

Abstract
Inflammatory bowel disease (IBD), encompassing Crohn's disease and ulcerative colitis, is associated with enhanced leukocyte infiltration to the gut, which is directly linked to the clinical aspects of these disorders. Thus, leukocyte trafficking is a major target for IBD therapy. Past and emerging techniques to study leukocyte trafficking both in vitro and in vivo have expanded our knowledge of the leukocyte migration process and the role of inhibitors. Various strategies have been employed to target chemokine- and integrin-ligand interactions within the multistep adhesion cascade and the S1P/S1PR1 axis in leukocyte migration. Though there is an abundance of preclinical data demonstrating efficacy of leukocyte trafficking inhibitors, many have yet to be confirmed in clinical studies. Vigilance for toxicity and further research is required into this complex and emerging area of IBD therapy.
1. Introduction
Leukocyte migration is fundamental to immunologic mobilization in response to insult and injury. A coordinated series of molecular events underpins the trafficking of lymphocytes and granulocytes into stressed tissue with participation of adhesion molecules, chemokines, and their receptors.
The inflammatory bowel diseases (IBDs), Crohn's disease (CD) and ulcerative colitis (UC), are distinct syndromes, but both are associated with enhanced leukocyte trafficking to the inflamed gut. Thus, targeting the multistep leukocyte adhesion cascade has been employed as a therapeutic strategy. Here, we focus on the contribution of leukocyte trafficking to the pathogenesis of IBD. An overview of strategies employed to target leukocyte recruitment and of emerging models used to test these targets is presented.
2. IBD
Crohn's disease and ulcerative colitis are recurring, relapsing, and remitting disorders characterized by chronic inflammation of the intestinal mucosa. Though they share some common clinical symptoms such as diarrhea and abdominal pain, CD and UC possess very distinct features. Crohn's disease manifests as a transmural inflammation that can potentially develop anywhere along the gastrointestinal tract but primarily occurs in the terminal ileum and proximal colon. Ulcerations, granulomas, and bowel fistulas are characteristic histopathological features. In contrast, ulcerative colitis seldom ulcerates and is a relatively superficial inflammation of the mucosa that is diffuse, continuous, and restricted to the colon, usually extending proximally from the rectum. It is characterized by significant goblet cell depletion and crypt abscesses, but granuloma development is not a feature. In both CD and UC, leukocyte infiltration into the inflamed intestine is fundamental to disease development and perpetuation. The infiltrated effector cells resist apoptosis and persistently release harmful inflammatory cytokines causing tissue damage. In CD, the characteristic granulomas form upon dense accumulation of activated T cells and macrophages. Ulcerative colitis is characterized by excessive mucosal infiltration of T cells and neutrophils, the latter forming the characteristic crypt abscesses [1]. The pivotal role of T cells, neutrophils, and their proinflammatory cytokines in the pathogenesis of IBD has been reviewed elsewhere [2, 3].
Genetic and environmental risk factors have been implicated in IBD (for reviews see Xavier and Podolsky [1], Melgar and Shanahan [4], and Cho and Brant [5]). Despite extensive research, current therapeutic options in IBD remain limited, often varying in their maintenance, toxicity, and tolerability [6–8]. Novel therapeutic strategies for IBD are needed.
3. Leukocyte Trafficking
3.1. In Vitro and In Vivo Models of Leukocyte Trafficking
3.1.1. Traditional Methods
The concept of lymphocyte recirculation and homing was first demonstrated by Gowans and Knight in 1964, when they followed the migration of radiolabeled lymphocytes in rats using autoradiography and scintography [9]. They and others found that while naïve lymphocytes migrated to all secondary lymphoid tissues, activated lymphocytes preferentially migrated back to the tissue in which they had been exposed to the antigen. In order to devise rational therapeutic strategies that target leukocyte migration, it is important to elucidate leukocyte trafficking patterns in vivo. Techniques employed to study leukocyte trafficking frequently involve ex vivo labeling of donor cells and adoptive transfer into recipient animals. Subsequently, the distribution of the labeled cell population in recipient tissues is assessed using a variety of imaging methods, such as histological analysis of fixed tissues. This technique has provided us with valuable mechanistic knowledge, but it does not permit direct examination of dynamic processes at single-cell level or provide temporal or spatial information within the physiological environment of lymphoid tissues. Furthermore, labeling of cells ex vivo has been associated with variable labeling efficiency, alteration of cellular functions, and label elution postadoptive transfer. Radiolabeling techniques, such as white cell scintigraphy, have been used to successfully study neutrophil migration in clinical IBD studies [10]. However, such methods are hampered by radioactive decay, poor resolution, and cellular toxicity [11]. Myeloperoxidase (MPO) assays are commonly used to study neutrophils and quantitate neutrophil influx, but they do not distinguish between neutrophils and macrophages and can be problematic to carry out. Similarly, while in vitro chemotaxis assays are regularly employed to analyze the effects of potential inhibitors on cell migration, there is no guarantee that the cells will respond in the same way to the test compound in vivo. Multiphoton intravital microscopy has been widely used to image the dynamic movement of lymphocytes by tracking fluorescently labeled cells in exposed or explanted lymph nodes (LNs) of living animals. This technique has provided valuable insights into the dynamics of T- and B-cell homing to LNs [12, 13] and allows single-cell tracking, in conjunction with high-resolution images. Nonetheless, intravital microscopy is an invasive technique, and the surgery required may interfere with the flow of blood and lymph creating experimental artifacts.
3.1.2. Molecular Imaging Techniques
Molecular imaging is defined as ‘‘the visualization, characterization and measurement of biological processes at the molecular and cellular levels in humans and other living systems” [14]. Over the past decade, a number of such techniques have been adapted to small animal imaging, offering dynamic imaging methods to localize leukocytes in vivo. Positron emission tomography (PET) [15] and single-photon emission-computed tomography (SPECT) [16, 17] use scintillation cameras and other devices to detect radioactive emission from radiolabeled cells within the body. Though these nuclear imaging methods have excellent tissue penetration and cell quantitation capability, they too are subject to the drawbacks associated with radiotracers and exogenous labeling of cells. Magnetic resonance imaging (MRI), which relies on the nuclear resonance of protons in tissues upon scanning with radio frequency radiation, has shown great promise to track the recruitment of, for example, antigen-specific CD8+ T cells to target tumors in vivo [18]. However, the time required for imaging using this technique makes it unsuitable for tracking fast-moving cells [19]. The substantial expense of MRI and its relatively poor sensitivity and quantitative capability have also hindered its use as a basic research tool. Optical fluorescence imaging has been frequently used to track T cells and has provided us with invaluable data on their migration patterns. The pitfalls with using fluorescent labels, such as CFSE and GFP, include signal loss due to label dilution upon cell division and limited sensitivity and specificity due to endogenous tissue autofluorescence and light scattering and absorption [20].
3.1.3. Bioluminescence Imaging
To illuminate cell trafficking in vivo and to test specific inhibitors of this migration, we used bioluminescence imaging (BLI) technology [21–23]. This form of optical imaging has several advantages over other techniques for tracking cell migration. It eliminates the necessity to prelabel cells, avoiding problems with exogenous cell labeling. It allows direct in vivo and ex vivo visualization, with no further processing of tissues required, and is, therefore, a less complicated and less labor intensive technique than most other in vitro/ex vivo methods. Unlike other molecular imaging methods, BLI combines high sensitivity with relatively low cost while providing quantitative, spatial, and temporal data. However, this technology has limitations. Since light transmission through animal tissues is wavelength dependent, loss of photon signal can occur with tissue depth and light sources closer to the surface of the animal can appear brighter. In addition, validation of potential therapeutics using BLI should be carried out in conjunction with in vitro mechanistic assays and in vivo efficacy studies. BLI has successfully monitored trafficking of bone marrow mononuclear cells in ischemic myocardium [24] and CD4+ T cells in an experimental model of multiple sclerosis (MS) [25]. These and other studies have shown robust and reliable correlation between cell numbers and bioluminescence signals. We employed BLI to track both neutrophils and lymphocytes in murine models of experimental colitis and to test potential inhibitors of their migration Figure 2 [21, 22].
