

Abstract
Maintenance of replication fork stability is of utmost importance for dividing cells to preserve viability and prevent disease. The processes involved not only ensure faithful genome duplication in the face of endogenous and exogenous DNA damage but also prevent genomic instability, a recognized causative factor in tumor development.
Here, we describe a simple and cost-effective fluorescence microscopy-based method to visualize DNA replication in the avian B-cell line DT40. This cell line provides a powerful tool to investigate protein function in vivo by reverse genetics in vertebrate cells1. DNA fiber fluorography in DT40 cells lacking a specific gene allows one to elucidate the function of this gene product in DNA replication and genome stability. Traditional methods to analyze replication fork dynamics in vertebrate cells rely on measuring the overall rate of DNA synthesis in a population of pulse-labeled cells. This is a quantitative approach and does not allow for qualitative analysis of parameters that influence DNA synthesis. In contrast, the rate of movement of active forks can be followed directly when using the DNA fiber technique2-4. In this approach, nascent DNA is labeled in vivo by incorporation of halogenated nucleotides (Fig 1A). Subsequently, individual fibers are stretched onto a microscope slide, and the labeled DNA replication tracts are stained with specific antibodies and visualized by fluorescence microscopy (Fig 1B). Initiation of replication as well as fork directionality is determined by the consecutive use of two differently modified analogues. Furthermore, the dual-labeling approach allows for quantitative analysis of parameters that influence DNA synthesis during the S-phase, i.e. replication structures such as ongoing and stalled forks, replication origin density as well as fork terminations. Finally, the experimental procedure can be accomplished within a day, and requires only general laboratory equipment and a fluorescence microscope.
Keywords: Molecular Biology, Issue 56, Genetics, DNA fiber analysis, replication speed, fork stalling, origin firing, termination
Protocol
The method described here was used in the following publication: Schwab et al., EMBO J., 29(4):806-185.
1. DT40 cell culture
Materials: Fetal bovine serum (FBS), chicken serum, penicillin/streptomycin, 2-mercaptoethanol, RPMI
Prepare cell culture medium: add 7% FBS, 3% chicken serum, 1x penicillin/streptomycin and 10 μM 2-mercaptoethanol to RPMI medium.
DT40 cells are grown in sterile cell culture flasks at 38 °C and 5% CO2 in a humidified incubator. Split cells every day to a density of 5 x 105 cells/ml6.
2. In vivo labeling
Materials: IdU, CldU
Prepare stock solutions of nucleotide analogues: dissolve IdU to 5 mM and CldU to 2.5 mM in medium. Heat both solutions briefly to 60 °C and vortex until the nucleotide analogues are dissolved completely.
Add IdU to a final concentration of 25 μM to exponentially growing DT40 cells and mix the cell suspension well. Incubate cells for 20 minutes at 38 °C and 5% CO2.
After incubation with the first label, add CldU to a final concentration of 250 μM and treat cells as described in the previous step.
Wash cells with ice cold PBS and resuspend them at a concentration of 7.5 x 105 cells/ml in cold PBS. Keep the labeled cells on ice.
The labeling procedure described is merely a suggestion and can be modified to address specific questions. Please refer to the discussion section for more information on experimental design.
3. Cell lysis and DNA spreading
Materials: Glass slides, fiber lysis solution
Prepare the fiber lysis solution: 50 mM EDTA and 0.5% SDS in 200 mM Tris-HCl, pH 7.5
Spot 2 μl of the cell suspension to one end of the glass slide. Air-dry for 5 minutes or until the volume of the drop is greatly reduced but not dry.
Pipet 7 μl lysis solution on top of the cell suspension and gently stir with the pipette tip to mix the solutions. Incubate for 2 minutes for cell lysis to proceed.
Tilt slides to 15° to allow the fibers to spread along the slide.
Once the fiber solution has reached the bottom of the slide, place it horizontally to air-dry. After drying, a thin, opaque line should be visible along the slide. At this point, the beginning of the stretched fibers should be marked with a pencil as after staining the line will not be visible any longer. This mark will later help to locate the fibers under the microscope.
