphite reaction can aid the maintenance of ssDNA. However, it is important to be aware that DNA degradation is greatly accelerated at these higher temperatures. Various reagents can also be added to disrupt re-annealing of the DNA strands, such as urea.
The concentration of DNA and its quality can also affect the efficiency of the bisulphite reaction and ultimate PCR yield. DNA degradation is a limitation of the bisulphite conversion protocol. The chemical treatment of the DNA introduces various strand breaks in ssDNA and some of the harsh conditions necessary for complete bisulphite conversion including high bisulphite concentration, long incubation times can accelerate DNA degradation. Under the standard reaction conditions described, DNA degradation is not a common problem however, if protocol modifications are introduced, such as for sequences that are refractory to conversion, for DNA templates that are degraded or of poor quality such as FFPE samples, then DNA degradation can become a significant limitation.
Formalin fixed, paraffin embedded (FFPE) and degraded DNA samples can be effectively bisulphite converted and amplified using this protocol, however modifications to ensure that the DNA is not further degraded are advised. The use of a carrier RNA such as tRNA is recommended. Also glycogen can be added as a carrier when DNA concentration is low (<200 ng). Because DNA isolated from FFPE tissues is already fragmented and further fragmentation takes place during deamination, care must be taken to minimize further degradation. Do not fragment DNA any further before denaturation and limit the incubation time for the bisulphite reaction to 4 hours. Design amplicons no larger than 300 bp due to fragmented DNA.
Another factor that can affect PCR amplification that bisulphite conversion of unmethylated cytosines reduces the complexity of the DNA template. The following primer design protocol outlined in section 4.1, increased primer length and nested PCR can all help to increase specificity and PCR yield.
Finally, since the DNA template is altered during bisulphite conversion, amplification bias can be a concern. Ensure that proportional PCR amplification of methylated and unmethylated templates is checked with a control 50/50 M/U DNA mix, as outlined in the protocol, see Figure 3. Also ensure that amplification of only converted templates is occurring using HpaIII restriction enzyme and gel electrophoresis (Figure 3). The PCR conditions may need to be optimized by varying the temperature and/or magnesium concentration or lengthening extension time. Some templates appear to be particularly problematic and primer position may need to be adjusted or degenerate primers may need to be used.
Next generation sequencing techniques are rapidly evolving to achieve a similar resolution to bisulphite genomic sequencing, and third generation single molecule sequencing has the potential to allow for analysis of both primary DNA and methylation without the need for bisulphite sequencing. However, widespread availability and specificity of these techniques remain some time away. Bisulphite genomic sequencing is the gold standard in methylation analysis and is a robust protocol. The method has progressed to the point where the common problems and limitations have been resolved and the applications have expanded from individual gene analyses to high throughput and whole genome analysis described by Clark et al. 20064. Troubleshooting guidelines and additional recommendations are outlined in Table 1. It is suggested that the optimum approach is to initially follow the standard protocol and only introduce modification if and when a problem arises.
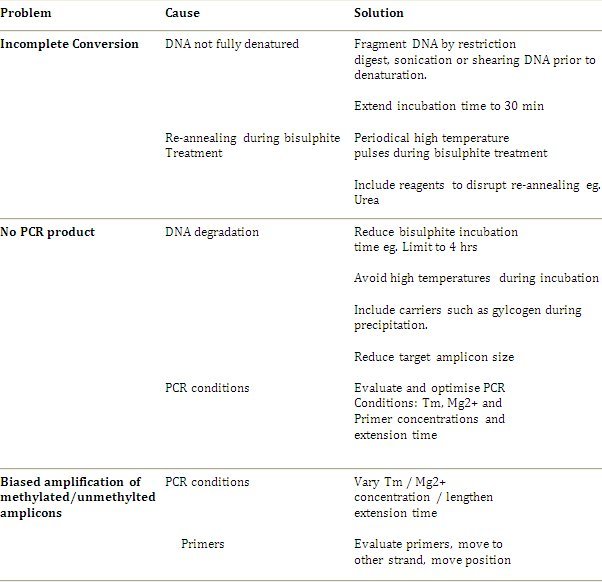 Table 1. Troubleshooting Guidelines
Table 1. Troubleshooting Guidelines
Disclosures
No conflicts of interest declared.
References
- Frommer M. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. U. S. A. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SJ. High sensitivity mapping of methylated cytosines. Nucleic. Acids. Res. 1994;22:2990–2997. doi: 10.1093/nar/22.15.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunau C, Clark SJ, Rosenthal A. Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic. Acids. Res. 2001;29:E65–E65. doi: 10.1093/nar/29.13.e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SJ. DNA methylation: bisulphite modification and analysis. Nat. Protoc. 2006;1:2353–2364. doi: 10.1038/nprot.2006.324. [DOI] [PubMed] [Google Scholar]