Figure 2.
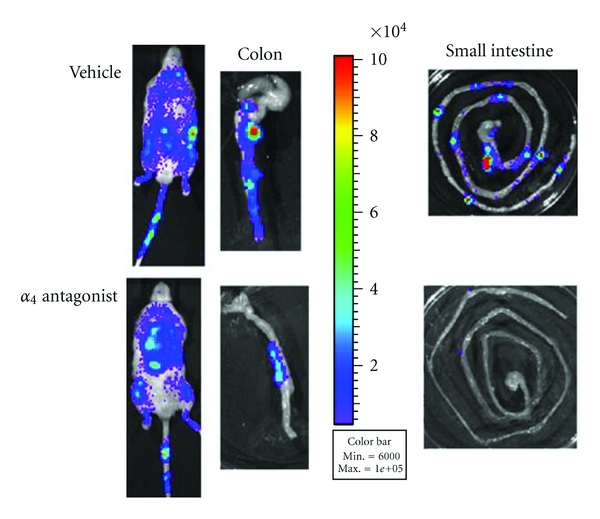
Inhibition of leukocyte migration by an α 4 integrin antagonist in experimental colitis. Leukocytes were isolated from mesenteric lymph nodes (MLNs) of β-actin luciferase mice and injected into recipient mice with dextran sodium sulphate- (DSS-) induced colitis. The recipient mice received vehicle or α 4 antagonist, 1 hour pre cell transfer. Whole body and organs ex vivo (colon and small intestine) were imaged using an IVIS 100 charge-coupled device imaging system 4 hours following transfer. The pseudocolored images represent light intensity, where red is the strongest, and violet is the weakest signal. Inhibition is detected in the colon and in Peyer's patches of the α 4 antagonist-treated mice.
3.2. The Leukocyte Trafficking Cascade
3.2.1. Selectins, Integrins, and Their Ligands
Circulating leukocytes are subjected to extreme conditions with the flow of blood exerting a shearing stress on the cells dislodging any that touch the vascular wall. To leave the circulation and home to specific tissues, leukocytes must engage several adhesion pathways involving intimate interaction with endothelial cells [26]. Whether during physiological recirculation or inflammatory conditions, the mechanisms involved in leukocyte trafficking are effectively the same. The leukocyte trafficking cascade is depicted in Figure 1. Leukocyte recruitment begins with tethering and rolling of the cells along the microvascular endothelium via three selectins: L-selectin, expressed by leukocytes, and E-selectin and P-selectin, expressed by inflamed endothelial cells on the blood vessel wall [27]. The ligand P-selectin glycoprotein ligand-1 (PSGL-1) binds all three selectins and plays an important role in leukocyte recruitment under inflammatory conditions [28]. L-selectin is constitutively present on T cells and interacts with its counter receptors, peripheral lymph node addressin (PNAd) and mucosal addressin cell adhesion molecule (MAdCAM)-1, acting as a mechanical anchor or tether to the high endothelial venules (HEVs). This allows the lymphocytes to roll along the vascular lining at a much slower pace than erythrocytes. Selectins engage rapidly and form strong bonds to secure contact. These bonds allow chemokines and their ligands to transmit activating signals for the next step in the migration cascade. Targeting selectins and their ligands as a treatment strategy for inflammatory disorders has been reviewed elsewhere [29].
Figure 1.
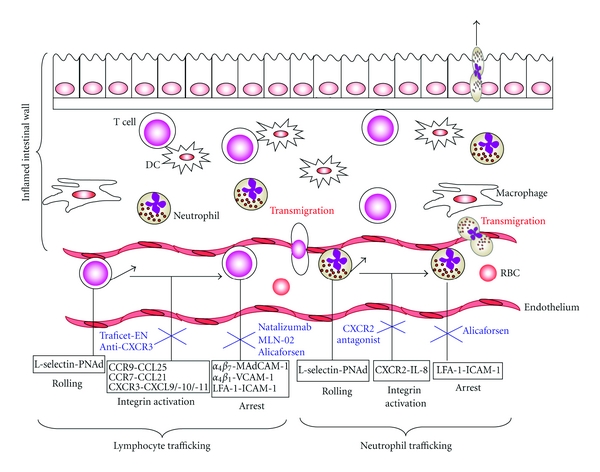
Schematic view of the leukocyte trafficking adhesion cascade in IBD. Leukocytes tether and roll along the vascular endothelium via selectin-mediated adhesion. They are then activated by chemokines into a high avidity, high affinity state so that integrin-mediated strong adhesion and arrest can take place. This prepares the leukocyte for transmigration through the blood vessel wall into the inflamed colon. Chemokine activation can be inhibited by various chemokine/chemokine receptor inhibitors such as the CCR9 small molecule antagonist Traficet-EN, a monoclonal antibody to CXCR3 or a CXCR2 antagonist (shown in blue). Additionally, antagonists of integrin firm adhesion include the anti- α 4 integrin monoclonal antibody Natalizumab, the selective α 4 β 7 small molecule antagonist MLN-02 and the antisense intercellular adhesion molecule-1 (ICAM-1) oligonucleotide Alicaforsen (shown in blue). KO, knock out; LFA-1, Lymphocyte function-associated antigen 1; MadCAM-1, mucosal addressin-cell adhesion molecule 1; PNAd, Peripheral lymph node addressin; RBC, red blood cell; V-CAM-1, vascular-cell adhesion molecule 1.
Selectin bonds are unable to arrest cells at the vessel wall. Firm leukocyte adhesion is achieved through bonds formed downstream by the secondary adhesion molecules, integrins. Integrins are diversely expressed on different leukocyte subpopulations and are composed of noncovalently linked α and β chains. The α 4 integrins, α 4 β 1 and α 4 β 7, play a regulatory role in lymphocyte homing and recruitment to inflammatory tissues, particularly to the inflamed intestine. Two decades ago, it was revealed that memory T cells from the gut preferentially homed to the gut [30]. This phenomenon is linked to the expression of unique adhesion molecules within the mucosa [31, 32]. Both α 4 β 1 and α 4 β 7 are expressed by lymphocytes that reside in the gut and gut-associated lymphoid tissues (GALTs), and their respective ligands vascular cell adhesion molecule (VCAM)-1 and MAdCAM-1 are expressed within the HEV's in Peyer's patches (PPs) and the flat-welled venules of the lamina propria [33]. While the α 4 β 7 -MAdCAM-1 interaction is restricted to leukocyte trafficking to the gut and GALT, the α 4 β 1-VCAM-1 pathway can also mediate leukocyte homing to the central nervous system, specifically to the inflamed brain [34]. Interestingly, mice deficient in the β 7 integrin gene are unable to form proper PPs and possess decreased numbers of lamina propria CD4+ T cells and B cells [35]. The β 2 integrins are also prominent participants in leukocyte trafficking, mediating firm adhesion, particularly in the case of neutrophils [36]. The β 2 integrin lymphocyte function-associated antigen (LFA)-1 is predominantly expressed by lymphocytes and neutrophils and binds to its endothelial cell ligands intercellular adhesion molecule (ICAM)-1 and ICAM-2. In addition to leukocyte arrest, integrins can participate in leukocyte rolling. Under inflammatory conditions, lymphocytes can skip the selectin-mediated phase and bind directly to endothelial cells via α 4 β 7 [37].
