

Abstract
Although it is increasingly affordable for emerging model organisms to obtain completely sequenced genomes, further in-depth gene function and expression analyses by RNA interference and stable transgenesis remain limited in many species due to the particular anatomy and molecular cellular biology of the organism. For example, outside of the crown group Caenorhabditis that includes Caenorhabditis elegans3, stably transmitted transgenic lines in non-Caenorhabditis species have not been reported in this specious phylum (Nematoda), with the exception of Strongyloides stercoralis4 and Pristionchus pacificus5. To facilitate the expanding role of P. pacificus in the study of development, evolution, and behavior6-7, we describe here the current methods to use microinjection for making transgenic animals and gene knock down by RNAi. Like the gonads of C. elegans and most other nematodes, the gonads of P. pacificus is syncitial and capable of incorporating DNA and RNA into the oocytes when delivered by direct microinjection. Unlike C. elegans however, stable transgene inheritance and somatic expression in P. pacificus requires the addition of self genomic DNA digested with endonucleases complementary to the ends of target transgenes and coinjection markers5. The addition of carrier genomic DNA is similar to the requirement for transgene expression in Strongyloides stercoralis4 and in the germ cells of C. elegans. However, it is not clear if the specific requirement for the animals' own genomic DNA is because P. pacificus soma is very efficient at silencing non-complex multi-copy genes or that extrachromosomal arrays in P. pacificus require genomic sequences for proper kinetochore assembly during mitosis. The ventral migration of the two-armed (didelphic) gonads in hermaphrodites further complicates the ability to inject both gonads in individual worms8. We also demonstrate the use of microinjection to knockdown a dominant mutant (roller,tu92) by injecting double-stranded RNA (dsRNA) into the gonads to obtain non-rolling F1 progeny. Unlike C. elegans, but like most other nematodes, P. pacificus PS312 is not receptive to systemic RNAi via feeding and soaking and therefore dsRNA must be administered by microinjection into the syncitial gonads. In this current study, we hope to describe the microinjection process needed to transform a Ppa-egl-4 promoter::GFP fusion reporter and knockdown a dominant roller prl-1 (tu92) mutant in a visually informative protocol.
Keywords: Developmental Biology, Issue 56, RNA interference, Pristionchus pacificus, microinjection, transgenesis, Caenorhabditis elegans, developmental biology, behavior, gene expression
Protocol
1. Transgenesis: DNA preparation
Dominant co-injection marker: pRL3 [Ppa-prl-1(tu92)] The pRL3 plasmid is a dominant co-injection marker for visually identifying successful transformation events. This plasmid encodes for a dominant mutant allele (tu92) of the Ppa-prl-1 gene closely related to the sqt-1 collagen gene in C. elegans and transforms the wildtype sinusoidal locomotion into clockwise twisting motions along the worm's body axis1,5. The transformed animal is very similar to the popular dominant selection marker rol-6 (su1006) used in C. elegans for the past 20 years.
Target reporter transgene: Ppa-egl-4p::gfp A gene of interest can be linked transcriptionally or translationally to a GFP coding sequence9. In this study, the Ppa-egl-4promoter::gfp (egl-4, cGMP dependent protein kinase) was used to determine if a 2 kb fragment upstream of the start of translation can confer GFP expression in P. pacificus PS312. This GFP vector contains a GFP coding region with modified introns and the multipurpose Ppa-rpl-23 3' UTR terminator5 kindly provided by Xiaoyue Wang and Ralf J. Sommer (Max-Planck Institute, Tuebingen, Germany, EU).
Genomic DNA: PS312 The addition of wildtype P. pacificus PS312 genomic DNA is required to obtain stable transgene inheritance, particularly from transgenic F1 animals to their F2 progeny5. It is not yet certain if the genomic DNA forms complex extra chromosomal arrays with the two transgenes (the co-injection marker pRL-3 and the Ppa-egl-4p::gfp reporter) similar to those observed in stable F2 transgenic lines in C. elegans3. However, the requirement to have the genomic DNA and transgenes to share identical cohesive overhangs strongly suggests the formation of complex extra-chromosomal arrays the host cells can recognize for proper DNA transmission (gDNA prepared using Sigma G1N10 kit).