4. Immunofluorescence staining
Materials: Methanol, acetic acid, HCl, 5% BSA in PBS, staining jar, anti-BrdU (mouse) antibody, anti-BrdU (rat) antibody, sheep anti-mouse Cy3 antibody, goat anti-rat Alexa Fluor 488 antibody, Vectashield mounting medium, coverslips, nail polish
Immerse slides in methanol/acetic acid (3:1) in a staining jar and incubate for 10 minutes.
Wash slides in distilled H2O, then immerse in 2.5 M HCl for 80 minutes.
Wash slides three times in PBS for 5 minutes.
Remove slides from the staining jar and collect excess PBS with a paper towel. Place the slides horizontally and pipet 5% BSA on top of each slide. Cover the slides gently with a coverslip to spread the BSA evenly over the slide and incubate for 20 minutes.
Dilute the primary antibodies in 5% BSA at the following concentrations: 1:25 anti-BrdU (mouse) and 1:400 anti-BrdU (rat).
Move the coverslip gently down the glass slide to remove it. Do not apply force if the cover slip sticks to the slide. The slide can be rehydrated in PBS until the coverslip becomes loose and can be removed with ease. Collect excess BSA with a paper towel and place the slides horizontally. Pipet 50 μl of the primary antibody solution onto each slide. Cover again with a coverslip to spread the antibody solution evenly over the slide and incubate in a humidified chamber for 2 hours.
After removing the coverslips, wash the slides three times in PBS for 5 minutes
Dilute the secondary antibodies in 5% BSA at the following concentrations: 1:500 sheep anti-mouse Cy3 and 1:400 goat anti-rat Alexa Fluor 488.
Apply 50 μl of the secondary antibody solution as described for the primary antibodies. Protect the slides from light and incubate for 1 hour.
After removing the coverslips, wash the slides three times in PBS for 5 minutes.
Add a drop of Vectashield mounting medium onto each slide and apply a coverslip. Gently press the coverslips and remove excess fluid around it with a paper towel. Seal coverslips with transparent nail polish and let them dry. Store the slides at -20 °C.
5. Image acquisition
Materials: fluorescence microscope, camera
Place a drop of immersion oil onto a slide close to the pencil mark and start locating the fibers. Typically, there is a main fiber bundle, but these fibers are too entangled and cannot be analyzed later. Move away from the main bundle to find areas where fibers are clearly separated from each other (Fig. 2). We would select pictures only using one color channel in order to avoid bias. Then, we would take approximately 10 pictures of each time point, concentration or cell line. However, the number of pictures depends on how many fibers can be counted on each picture. Move along the slide to take the different pictures, as one area of a slide may not provide representative fiber lengths or replication structures.
6. Data analysis
Materials: Picture analysis program, e.g. ImageJ (http://rsbweb.nih.gov/ij/)
Import pictures into a picture analysis program. Measure the lengths of the fiber tracts and/or count the different replication structures (Fig 3). Only count fibers which are clearly visible and which do not extend over the edge of the picture. We typically measure the length of about 100 fibers and count 150-200 replication structures and we would repeat individual experiments at least three times.
7. Representative Results:
Newly replicated DNA can be visualized as lines of antibody-labeled nucleotide analogues. In our experiments an ongoing fork is represented as adjacent red and green signals (Fig 3). The double labeling protocol also allows us to define four major classes of replication structures: 1) new initiation events can be divided into origins that have fired while the cells were incubated with the first label and origins that have fired during the incubation with the second label. The former consist of neighboring green-red-green signals and the latter of a green line only. 2) termination events manifest as adjoining red-green-red signals. 3) interspersed origins are sites in the genome with closely spaced origins. Such sites consist of consecutive origins and termination signals. 4) depending on the experimental design, stalled/collapsed forks can either be defined as a red only signal7 or a red line followed by a short green tract8,9.
Using the conditions described here, wild type DT40 cells have an average fork speed of 0.4 μm/min. We can detect approximately 63% ongoing forks, 10% origins, 16% stalled forks (red only tracts), 8% terminations and 3% interspersed fibers.