Abundant evidence reveals that IBD is associated with enhanced leukocyte trafficking to the gut mucosa and altered expression of adhesion molecules [38]. Cytokines such as interleukin (IL)-1β, IL-6, and TNF-α, produced upon stimulation of innate immune cells at inflammatory sites [39], upregulate adhesion molecules and chemokines, enhance leukocyte recruitment, and amplify the inflammatory cascade. Expression of MAdCAM-1 is upregulated in animal models of colitis [40, 41] and in active IBD [42–44]. ICAM-1 and LFA-1 have also been implicated in a number of experimental animal models of IBD [45, 46]. The importance of adhesion molecules in IBD is evident from preclinical colitis studies, where their blockade ameliorated disease severity [47]. Table 1 summarizes preclinical and clinical data reported so far. Similar results have been reported in animal models of autoimmune disease including MS and rheumatoid arthritis [48].
Table 1.
Targeting leukocyte trafficking in inflammatory bowel disease.
| Target | Drug type | Preclinical efficacy | Therapeutic | Clinical efficacy | |
|---|---|---|---|---|---|
| Adhesion molecules | |||||
| α 4 Integrins/ligands | Antisense MAdCAM-1 oligonucleotide | TNBS colitis [49, 50] | Natalizumab (humanized IgG4 mAb anti-α 4 integrin) | CD (Phase IV) [51, 52] | Prevented relapse, induced remission |
| Anti-VCAM-1 mAb | UC [53] | Pilot study | |||
| Anti-MAdCAM-1 mAb | DSS colitis [54, 55] | AJM300 (orally available anti-α 4 integrin mAB) | CD (Phase II) [56] | Reduced disease activity, good safety profile | |
| Small molecule α 4 integrin antagonist | DSS colitis [22] | ||||
| Anti-β 7 and anti-MAdCAM-1 mAb | T-cell transfer colitis [57] | Vedolizumab/MLN-02 (humanized IgG4 mAb α 4 β 7 integrin) | CD [58], UC Phase II [59, 60] | Induced clinical response and remission, good safety profile | |
| Anti-MAdCAM-1 mAb | SAMP1/Yit mice [61] | ||||
| Anti-α 4 β 7 mAb | Cotton top tamarin model [62] | ||||
|
| |||||
| ICAM-1/LFA-1 | Anti-ICAM-1 mAb Antisense ICAM-1 oligonucleotide | DSS colitis [63, 64] | Alicaforsen (ISIS2303) (antisense ICAM-1 oligonucleotide) | UC (Phase II) [65, 66] | Reduced disease activity, good safety profile |
| Anti-ICAM-1 mAb | SAMP1/Yit mice [67] | ||||
|
| |||||
| Chemokines | |||||
| CCR9/CCL25 | Anti-CCR9/CCL25 mAb Traficet-EN (CCX282-B) | SAMP1/Yit mice [68] TNF (DeltaARE) mice [69] | Traficet-EN/CCX282-B (small molecule CCR9 antagonist) | CD (Phase III) [70] | Induced clinical remission, good safety profile |
| CXCR3/CXCL10 | Anti-CXCL10 mAb | IL-10 KO [71, 72] DSS colitis [73] | MDX-1100 (humanized anti-CXCL10 mAb) | UC (Phase II) | NCT00295282 NCT00656890 |
CD: Crohn's disease; DSS: dextran sodium sulphate; ICAM-1: intercellular adhesion molecule 1; MadCAM: mucosal addressin-cell adhesion molecule 1; senescence accelerated mice (SAMP1/Yit); TNBS: trinitrobenzene sulfonic acid; UC: ulcerative colitis; V-CAM-1: vascular-cell adhesion molecule 1.
3.2.2. Chemokines and Their Receptors
Chemokines mediate cell migration under normal physiological conditions and leukocyte recruitment to tissues during innate and adaptive immune responses. These small heparin-binding proteins come from a diverse family that is classified into four major subfamilies, CC, CXC, C, and CX3C, based on structural and functional differences. The two most important subgroups in terms of leukocyte trafficking to inflamed tissues are the CC chemokines for dendritic cell (DC) and lymphocyte recruitment and the CXC chemokines for recruitment of neutrophils and monocytes. Chemokines exert their biological effects on target cells by binding to specific G-protein-coupled transmembrane receptors (GPCRs) on the cell surface and activating an intracellular signaling cascade. Consequently, an activating signal is sent to the integrin switching it into a high-affinity/high-avidity state so that the rolling leukocyte can arrest itself and firmly adhere to the HEVs, a step which is essential for leukocyte extravasation into the target tissue [74]. For example, binding of CCR7 on naïve T cells to its chemokine ligand CCL21 on HEVs in turn activates binding of α 4 β 7 and LFA-1 to their endothelial ligands MAdCAM-1 and ICAM-1, respectively [38]. In addition, the combined expression of chemokine receptors and adhesion molecules by naïve and memory T lymphocytes govern their selective homing patterns. For instance, while L-selectin and CCR7 regulate naïve T-cell migration to peripheral LNs, expression of α 4 β 7 in conjunction with CCR9 allows T-cell migration to the skin and gut. The encounter between CCR7 and its ligands CCL19 and CCL21 bridges the gap between innate and adaptive immune responses. Up-regulation of CCR7 on antigen-laden DCs facilitates their migration into LNs for T-cell priming. In addition, enhanced expression and binding of CCR7 on naïve T cells to CCL19 on DCs and CCL21 on HEVs mediate transmigration from peripheral tissues into LNs. In contrast, downregulation of CCR7 allows activated T cells to exit the LN area and migrate to target tissues to carry out effector functions [75, 76]. Furthermore, elevated expression of CXCR5, the ligand of CXCL13, on certain CD4+ T cells directs their migration to the follicle to provide B-cell help [77]. Upregulated mucosal expression of numerous chemokines and their counter receptors including CXCL8 (IL-8)/CXCR2, CXCL9,10,11/CXCR3, CCL25/CCR9, CCL19,21/CCR7, and CCL20/CCR6 is evident in active IBD and in models of colitis [78–80]. Thus, chemokines orchestrate the activation, recruitment, and retention of leukocytes, and, the more insight we gain into their vital role, the more attractive they become as potential therapeutic targets.
3.2.3. The Role of DCs in Lymphocyte Homing to the Gut
The specific homing receptors expressed by activated T cells are determined by DCs. Several murine studies show that DCs from mesenteric lymph nodes (MLNs) or PPs imprint gut tropism on antigen-experienced T cells, by inducing expression of the gut homing receptors α 4 β 7 and CCR9 [81–83]. In the “steady state” mouse intestine, the ability to confer gut homing specificity is restricted to the CD103+ intestinal DC subset [84–86]. The vitamin A metabolite retinoic acid (RA) plays a central role in the process of DC imprinting within lymphoid tissues. For instance, mice deficient in vitamin A have decreased numbers of T cells in the intestine [37, 87, 88]. However, the dependency of DC-T-cell imprinting on retinoic acid receptor (RAR) signaling is complex and not yet fully characterized [89]. Also, the key factors that induce DC imprinting activity have yet to be identified; for example, germ-free studies have indicated that gut bacteria are not required for intestinal DC imprinting of α 4 β 7 expression [90]. The specific role played by intestinal DC imprinting in IBD remains elusive, but presumably it influences the increased numbers of α 4 β 7 + and CCR9+ lymphocytes evident in the inflamed intestine [91]. Since the CD103+ intestinal DC subset favors the generation of Foxp3+ T-regulatory cells (Tregs) over Th17 cells, via a TGF-β- and RA-dependent mechanism [92], targeting leukocyte trafficking at the DC imprinting level may represent a potential therapeutic strategy for IBD. Notably, a diet low in vitamin A protects against colitis in mice, and this protection is associated with increased levels of Tregs in the gut mucosa. In this case, the reduced availability of RAR ligands affects lymphocyte homing to the gut by decreasing entry of α 4 β 7 and CCR9+ T cells in favor of Tregs [93].