DNA digestion Restriction enzymes are used to linearize the Ppa-egl-4p::gfp vector DNA and to produce sticky overhangs compatible with the co-injection marker pRL3 and PS312 gDNA. Mix by tapping, not vortexing. Incubate for one hour at 37°C.
| pRL3 | 4 μg |
| 10xbuffer | 10 μl |
| PstI (10 U/μl) | 4 μl |
| dH2O | ~ |
| Total: | 100 μL |
| Ppa-egl-4p::gfp | 4 μg |
| 10x buffer | 10 μl |
| SalI (10 U/μl) | 4 μl |
| dH2O | ~ |
| Total: | 100 μl |
| gDNA PS312 | 10 μg |
| 10x buffer | 10 μl |
| Pst I and Sal I (10 U/μl) | 8 μl |
| dH2O | ~ |
| Total: | 100 μl |
Table 1. Mixture for Restriction Enzyme Digest.
- DNA precipitation and preparation
- Add 1:10 sodium acetate (3 M NaCOOCH3 pH 5.2) and 2.5X the volume of 100% ethanol to the to digested product; add 20 ng/μl of glycogen to see pellet easier.
- Centrifuge at 14,000 rpm for 15 minutes at 4°C.
- Decant the supernatant by tipping slowly the centrifuge tube upside down and then tapping it on a tissue paper, making sure that the pellet is secure at the bottom corner of the tube and free of ethanol.
- Add 1 ml of 75% ethanol.
- Immediately spin at 14,000 rpm for 5 minutes at 4°C.
- Decant the supernatant by slowly tipping the centrifuge tube upside down and then tapping it on a tissue paper making sure that the pellet it secure at the bottom corner of the tube and free of ethanol.
- Dry pellet in vacuum centrifuge at 14,000 rpm for 5 minutes at 25-30°C, until dry and completely free of ethanol.
- Add 30 μl of TE or sterile dH2O and leave overnight at 4°C before mixing briefly with a vortex.
- Table 2 describes how to create the injection mixture from each purified DNA for injection.
- Store at -20°C. After thawing the mixture for injection, spin down the tube at 14,000 rpm for 5 minutes to rid debris particles that may clog the injection needle.
| Genetic material | Concentration |
| Ppa-egl-4p::gfp (SalI) | >5 ng/μl (0.1-10ng/μl) |
| pRL3 (PstI) | >1 ng/μl |
| gDNA PS312 (PstI + SalI) | >60 ng/μl |
| dH2O | ~ |
| Total: | 30 μl |
Table 2.Ppa-prl-1 injection mixture
2. RNA Interference: In vitro double stranded RNA synthesis
Use PCR to amplify the gene of interest. RHL091 5'- agtggatccGAAGGTCCATACGGGAGC-3' (BamHI site) and RHL092 5'- tatctgcagGTGAGGAGTACCAGGAGAG-3' (PstI site) were used to amplify a 464 bp genomic fragment (position 3-466) from the pRL3 vector containing the Ppa-prl-1(tu92) allele.
Digest the PCR product with BamHI and PstI.
Ligate the fragment to BamHI/PstI digested pL4440 RNAi feeding vector2.
Transform the inserted vector into competent cells and grow them on Ampicillin LB media plates (50 μg/ml).
Select the successfully inserted vector by PCR using the T7 primers.
Use the T7 flanked PCR product containing the prl-1 gene to make double-stranded RNA dsRNA) using the BLOCK-IT RNAi synthesis kit (Invitrogen).
The dsRNA concentration is best if >200 ng/μl, up to 1 μg/μl for injection.
Store at -20°C. After thawing the mixture for injection, spin down the tube at 14,000 rpm for 5 minutes to rid of debris particles that may clog the injection needle.