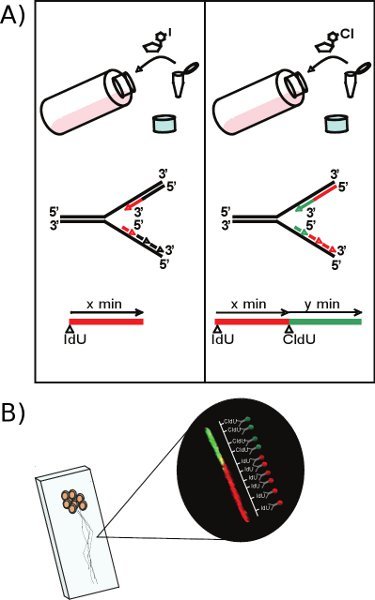 Figure 1. (A) The left hand side of the cartoon depicts the first step of the DNA labeling procedure: adding IdU to exponentially growing DT40 cells leads the nucleotide analogue to be readily incorporated into the newly synthesized leading and lagging DNA strands. This can be visualized by using a specific antibody, and will appear as a red line when viewed under the microscope. Subsequently, the same procedure is repeated with CldU as shown on the right side of the figure. After incubation with the second nucleotide analogue, an active fork will be visible as an adjoining red-green tract. The average fiber length is proportional to the incubation time of the nucleotide analogues. (B) DNA from lysed DT40 cells is stretched by gravity onto a microscope slide and the incorporated nucleotide analogues are visualized by use of specific antibodies and fluorescence microscopy.
Figure 1. (A) The left hand side of the cartoon depicts the first step of the DNA labeling procedure: adding IdU to exponentially growing DT40 cells leads the nucleotide analogue to be readily incorporated into the newly synthesized leading and lagging DNA strands. This can be visualized by using a specific antibody, and will appear as a red line when viewed under the microscope. Subsequently, the same procedure is repeated with CldU as shown on the right side of the figure. After incubation with the second nucleotide analogue, an active fork will be visible as an adjoining red-green tract. The average fiber length is proportional to the incubation time of the nucleotide analogues. (B) DNA from lysed DT40 cells is stretched by gravity onto a microscope slide and the incorporated nucleotide analogues are visualized by use of specific antibodies and fluorescence microscopy.
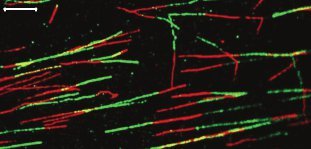 Figure 2. A representative fluorescence image shows fibers from wild type DT40 cells subsequently labeled with IdU and CldU for 20 minutes each. Most of the fibers are well separated from each other and can thus be easily analyzed. The white bar represents 10 μm.
Figure 2. A representative fluorescence image shows fibers from wild type DT40 cells subsequently labeled with IdU and CldU for 20 minutes each. Most of the fibers are well separated from each other and can thus be easily analyzed. The white bar represents 10 μm.
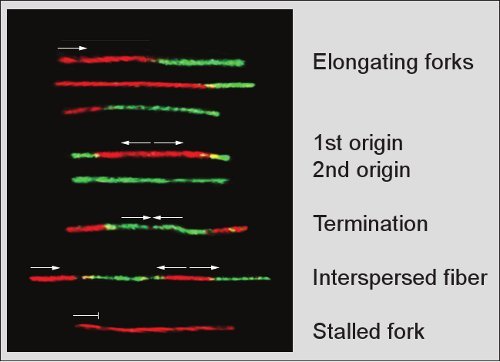 Figure 3. The double-labeling fiber technique allows one to distinguish between different replication structures; 1 - actively replicating forks, 2 - new sites of DNA replication (firing of new origins), 3 - fork terminations (two converging forks), 4 - interspersed fibers (sites of closely spaced origins) and 5 - stalled forks (red only signal).
Figure 3. The double-labeling fiber technique allows one to distinguish between different replication structures; 1 - actively replicating forks, 2 - new sites of DNA replication (firing of new origins), 3 - fork terminations (two converging forks), 4 - interspersed fibers (sites of closely spaced origins) and 5 - stalled forks (red only signal).