3.2.4. S1P and Control of Leukocyte Egress from Tissues
While chemokines control naïve T-cell migration to and within LNs, the natural bioactive lipid sphingosine-1-phosphate (S1P) regulates lymphocyte egress. S1P is formed upon the phosphorylation of sphingosine by sphingosine kinase and mediates a number of fundamental biological events including endothelial barrier enhancement, lymphocyte differentiation and immune cell trafficking [94]. Levels of S1P in the blood and lymph are constitutively high while they are low in tissues, increasing substantially upon inflammation. Although synthesized in most cells, tissue levels of S1P are tightly controlled due to intracellular degradation by S1P lyase or phosphorylation by S1P phosphatases. Five S1P receptors have been described so far, S1PR1–5, and are expressed in a cell-type specific manner within different tissues [95]. S1PR1–3 are ubiquitously expressed in mammals [96], while S1PR4 expression is restricted to lymphoid tissues [97] and lung, and S1PR5 to brain, skin, and natural killer (NK) cells [98]. The S1P/S1PR1 axis is essential for lymphocyte egress from the thymus and spleen into the blood and from the LNs into the lymph [99]. During T-cell priming, elevated expression of CD69 on activated lymphocytes promotes the temporary downregulation of S1PR1 expression on the cell membrane, disabling ligation to S1P and consequently trapping them within the LNs [100]. Concurrent binding of CCR7 to its ligand CCL21 mediates competitive retention signals. Following clonal expansion, CCR7 expression is lost and S1PR1 expression is upregulated once again, allowing the effector T cells to leave the lymphoid tissues, reenter the systemic circulation, and rapidly migrate to sites of inflammation [101]. The S1P/S1PR interaction also regulates the movement of DCs [102], neutrophils [103], and NK cells [104]. S1P signaling and functions in immunity have been reviewed elsewhere [105].
The discovery that the S1P/S1PR1 axis is essential for lymphocyte egress added a new potential target for blocking leukocyte migration in inflammatory diseases. Disruption of this S1P gradient has been reported in numerous inflammatory and/or autoimmune disorders including asthma [106] and rheumatoid arthritis [107]. The novel immunosuppressant FTY720 (Fingolimod) [108] is structurally similar to S1P and poses as an S1P analog. Like S1P, it is phosphorylated in vivo and binds with high affinity to 4 of the 5 S1P receptors S1PR1, S1PR3, S1PR4, and S1PR5 [109]. FTY720 interferes with S1P signaling and blocks the response of lymphocytes to egress signals from the lymphoid organs, sequestering them within the LNs and PPs. The result is a rapid and dramatic peripheral blood lymphopenia with depletion of circulating T and B cells. In contrast, FTY720 increases the number of DCs in the blood and simultaneously reduces their numbers in secondary lymphoid organs. In addition, it can modulate DC cytokine signaling potentially affecting T-cell responses [110]. The mechanism of action of FTY720 is complex, and it is currently unclear whether it acts as an agonist or functional antagonist or both during regulation of lymphocyte recirculation in vivo. Since FTY720 inhibits cell migration to inflammatory sites, it has shown great potential as a treatment for inflammatory disorders [108]. In clinical studies, FTY720 successfully prevented kidney transplant rejection [111–114] and proved highly effective in treating MS. It was recently approved by the US Food and Drug Administration (FDA) as a first-line treatment for relapsing forms of MS. In terms of IBD, FTY720 ameliorated experimental colitis arising as a result of chemical induction [115–117], T-cell transfer [118], and IL-10 deficiency [119], suggesting it may be a potential candidate for IBD treatment. However, reports of side effects such as bradycardia and increased susceptibility to opportunistic infections [108] dictate that the use of FTY720 therapeutically should be approached with caution and that perhaps using more selective drugs to target the S1P receptor pathway may be a safer option and yield less side effects. Indeed, the specific agonist of S1PR1, SEW2871, has shown promise in preclinical kidney transplantation studies [120] and exhibited anti-inflammatory effects in mice administered TNF-α [121]. In addition, another selective S1PR1 agonist KRP-203 showed therapeutic potential in IL-10-deficient mice [122].
4. Preclinical and Clinical Evidence of Targeting Adhesion Molecules
Figure 1 and Table 1 summarize the data presented under Sections 4 and 5.
4.1. Natalizumab
Promising preclinical data [22, 49, 50, 54, 55, 57, 61, 62] (also see Table 1) led to the use of humanized α 4 integrin antibodies in clinical trials. The most well-known anti-α 4 drug is Natalizumab (Tysabri), a humanized pan-α 4 monoclonal antibody. Natalizumab blocks the ability of α 4 β 1 and α 4 β 7 to bind to their respective ligands on the endothelium, preventing lymphocyte transendothelial migration. This α 4 antagonist was approved by the FDA in 2004 and is highly effective in treating the symptoms of MS [123, 124] and in preventing relapse and increasing remission rates in sufferers with moderate to severe CD [51, 52]. Natalizumab therapy has been associated with cases of progressive multifocal leukoencephalopathy (PML) in a small number of patients, which is induced by the JC virus, an opportunistic infection of the brain [125]. Though rare, PML is a serious and often fatal disease. The approval and relative success of Natalizumab have heightened interest and greatly encouraged further research into targeting integrins and their counter adhesion molecule ligands as a novel treatment strategy for chronic inflammatory diseases, including IBD.
4.2. MLN-02
Vedolizumab (MLN-02) is a recombinant humanized IgG1 monoclonal antibody selective for the gut-specific integrin α 4 β 7. By binding to α 4 β 7, MLN-02 inhibits the adhesion and migration of leukocytes into the gastrointestinal tract, preventing intestinal inflammation. MLN-02 treatment ameliorated disease in the cotton top tamarin model of colitis [126] and safely and effectively induced clinical response and remission in two double-blinded placebo-controlled clinical trials of patients with active CD and UC [58–60]. The selectivity of MLN-02 makes it less likely to impair systemic immunity and more attractive as a therapeutic target for IBD.
4.3. Alicaforsen- (ISIS2302-) and LFA-1-Targeting Drugs
Alicaforsen, a human ICAM-1 antisense oligonucleotide, inhibits ICAM-1 production preventing T-cell adhesion, extravasation, and subsequent migration to inflamed areas [127]. Blocking ICAM-1 ameliorated colitis in a number of preclinical models [63, 64, 67]. In clinical studies, this approach has had variable success and in general has yielded disappointing results in the treatment of CD [128–130]. However, there have been promising results with an enema formulation of Alicaforsen in the treatment of UC [65] and refractory pouchitis [131]. Efalizumab, a humanized monoclonal IgG1 antibody treatment for plaque psoriasis, is FDA approved and also acts by blocking the LFA-1/ICAM-1 interaction. By doing this it inhibits T-cell migration to the inflamed dermal and epidermal tissues. However, similar to Natalizumab, serious adverse effects, such as the Epstein-Barr virus-associated B-cell lymphoma development, were reported following treatment [132].
4.4. Small Molecule Antagonists
The immunogenicity of antibody therapies has increased research into the use of nonpeptide small molecule antagonists to block leukocyte trafficking. Such therapeutics are less likely to elicit the undesirable and serious immunogenic responses associated with monoclonal antibody therapy, and, unlike antibodies, they can be taken orally and are less expensive to produce. We analyzed the leukocyte trafficking blockade effect of a small molecule α4 integrin antagonist in a preclinical model of IBD. We confirmed the therapeutic efficacy of the compound in dextran sodium sulphate- (DSS-) induced acute colitis and demonstrated its ability to inhibit leukocyte trafficking to the inflamed gastrointestinal tract in vivo using bioluminescence imaging, as shown in Figure 2 [22]. Previous studies using small molecule integrin antagonists in other models of inflammatory disease have also shown promising results [133, 134].