3. Microinjection: Protocol for injection of transgenes and/or dsRNA
- Injection Needles
- Set the Narishige needle puller (model #: PC-10) temperature:
- Turn bottom knob to "No.2 Heater."
- Turn the "No.2 Heater Adj." knob until panel reads 55.0-60.0°C for tapered needle tip of varying lengths.
- Turn the bottom knob back to "Step 1" (Pulls needle in one step).
- Load needle into the chamber and make sure it is secure.
- Press the "Start" button (red button) and allow needle to be pulled with all the weights (this should make two identical needles in opposite directions).
- Remove needle from the chamber and store in a plastic culture plate secured in place by Play-Doh to prevent dust from accumulating on the needles.
- Mounting Pads
- In a 1.5 ml centrifuge tube, make a 2% Noble Agar solution by adding 0.02 g of Noble Agar to 1 ml of H2O. The agar can be stored at 4°C for up to a year.
- Mix thoroughly and melt agar in a heat block set to >88°C.
- Using a 1 ml micropipette, place a drop of liquid 2% Noble Agar to the middle of the glass slide (Fisher, 12-544-F).
- Flatten by placing another glass slide on top of the drop.
- Repeat "Drop Step" until there is a layer of five-glass slide.
- Remove each glass slide by sliding the glass slides off each other.
- Dry flatten agar pad on glass slides in a vacuum oven at 70°C for 4 hours to overnight (or at room temperature overnight).
- Make multiple Injection pads and store for future use.
Microinjection into hermaphroditic gonads
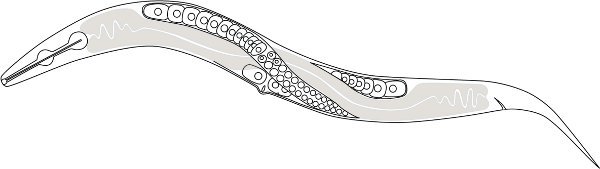 Figure 1. A schematic drawing of the P. pacificus anatomy. The clear nucleated cells are female germ cells (oocytes) and the gray shaded part is the intestine. Only one distal anterior gonad is shown but notice the two distal gonads cross each other dorsally near mid-body.
Figure 1. A schematic drawing of the P. pacificus anatomy. The clear nucleated cells are female germ cells (oocytes) and the gray shaded part is the intestine. Only one distal anterior gonad is shown but notice the two distal gonads cross each other dorsally near mid-body.
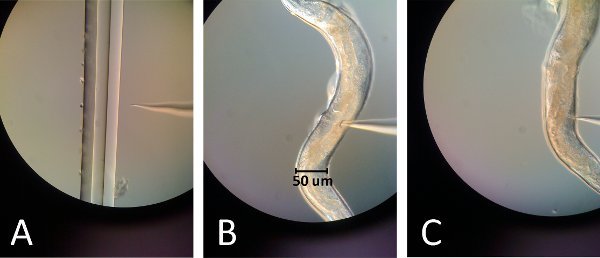 Figure 2. Images of microinjection. (A) The injection needle tip is in focus with the far right line of the pulled capillary piece. By lightly touching the tip of the needle to that edge, the needle tip should break while still retaining a sharp point. (B) The needle inserts into the top gonad just above the gut curvature for the first injection. (C) The needle inserts into the bottom gonad just below the gut curvature for the second injection.
Figure 2. Images of microinjection. (A) The injection needle tip is in focus with the far right line of the pulled capillary piece. By lightly touching the tip of the needle to that edge, the needle tip should break while still retaining a sharp point. (B) The needle inserts into the top gonad just above the gut curvature for the first injection. (C) The needle inserts into the bottom gonad just below the gut curvature for the second injection.
- Make sure the contrast and DIC shearing setting is ideal for viewing the hermaphrodite gonads.
- Load 1.0-2.0 μl of injection mixture into the pulled capillary needle.