Discussion
We describe a method that allows for quantitative analysis of parameters that influence DNA synthesis during S-phase at a single molecule level. Over the past ten years, different versions of the DNA fiber fluorography technique were developed to visualize the movement of individual replication forks within living cells. The critical parameter in all these techniques is the procedure used to achieve the best possible stretching of DNA on glass surfaces providing well-separated DNA fibers. A crucial step to achieve this is the incubation time of the cell suspension on the microscope slide as well as lysis time. These parameters depend on the temperature and airflow in a particular lab and should be determined empirically. The practical part of a DNA fiber experiment is typically accomplished within one day. However, subsequent picture acquisition and analysis is more time consuming and requires practice.
The labeling protocol can be adapted to answer various scientific problems: using an agent that interferes with replication during the second pulse labeling monitors fork elongation/stalling in response to replicative stress. In this scenario, the first label does not need to be washed out as the second nucleotide is added in excess. Alternatively, drugs that impede replication can be used in combination or just after addition of the first label. This requires the first label and the drug to be removed before the second nucleotide analogue is applied. This procedure allows one to determine the importance of various DNA repair factors on replicative fork stability/recovery under conditions of replicative stress (i.e fork stalling and/or collapse). Noteworthy, a more detailed analysis of particulars of the DNA replication program could be performed with the use of an alternative approaches combining DNA combing and the fluorescence in situ hybridization. This however, is technically more challenging and requires expensive equipment.
The ability to qualitatively and quantitatively analyze particulars of DNA replication provides an essential tool for investigating defects in DNA replication itself as well as the pathways that suppress genomic instability associated with replication forks. Undoubtedly, this assay will further help to shed light on the mechanisms of replication-mediated DNA repair that allow normal cells to respond/adapt to environmental changes as well as how cancer cells utilize these mechanisms to resist the toxicity of drugs targeting replication forks.
Disclosures
No conflicts of interest declared.
Acknowledgments
We thank Dr T. Helleday and Dr E. Petermann for help with the fiber technique as well as members of the lab for helpful discussion. WN is supported by a Senior International Research Fellowship from the Association for International Cancer Research and by the Polish State Committee for Scientific Research grant (N301 165935).
References
- Caldwell RB, Fiedler P, Schoetz U, Buerstedde JM. Gene function analysis using the chicken B-cell line DT40. Methods. Mol. Biol. 2007;408:193–210. doi: 10.1007/978-1-59745-547-3_11. [DOI] [PubMed] [Google Scholar]
- Petes TD, Williamson DH. Fiber autoradiography of replicating yeast DNA. Exp. Cell. Res. 1975;95:103–110. doi: 10.1016/0014-4827(75)90614-x. [DOI] [PubMed] [Google Scholar]
- Takeuchi F. Altered frequency of initiation sites of DNA replication in Werner's syndrome cells. Hum. Genet. 1982;60:365–368. doi: 10.1007/BF00569220. [DOI] [PubMed] [Google Scholar]
- Jackson DA, Pombo A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J. Cell. Biol. 1998;140:1285–1295. doi: 10.1083/jcb.140.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab RA, Blackford AN, Niedzwiedz W. ATR activation and replication fork restart are defective in FANCM-deficient cells. EMBO. J. 2010;29:806–818. doi: 10.1038/emboj.2009.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribasak H, Arakawa H. Basic cell culture conditions. Subcell. Biochem. 2006;40:345–346. doi: 10.1007/978-1-4020-4896-8_21. [DOI] [PubMed] [Google Scholar]
- Conti C, Seiler JA, Pommier Y. The mammalian DNA replication elongation checkpoint: implication of Chk1 and relationship with origin firing as determined by single DNA molecule and single cell analyses. Cell. Cycle. 2007;6:2760–2767. doi: 10.4161/cc.6.22.4932. [DOI] [PubMed] [Google Scholar]
- Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmunds CE, Simpson LJ, Sale JE. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol. Cell. 2008;30:519–529. doi: 10.1016/j.molcel.2008.03.024. [DOI] [PubMed] [Google Scholar]