5. Preclinical and Clinical Evidence of Targeting Chemokines
5.1. CCR9
The chemokine CCR9 is exclusively expressed by gut homing leukocytes and interaction with its counter ligand CCL25 is essential for T-cell homing to the small intestine [135]. The CCR9/CCL25 interaction specifically contributes to the pathophysiology of small bowel CD [136]. Antibody blockade of this interaction reduced inflammation in early stages of chronic ileitis in senescence accelerated (SAMP1/Yit) mice [68]. Additionally, pre- or post administration of a small molecule CCR9 antagonist (CCX282-B/Traficet-EN) reduced gut inflammation in TNFΔARE mice, an experimental model of CD [69]. Interestingly, Wermers et al. recently demonstrated that blockade of CCR9 exacerbated chronic ileitis in these mice, by inhibiting recruitment of Tregs to the small intestinal lamina propria and MLNs [137]. The exact role of CCR9/CCL25 in large intestinal inflammation remains unclear, and studies have yielded conflicting results. Preliminary clinical data using Traficet-EN demonstrated a beneficial therapeutic effect in both patients with ileal and colonic CD, by significantly reducing proinflammatory cytokine levels and disease scores and maintaining clinical remission [70]. This is surprising, since there is little or no expression of CCL25 in the colon [138, 139]. However, recent data showed that, although colonic levels of CCL25 are low in healthy mice, they are significantly upregulated upon DSS-induced colitis. In addition, CCR9 knock-out (KO) mice with acute DSS colitis exhibit enhanced severity of clinical symptoms and tissue injury and display delayed recovery. Exacerbation of disease was associated with an imbalance in DC subpopulations and increased macrophage infiltration into the colon [138]. These data suggest that use of CCR9 blockade therapy in, for example, strictly colonic UC, could have detrimental effects.
5.2. CXCR3
CXCR3 is expressed by monocytes, T cells, and NK cells and can mediate their recruitment to inflammatory sites by binding to its ligands CXCL9, CXCL10, and CXCL11. CXCR3 engagement with these chemokines mediates the rapid arrest of effector T cells in vitro [140] and selectively mobilizes high-CXCR3-expressing Th1 cells to sites of mucosal inflammation [141]. Expression of CXCR3 and its chemokine ligands is elevated in both preclinical and clinical models of IBD [142]. CXCL10 is considered the most crucial and potent chemokine in CXCR3-mediated chemotaxis, as it is highly upregulated and its expression robustly correlates with disease severity in inflammatory disorders such as IBD, MS, and arthritis [143–145]. Moreover, the ability of CXCL10 to preferentially attract Th1 cells emphasizes its contribution to these diseases [146]. CXCL10 antagonism prevented or ameliorated inflammation in numerous preclinical models of inflammatory disease [142]. More specifically, neutralization of CXCL10 using monoclonal antibody therapy proved effective in various experimental models of IBD [71–73]. In contrast, in a more recent study, though antibody blockade of CXCL10 reduced intestinal epithelial cell proliferation and CXCR3+ cell migration in vitro and in vivo, it had no significant effect on disease in several preclinical models including IBD, arthritis, and MS [147]. The reasons for the discrepancies between these studies are unclear, but differing methods of antagonism and disease induction may play a part. For instance, Byrne et al. used T-cell (CD4+CD45RBHi) transfer to induce colitis, while earlier studies employed the IL-10KO and DSS-induced colitis models. Future clinical trials are likely to resolve these uncertainties (NCT01294410).
5.3. CXCR2
Since neutrophil influx into the intestinal mucosa and resulting tissue damage is a major characteristic of active IBD, especially UC, neutrophil-specific chemokine receptors, and their ligands also represent potential therapeutics. Neutrophils exclusively use integrins of the β 2 family to arrest on the endothelium and antibodies against these integrins reduced tissue damage in experimental colitis [148]. Engagement of the chemokines human IL-8/CXCL8 and the murine functional homologs CXCL1 and CXCL2 with their receptors CXCR1 and CXCR2 triggers numerous signal transduction cascades, which in turn activate neutrophil recruitment to target tissues [149]. Inflammatory mediators such as bacterial lipopolysaccharide (LPS), TNF-α, and IL-1 stimulate the production of IL-8. Upregulation of these chemokines in conjunction with polymorphonuclear cells (PMN) infiltration into the inflamed intestinal mucosa correlates well with the degree of active inflammation and tissue injury in human and experimental models of IBD [78, 150, 151]. CXCR2 is a well-established mediator of PMN recruitment in preclinical models of inflammatory disease [152–154]. A small molecule CXCR2 antagonist (SB225002) was effective in ameliorating trinitrobenzene sulfonic acid- (TNBS-) induced colitis in mice, as was an anti-CXCL1 antibody [155]. We used bioluminescence imaging of adoptively transferred luciferase-expressing neutrophils to study the kinetics of neutrophil migration in acute DSS-induced colitis. This enabled demonstration of preferential recruitment of the neutrophils to the inflamed colon and the blockade effect of an anti-CXCL1 antibody on the trafficking neutrophils [21].
5.4. The “Redundancy” Issue
The promise of drug inhibition of chemokines and their receptors in IBD has not yet been realized. The chemokine system is “redundant,” and the same biological function can be carried out by several chemokines and their receptors in vivo. This questions their suitability as anti-inflammatory drug targets. However, it was recently pointed out that the lack of progress in chemokine strategies may not be due to “redundancy,” but rather to the shortcomings in the approaches employed to target them [156]. Clinical efficacy of a small molecule CCR9 antagonist was demonstrated in patients with moderate to severe CD [157], proving that targeting one chemokine receptor can be therapeutically successful in the treatment of chronic inflammation.
6. Future Directions: MicroRNAs
Recent studies suggest that microRNAs (miRNAs) play a specific role in the posttranscriptional regulation of leukocyte trafficking. miRNAs are small (21−23 nucleotide) noncoding RNAs that control gene expression. By targeting complementary messenger RNAs (mRNAs) for degradation or translational repression, they suppress the expression of protein-coding genes. Blockade of miR-126 function using an antagomir, a single-stranded antisense-like molecule, suppressed the Th2 response and subsequently the development of disease in an experimental model of allergic asthma [158]. Interestingly, miR-126 was identified as one of a set of miRNAs expressed in endothelial cells [159, 160] and was shown to inhibit TNF-induced endothelial expression of VCAM-1, thus blocking leukocyte adhesion via its lymphocyte integrin ligand α 4 β 1 [161]. In terms of neutrophil trafficking, downregulation of E-selectin and ICAM-1 by miR-31 and miR-17-3, respectively, controlled neutrophil binding to endothelial cells [162]. Inhibition of these miRNAs using specific antagonists increased neutrophil adhesion to endothelial cells in vitro, while transfecting with mimics (agonists) of these miRNAs had the opposite effect. This study suggests that miRNAs negatively regulate inflammatory processes. In another study, miR-7 downregulated expression of CD98, a lymphocyte receptor that regulates integrin signaling [163]. CD98 levels were increased in the inflamed colons of patients with CD, while miR-7 levels were decreased. Taken together, the biological importance of miRNAs in the pathogenesis of IBD is becoming clearer, and targeting miRNAs in the context of leukocyte trafficking may be a safer approach for future therapeutic opportunities.