- Place the needle in the microinjection manipulator; pressure gauges of the nitrogen tank should be set to ideal conditions (10-20 psi for 0.5-1 sec).
- Break the needle tip by touching it slightly to edge of a thinly pulled capillary tube placed on a slide (figure 2A).
- Needle breaker slide: the thinnest part of a pulled capillary placed on a glass slide in a drop of oil
- Once the needle is broken it can be place at an idle position while you pick your worm to the pad (P. pacificus have a 5 minutes window for injection before they dry-out and die).
- Using a plate full of J4-young adult worms, pour enough paraffin oil (heavy) to just cover all the worms in OP50 bacterial lawn.
- Pick a worm and place it on the injection pad.
- Place the glass slide with the worm on the microscope; position the worm's gonad in direction of the needle and the vulva away from the needle (Figure 2B, 2C).
- Lower the needle in to position of the microscope view.
- Position the top gonad and the needle in the same focal plane (Figure 2B).
- Poke the worm gently and pump in the injection mixture; ooctyes separating towards the distal tip indicate a successful injection (To optimize transgene or dsRNA delivery, you should upon injection see a wave of expansion in the gonad that continues until it is close to the distal tip).
- Reposition the needle to inject to the lower gonad and repeat injection; however since this gonad is under the gut the spreading of the oocytes will not be seen and is thus more difficult to achieve (Figure 2C).
- Normal looking worm indicates a successful injection after the injection; there should not be any damaged gonads (protruding out from the vulva or popped inside the body cavity).
- Place the needle back in idle position.
- Prepare to rescue the worm.
- Add a drop (0.5 μl) of M9 buffer onto the pad where the injected worm is placed.
- Using some OP50 bacteria on your pick, touch the worm with the OP50, and it should stick. We do not use mouth pipette.
- Place the worm onto plate seeded with 50 μl spots of OP50, 2-4 worms can be rescued on such plates.
- Post-injection Storage & Screening for Transgenesis
- Store injected worms (P0) at 20°C for ~4 days to lay eggs and grow up F1.
- Screen worms on the fourth day for selected phenotype (To screen for transgenesis, search for rolling animals expressing the Ppa-prl-1 dominant marker).
- Pick single independent F1's animals with the observed phenotype to new seeded plate and store at 25°C; this temperature encourages the formation of stable lines from F1's.
- Single F2 from independent F1 lines. Individual F2 lines have similar transgene transmission rates. We often obtain >15 F2 lines per transformed F1 animal.
- Store subsequent generations at 25°C.
- The transmission rate of rollers usually remains constant after the F4 generation, thus it is necessary at this time to determine the expression level of the target transgene (Ppa-egl-4p::gfp) for each individual line. GFP expression may not correlate with the extent of penetrance of the dominant roller phenotype.
- Post-injection Storage & Screening for RNAi phenotypes
- Store injected worms (P0) at 20°C for ~4 days to lay and grow F1.
- Screen F1 worms on the fourth day for selected phenotype (To screen for RNAi knockdown phenotype, search for penetrant phenotypes that may be associated with loss of target gene activity, in our case the loss of the Ppa-prl-1 phenotype).
- In our experience, the non-roller knockdown phenotype is lost in >97% of the F2.
4. Representative results:
1. Result of Transgenesis
| Session | Injected P0 | F1 Rollers | % transgenic F2 | Average % transmission after F2 |
| 1 | 60 | 2 | (line 1) 0% (line 2) 13% | NA 13%±10 |
| 2 | 40 | 1* | (line 3) 0% | NA |
| 3 | 40 | 0 | 0% | NA |
| 4 | 40 | 1 | (line 4) 0% | NA |
| 5 | 20 | 0 | 0% | NA |
| 6 | 40 | 1 | (line 5) 26% | 22%±10 |
* a male roller that did not cross; NA: Not applicable
Table 3. Results of injected PS312 with Ppa-egl-4p::gfp (Sal I digested), pRL3(roller) (Pst I digested) , and gDNA PS312 (PstI and SalI digested).
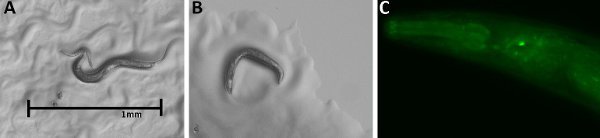 Figure 3. (A) wildtype (B, C) A stable F4
pRL3; Ppa-egl-4p::gfp transgenic line showing head neuron GFP expression.
Figure 3. (A) wildtype (B, C) A stable F4
pRL3; Ppa-egl-4p::gfp transgenic line showing head neuron GFP expression.
2. Result of RNA interference
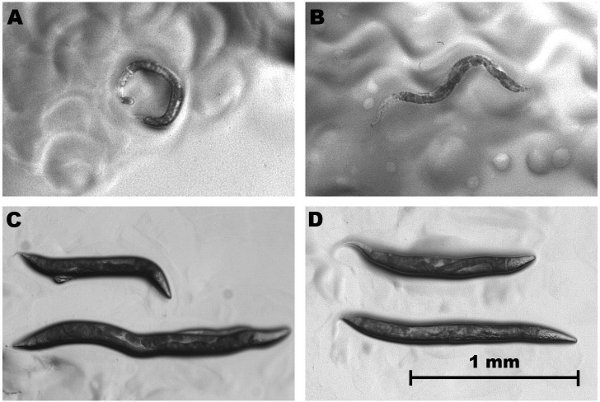 Figure 4. RNA injected prl-1 mutants show complete knockdown of "rolling" locomotion. (A) prl-1 roller gain-of-function mutant tu92. (B) "knocked down" prl-1 mutant exhibit normal wildtype posture and locomotion. (C) Injected prl-1 (bottom) also has longer body than prl-1 mutant (top). (D) The longer body of injected prl-1 (bottom) is also longer than the wildtype PS312 (top). The longer body phenotype was also observed in the rol-5 (sqt-1) RNAi knockdown of C. elegans N2 (data not shown).
Figure 4. RNA injected prl-1 mutants show complete knockdown of "rolling" locomotion. (A) prl-1 roller gain-of-function mutant tu92. (B) "knocked down" prl-1 mutant exhibit normal wildtype posture and locomotion. (C) Injected prl-1 (bottom) also has longer body than prl-1 mutant (top). (D) The longer body of injected prl-1 (bottom) is also longer than the wildtype PS312 (top). The longer body phenotype was also observed in the rol-5 (sqt-1) RNAi knockdown of C. elegans N2 (data not shown).
| roll | non-roll | |
| A prl-1 dsRNA (200 ng/μl) | 13 | 21 |
| B prl-1 dsRNA (1000 ng/μl) | 9 | 16 |
| control | 20 | 2 |
Table 4. Summary of Ppa-prl-1 dsRNA injections. (A) [dsRNA] = 200 ng/μl; (B) [dsRNA] = 1000 ng/μl. P = 0.0019 and 0.041 by Fisher's Exact Test, two-tailed, for 200 and 1000 ng/μl injections, respectively.
Discussion
P. pacificus populations are found in close association with various scarab beetle species worldwide and is a model nematode intermediate between free living and parasitic nematodes. The strength of the P. pacificus as an emerging model organism lay in the integration of its genetic and physical maps that promote positional mapping of mutants isolated from unbiased forward genetic screens (i.e. not just for candidate genes previously characterized in C. elegans)6,10. However, C. elegans genetic techniques are not readily transferable to P. pacificus due to the significant differences in organ morphology, primary DNA sequences, as well as response to foreign DNA. Our present study illustrates in detail how to introduce stable reporter genes previously described by Schlager et al5 and how to knock down the same dominant roller mutant by RNAi. Our protocol does not presume prior knowledge of transgenesis or RNAi in C. elegans.