7. Concluding Remarks
Targeting leukocyte migration is a realistic strategy, and Natalizumab shows proof of principle. Compounds inhibiting a single chemokine, such as CCR9, have also shown promise, but awareness is necessary. Targeting molecules such as miRNAs, histone deacetylases [164], or the family of bromodomain proteins [165], which regulate gene expression programs that govern endothelial cell function in inflammatory settings, may represent a new generation of drugs for IBD.
Acknowledgments
The authors are supported, in part, by the Science Foundation Ireland (SFI), the Higher Education Authority of Ireland, the European Union, and GlaxoSmithKline Ltd (GSK). The content of this paper was neither influenced nor constrained by this fact.
Abbreviations
- BLI:
Bioluminescent imaging
- CD:
Crohn's disease
- DC:
Dendritic cell
- DSS:
Dextran sodium sulphate
- GALT:
Gut-associated lymphoid tissues
- GPCRs:
G-protein-coupled transmembrane receptors
- HEV:
High endothelial venules
- ICAM-1:
Intercellular adhesion molecule 1
- IBD:
Inflammatory bowel disease
- IL:
Interleukin
- KO:
Knock out
- LFA-1:
Lymphocyte-function-associated antigen 1
- LN:
Lymph node
- LPS:
Lipopolysaccharide
- MadCAM:
Mucosal addressin-cell adhesion molecule 1
- MLNs:
Mesenteric lymph nodes
- MPO:
Myeloperoxidase
- MRI:
Magnetic resonance imaging
- mRNA:
Messenger RNA
- miRNAs:
MicroRNAs
- MS:
Multiple sclerosis
- NK:
Natural killer
- PET:
Positron emission tomography
- PMN:
Polymorphonuclear cells
- PNAd:
Peripheral lymph node addressin
- PPs:
Peyer's patches
- PSGL-1:
P-selectin glycoprotein ligand 1
- RA:
Retinoic acid
- RAR:
Retinoic acid receptor
- SPECT:
Single photon emission computed tomography
- S1P:
Sphingosine-1-phosphate
- TNF-α:
Tumor necrosis factor-α
- TNBS:
Trinitrobenzene sulfonic acid
- Tregs:
T-regulatory cells
- UC:
Ulcerative colitis
- V-CAM-1:
Vascular-cell adhesion molecule 1.
References
- 1.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448(7152):427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 2.Naito Y, Takagi T, Yoshikawa T. Molecular fingerprints of neutrophil-dependent oxidative stress in inflammatory bowel disease. Journal of Gastroenterology. 2007;42(10):787–798. doi: 10.1007/s00535-007-2096-y. [DOI] [PubMed] [Google Scholar]
- 3.Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140(6):1756–1767. doi: 10.1053/j.gastro.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Melgar S, Shanahan F. Inflammatory bowel disease-from mechanisms to treatment strategies. Autoimmunity. 2010;43(7):463–477. doi: 10.3109/08916931003674709. [DOI] [PubMed] [Google Scholar]
- 5.Cho JH, Brant SR. Recent insights into the genetics of inflammatory bowel disease. Gastroenterology. 2011;140(6):1704–1712. doi: 10.1053/j.gastro.2011.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Targan SR. Current limitations of IBD treatment: where do we go from here? Annals of the New York Academy of Sciences. 2006;1072:1–8. doi: 10.1196/annals.1326.032. [DOI] [PubMed] [Google Scholar]
- 7.Rutgeerts P, Vermeire S, Van Assche G. Biological therapies for inflammatory bowel diseases. Gastroenterology. 2009;136(4):1182–1197. doi: 10.1053/j.gastro.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 8.Waldner MJ, Neurath MF. Novel cytokine-targeted therapies and intestinal inflammation. Current Opinion in Pharmacology. 2009;9(6):702–707. doi: 10.1016/j.coph.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 9.Gowans JL, Knight EJ. The route of re-circulation of lymphocytes in the rat. Proceedings of the Royal Society of London Series B. 1964;159:257–282. doi: 10.1098/rspb.1964.0001. [DOI] [PubMed] [Google Scholar]
- 10.Bennink RJ, Peeters M, Rutgeerts P, Mortelmans L. Evaluation of early treatment response and predicting the need for colectomy in active ulcerative colitis with 99mTc-HMPAO white blood cell scintigraphy. Journal of Nuclear Medicine. 2004;45(10):1698–1704. [PubMed] [Google Scholar]
- 11.Parish CR. Fluorescent dyes for lymphocyte migration and proliferation studies. Immunology and Cell Biology. 1999;77(6):499–508. doi: 10.1046/j.1440-1711.1999.00877.x. [DOI] [PubMed] [Google Scholar]
- 12.Witt CM, Robbins K. Tracking thymocyte migration in situ. Seminars in Immunology. 2005;17(6):421–430. doi: 10.1016/j.smim.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 13.Miller MJ, Wei SH, Cahalan MD, Parker I. Autonomous T-cell trafficking examined in vivo with intravital two-photon microscopy. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(5):2604–2609. doi: 10.1073/pnas.2628040100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mankoff DA. A definition of molecular imaging. Journal of Nuclear Medicine. 2007;48(6):18–21. [PubMed] [Google Scholar]
- 15.Herschman HR. Micro-PET imaging and small animal models of disease. Current Opinion in Immunology. 2003;15(4):378–384. doi: 10.1016/s0952-7915(03)00066-9. [DOI] [PubMed] [Google Scholar]
- 16.Nuyts J, Vunckx K, Defrise M, Vanhove C. Small animal imaging with multi-pinhole SPECT. Methods. 2009;48(2):83–91. doi: 10.1016/j.ymeth.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 17.Bennink RJ, Hamann J, De Bruin K, Ten Kate FJW, Van Deventer SJH, Te Velde AA. Dedicated pinhole SPECT of intestinal neutrophil recruitment in a mouse model of dextran sulfate sodium-induced colitis. Journal of Nuclear Medicine. 2005;46(3):526–531. [PubMed] [Google Scholar]
- 18.Kircher MF, Allport JR, Graves EE, et al. In vivo high resolution three-dimensional imaging of antigen-specific cytotoxic T-lymphocyte trafficking to tumors. Cancer Research. 2003;63(20):6838–6846. [PubMed] [Google Scholar]
- 19.Millington OR, Zinselmeyer BH, Brewer JM, Garside P, Rush CM. Lymphocyte tracking and interactions in secondary lymphoid organs. Inflammation Research. 2007;56(10):391–401. doi: 10.1007/s00011-007-7017-2. [DOI] [PubMed] [Google Scholar]
- 20.Troy T, Jekic-McMullen D, Sambucetti L, Rice B. Quantitative comparison of the sensitivity of detection of fluorescent and bioluminescent reporters in animal models. Molecular Imaging. 2004;3(1):9–23. doi: 10.1162/15353500200403196. [DOI] [PubMed] [Google Scholar]
- 21.Murphy CT, Moloney G, Hall LJ, et al. Use of bioluminescence imaging to track neutrophil migration and its inhibition in experimental colitis. Clinical and Experimental Immunology. 2010;162(1):188–196. doi: 10.1111/j.1365-2249.2010.04234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murphy CT, Moloney G, MacSharry J, et al. Technical Advance: function and efficacy of an α4-integrin antagonist using bioluminescence imaging to detect leukocyte trafficking in murine experimental colitis. Journal of Leukocyte Biology. 2010;88(6):1271–1278. doi: 10.1189/jlb.0909627. [DOI] [PubMed] [Google Scholar]