The F1 transformation rate shown in this study (˜2%) is comparable to previous results using the prl-1 marker in P. pacificus (2-10%)5, but less than the rate found in S. stercoralis (3-22%)4. There is still much potential for improvement of transgenic technology in P. pacificus. Even using the prl-1(tu92) dominant roller marker homologous to the commonly used rol-6 (su1006) allele in C. elegans, the effectiveness of transgenesis in P. pacificus is significantly less than those first described for C. elegans by Mello and co-workers3, in which multiple transgenic F1 progeny can be obtained per injected animal in expert hands. Several factors may explain this difference: (1) The lower number of oocytes in diakinesis in the P. pacificus female germline (average 1 per gonad)8 may reflect a lower rate of mitotic germ cells transitioning to meiosis in P. pacificus compared to C. elegans11. Hence, fewer oocytes can take up DNA and RNA following each injection. The overall lower brood size per hermaphrodite in P. pacificus PS312 (˜200) compared C. elegans N2 (˜300) may also exacerbate the lower number of the transgenic F1 per injected animal. (2) The requirement for genomic DNA in the injection mix for transmission of foreign DNA from F1 to F2 suggest a stronger gene silencing mechanism may also be involved in P. pacificus than in C. elegans. Nevertheless, P. pacificus transgene expression does not seem to undergo the extreme gene silencing found in S. stercoralis in which transgene expression is limited to F1 animals4. We are currently characterizing the expression of PPa-egl-4p::gfp lines in F6 animals. One straightforward method to improve rates of transgenesis is to increasing the concentration of the roller co-injection marker (currently 1 ng/μl of pRL3 compared to 25-50 ng/μl of pRF4 in C. elegans).
The ability to manipulate gene function at the organismal level by RNAi and transgenesis are the twin pillars of technology that elevate C. elegans above many other model organisms. We hope our current study will greatly enhance the P. pacificus model for genetics studies in development and behavior by providing easily accessible instructions for transgenesis and RNAi.
Disclosures
No conflicts of interest declared.
Acknowledgments
The authors are very grateful to RJ Sommer and X Wang for assistance with microinjection, as well as insightful comments from the anonymous reviewers. This work is supported by NIH grant SC2GM089602.
References
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Ahringer J. Reverse Genetics. Wormbook. 2006.
- Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junio AJ, Li X, Massery HC, Nolan TJ, Lamitina ST, Sundaram MV, Lok JB. Strongyloides stercoralis: cell- and tissue-specific transgene expression and co-transformation with vector constructs incorporating a common multifunctional 3' UTR. Exp. Parasitology. 2008;118:253–265. doi: 10.1016/j.exppara.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlager B, Wang X, Braach G, Sommer RJ. Molecular cloning of a dominant roller mutant and establishment of DNA-mediated transformation in the nematode Pristionchus pacificus. Genesis. 2009;47:300–304. doi: 10.1002/dvg.20499. [DOI] [PubMed] [Google Scholar]
- Hong RL, Sommer RJ. Pristionchus pacificus: a well-rounded nematode. Bioessays. 2006;28:651–659. doi: 10.1002/bies.20404. [DOI] [PubMed] [Google Scholar]
- Hong RL, Sommer RJ. Chemoattraction in Pristionchus nematodes and implications for insect recognition. Curr. Biol. 2006;16:2359–2365. doi: 10.1016/j.cub.2006.10.031. [DOI] [PubMed] [Google Scholar]
- Rudel D, Riebesell M, Sommer RJ. Gonadogenesis in Pristionchus pacificus and organ evolution: development, adult morphology and cell-cell interactions in the hermaphrodite gonad. Dev. Biol. 2005;277:200–221. doi: 10.1016/j.ydbio.2004.09.021. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;11:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- Chaudhuri J, Parihar M, Pires-daSilva A. An Introduction to Worm Lab: from Culturing Worms to Mutagenesis. J. Vis. Exp. 2011. pp. e2293–e2293. [DOI] [PMC free article] [PubMed]
- Evans TC. Transformation and Microinjection. Wormbook. 2006.