- 23.Sadikot RT, Blackwell TS. Bioluminescence imaging. Proceedings of the American Thoracic Society. 2005;2(6):537–540. doi: 10.1513/pats.200507-067DS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sheikh AY, Lin SA, Cao F, et al. Molecular imaging of bone marrow mononuclear cell homing and engraftment in ischemic myocardium. Stem Cells. 2007;25(10):2677–2684. doi: 10.1634/stemcells.2007-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Costa GL, Sandora MR, Nakajima N, et al. Adoptive immunotherapy of experimental autoimmune encephalomyelitis via T-cell delivery of the IL-12 p40 subunit. Journal of Immunology. 2001;167(4):2379–2387. doi: 10.4049/jimmunol.167.4.2379. [DOI] [PubMed] [Google Scholar]
- 26.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nature Reviews Immunology. 2007;7(9):678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 27.Ley K, Kansas GS. Selectins in T-cell recruitment to non-lymphoid tissues and sites of inflammation. Nature Reviews Immunology. 2004;4(5):325–335. doi: 10.1038/nri1351. [DOI] [PubMed] [Google Scholar]
- 28.Sperandio M. Selectins and glycosyltransferases in leukocyte rolling in vivo. FEBS Journal. 2006;273(19):4377–4389. doi: 10.1111/j.1742-4658.2006.05437.x. [DOI] [PubMed] [Google Scholar]
- 29.Rossi B, Constantin G. Anti-selectin therapy for the treatment of inflammatory diseases. Inflammation and Allergy Drug Targets. 2008;7(2):85–93. doi: 10.2174/187152808785107633. [DOI] [PubMed] [Google Scholar]
- 30.Jalkanen S. Lymphocyte traffic to mucosa-associated lymphatic tissues. Immunologic Research. 1991;10(3-4):268–270. doi: 10.1007/BF02919705. [DOI] [PubMed] [Google Scholar]
- 31.Pierce NF, Cray WC. Determinants of the localization, magnitude, and duration of a specific mucosal IgA plasma cell response in enterically immunized rats. Journal of Immunology. 1982;128(3):1311–1315. [PubMed] [Google Scholar]
- 32.Xu B, Wagner N, Pham LN, et al. Lymphocyte homing to bronchus-associated lymphoid tissue (BALT) is mediated by L-selectin/PNAd, α4β1 integrin/VCAM-1, and LFA-1 adhesion pathways. Journal of Experimental Medicine. 2003;197(10):1255–1267. doi: 10.1084/jem.20010685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Butcher EC, Williams M, Youngman K, Rott L, Briskin M. Lymphocyte trafficking and regional immunity. Advances in Immunology. 1999;(72):209–253. doi: 10.1016/s0065-2776(08)60022-x. [DOI] [PubMed] [Google Scholar]
- 34.Kerfoot SM, Kubes P. Overlapping roles of P-selectin and α4 integrin to recruit leukocytes to the central nervous system in experimental autoimmune encephalomyelitis. Journal of Immunology. 2002;169(2):1000–1006. doi: 10.4049/jimmunol.169.2.1000. [DOI] [PubMed] [Google Scholar]
- 35.Wagner N, Lohler J, Kunkel EJ, et al. Critical role for β7 integrins in formation of the gut-associated lymphoid tissue. Nature. 1996;382(6589):366–370. doi: 10.1038/382366a0. [DOI] [PubMed] [Google Scholar]
- 36.Chesnutt BC, Smith DF, Raffler NA, Smith ML, White EJ, Ley K. Induction of LFA-1-dependent neutrophil rolling on ICAM-1 by engagement of E-selectin. Microcirculation. 2006;13(2):99–109. doi: 10.1080/10739680500466376. [DOI] [PubMed] [Google Scholar]
- 37.Mora JR. Homing imprinting and immunomodulation in the gut: role of dendritic cells and retinoids. Inflammatory Bowel Diseases. 2008;14(2):275–289. doi: 10.1002/ibd.20280. [DOI] [PubMed] [Google Scholar]
- 38.Koboziev I, Karlsson F, Grisham MB. Gut-associated lymphoid tissue, T-cell trafficking, and chronic intestinal inflammation. Annals of the New York Academy of Sciences. 2010;1207:E86–93. doi: 10.1111/j.1749-6632.2010.05711.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. Journal of Clinical Investigation. 2007;117(3):514–521. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kawachi S, Jennings S, Panes J, et al. Cytokine and endothelial cell adhesion molecule expression in interleukin-10-deficient mice. American Journal of Physiology. 2000;278(5):G734–G743. doi: 10.1152/ajpgi.2000.278.5.G734. [DOI] [PubMed] [Google Scholar]
- 41.Rivera-Nieves J, Olson T, Bamias G, et al. L-selectin, α4β1, and α 4β7 integrins participate in CD4+ T-cell recruitment to chronically inflamed small intestine. Journal of Immunology. 2005;174(4):2343–2352. doi: 10.4049/jimmunol.174.4.2343. [DOI] [PubMed] [Google Scholar]
- 42.Souza HS, Elia CCS, Spencer J, MacDonald TT. Expression of lymphocyte-endothelial receptor-ligand pairs, α4β7/MAdCAM-1 and OX40/OX40 ligand in the colon and jejunum of patients with inflammatory bowel disease. Gut. 1999;45(6):856–863. doi: 10.1136/gut.45.6.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arihiro S, Ohtani H, Suzuki M, et al. Differential expression of mucosal addressin cell adhesion molecule-1 (MAdCAM-1) in ulcerative colitis and Crohn’s disease. Pathology International. 2002;52(5-6):367–374. doi: 10.1046/j.1440-1827.2002.01365.x. [DOI] [PubMed] [Google Scholar]
- 44.Briskin M, Winsor-Hines D, Shyjan A, et al. Human mucosal addressin cell adhesion molecule-1 is preferentially expressed in intestinal tract and associated lymphoid tissue. American Journal of Pathology. 1997;151(1):97–110. [PMC free article] [PubMed] [Google Scholar]
- 45.Abdelbaqi M, Chidlow JH, Matthews KM, et al. Regulation of dextran sodium sulfate induced colitis by leukocyte beta 2 integrins. Laboratory Investigation. 2006;86(4):380–390. doi: 10.1038/labinvest.3700398. [DOI] [PubMed] [Google Scholar]
- 46.Pavlick KP, Ostanin DV, Furr KL, et al. Role of T-cell-associated lymphocyte function-associated antigen-1 in the pathogenesis of experimental colitis. International Immunology. 2006;18(2):389–398. doi: 10.1093/intimm/dxh378. [DOI] [PubMed] [Google Scholar]
- 47.Rivera-Nieves J, Gorfu G, Ley K. Leukocyte adhesion molecules in animal models of inflammatory bowel disease. Inflammatory Bowel Diseases. 2008;14(12):1715–1735. doi: 10.1002/ibd.20501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simmons DL. Anti-adhesion therapies. Current Opinion in Pharmacology. 2005;5(4):398–404. doi: 10.1016/j.coph.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 49.Goto A, Arimura Y, Shinomura Y, Imai K, Hinoda Y. Antisense therapy of MAdCAM-1 for trinitrobenzenesulfonic acid-induced murine colitis. Inflammatory Bowel Diseases. 2006;12(8):758–765. doi: 10.1097/00054725-200608000-00013. [DOI] [PubMed] [Google Scholar]
- 50.Sans M, Panes J, Ardite E, et al. VCAM-1 and ICAM-1 mediate leukocyte-endothelial cell adhesion in rat experimental colitis. Gastroenterology. 1999;116(4):874–883. doi: 10.1016/s0016-5085(99)70070-3. [DOI] [PubMed] [Google Scholar]
- 51.MacDonald JK, McDonald JW. Natalizumab for induction of remission in Crohn’s disease. Cochrane Database of Systematic Reviews. 2007;(1, article CD006097) doi: 10.1002/14651858.CD006097.pub2. [DOI] [PubMed] [Google Scholar]
- 52.Targan SR, Feagan BG, Fedorak RN, et al. Natalizumab for the treatment of active Crohn’s Disease: results of the ENCORE Trial. Gastroenterology. 2007;132(5):1672–1683. doi: 10.1053/j.gastro.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 53.Gordon FH, Hamilton MI, Donoghue S, et al. A pilot study of treatment of active ulcerative colitis with natalizumab, a humanized monoclonal antibody to alpha-4 integrin. Alimentary Pharmacology and Therapeutics. 2002;16(4):699–705. doi: 10.1046/j.1365-2036.2002.01205.x. [DOI] [PubMed] [Google Scholar]
- 54.Farkas S, Hornung M, Sattler C, et al. Blocking MAdCAM-1 in vivo reduces leukocyte extravasation and reverses chronic inflammation in experimental colitis. International Journal of Colorectal Disease. 2006;21(1):71–78. doi: 10.1007/s00384-004-0709-y. [DOI] [PubMed] [Google Scholar]
- 55.Kato S, Hokari R, Matsuzaki K, et al. Amelioration of murine experimental colitis by inhibition of mucosal addressin cell adhesion molecule-1. Journal of Pharmacology and Experimental Therapeutics. 2000;295(1):183–189. [PubMed] [Google Scholar]
- 56.Takazoe M, Watanabe M, Kawaguchi T, et al. S1066 Oral Alpha-4 Integrin Inhibitor (AJM300) in patients with active Crohn's disease—a Randomized, Double-Blind, Placebo-Controlled Trial. Gastroenterology. 2009;136:p. A-181. [Google Scholar]
- 57.Picarella D, Hurlbut P, Rottman J, Shi X, Butcher E, Ringler DJ. Monoclonal antibodies specific for β7 integrin and mucosal addressin cell adhesion molecule-1 (MAdCAM-1) reduce inflammation in the colon of scid Mice reconstituted with CD45RBhigh CD4+ T cells. Journal of Immunology. 1997;158(5):2099–2106. [PubMed] [Google Scholar]
- 58.Feagan BG, Greenberg GR, Wild G, et al. Treatment of ulcerative colitis with a humanized antibody to the α4β7 integrin. The New England Journal of Medicine. 2005;352(24):2499–2507. doi: 10.1056/NEJMoa042982. [DOI] [PubMed] [Google Scholar]
- 59.Behm BW, Bickston SJ. Humanized antibody to the alpha4beta7 integrin for induction of remission in ulcerative colitis. Cochrane Database of Systematic Reviews. 2009;(1, article CD007571) doi: 10.1002/14651858.CD007571. [DOI] [PubMed] [Google Scholar]
- 60.Feagan BG, Greenberg GR, Wild G, et al. Treatment of active Crohn's disease With MLN0002, a humanized antibody to the α4β7 integrin. Clinical Gastroenterology and Hepatology. 2008;6(12):1370–1377. doi: 10.1016/j.cgh.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 61.Matsuzaki K, Tsuzuki Y, Matsunaga H, et al. In vivo demonstration of T lymphocyte migration and amelioration of ileitis in intestinal mucosa of SAMP1/Yit mice by the inhibition of MAdCAM-1. Clinical and Experimental Immunology. 2005;140(1):22–31. doi: 10.1111/j.1365-2249.2005.02742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Podolsky DK, Lobb R, King N, et al. Attenuation of colitis in the cotton-top tamarin by anti-α4 integrin monoclonal antibody. Journal of Clinical Investigation. 1993;92(1):372–380. doi: 10.1172/JCI116575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taniguchi T, Tsukada H, Nakamura H, et al. Effects of the anti-ICAM-1 monoclonal antibody on dextran sodium sulphate-induced colitis in rats. Journal of Gastroenterology and Hepatology. 1998;13(9):945–949. doi: 10.1111/j.1440-1746.1998.tb00766.x. [DOI] [PubMed] [Google Scholar]
- 64.Bennett CF, Kornbrust D, Henry S, et al. An ICAM-1 antisense oligonucleotide prevents and reverses dextran sulfate sodium-induced colitis in mice. Journal of Pharmacology and Experimental Therapeutics. 1997;280(2):988–1000. [PubMed] [Google Scholar]
- 65.Van Deventer SJ, Wedel MK, Baker BF, Xia S, Chuang E, Miner PB., Jr. A Phase II dose ranging, double-blind, placebo-controlled study of alicaforsen enema in subjects with acute exacerbation of mild to moderate left-sided ulcerative colitis. Alimentary Pharmacology and Therapeutics. 2006;23(10):1415–1425. doi: 10.1111/j.1365-2036.2006.02910.x. [DOI] [PubMed] [Google Scholar]
- 66.Van Deventer SJH, Tami JA, Wedel MK. A randomised, controlled, double blind, escalating dose study of alicaforsen enema in active ulcerative colitis. Gut. 2004;53(11):1646–1651. doi: 10.1136/gut.2003.036160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Burns RC, Rivera-Nieves J, Moskaluk CA, Matsumoto S, Cominelli F, Ley K. Antibody blockade of ICAM-1 and VCAM-1 ameliorates inflammation in the SAMP-1/Yit adoptive transfer model of Crohn’s disease in mice. Gastroenterology. 2001;121(6):1428–1436. doi: 10.1053/gast.2001.29568. [DOI] [PubMed] [Google Scholar]
- 68.Rivera-Nieves J, Ho J, Bamias G, et al. Antibody Blockade of CCL25/CCR9 Ameliorates Early but not Late Chronic Murine Ileitis. Gastroenterology. 2006;131(5):1518–1529. doi: 10.1053/j.gastro.2006.08.031. [DOI] [PubMed] [Google Scholar]
- 69.Walters MJ, Wang Y, Lai N, et al. Characterization of CCX282-B, an orally bioavailable antagonist of the CCR9 chemokine receptor, for treatment of inflammatory bowel disease. Journal of Pharmacology and Experimental Therapeutics. 2010;335(1):61–69. doi: 10.1124/jpet.110.169714. [DOI] [PubMed] [Google Scholar]
- 70.Eksteen B, Adams DH. GSK-1605786, a selective small-molecule antagonist of the CCR9 chemokine receptor for the treatment of Crohn’s disease. IDrugs. 2010;13(7):472–481. [PubMed] [Google Scholar]
- 71.Hyun JG, Lee G, Brown JB, et al. Anti-interferon-inducible chemokine, CXCL10, reduces colitis by impairing T helper-1 induction and recruitment in mice. Inflammatory Bowel Diseases. 2005;11(9):799–805. doi: 10.1097/01.mib.0000178263.34099.89. [DOI] [PubMed] [Google Scholar]
- 72.Singh UP, Singh S, Taub DD, Lillard JW. Inhibition of IFN-γ-inducible protein-10 abrogates colitis in IL-10-/- mice. Journal of Immunology. 2003;171(3):1401–1406. doi: 10.4049/jimmunol.171.3.1401. [DOI] [PubMed] [Google Scholar]
- 73.Sasaki S, Yoneyama H, Suzuki K, et al. Blockade of CXCL 10 protects mice from acute colitis and enhances crypt cell survival. European Journal of Immunology. 2002;32(11):3197–3205. doi: 10.1002/1521-4141(200211)32:11<3197::AID-IMMU3197>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 74.Zhong W, Kolls JK, Chen H, McAllister F, Oliver PD, Zhang Z. Chemokines orchestrate leukocyte trafficking in inflammatory bowel disease. Frontiers in Bioscience. 2008;13(5):1654–1664. doi: 10.2741/2789. [DOI] [PubMed] [Google Scholar]
- 75.Förster R, Schubel A, Breitfeld D, et al. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99(1):23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 76.Bromley SK, Thomas SY, Luster AD. Chemokine receptor CCR7 guides T-cell exit from peripheral tissues and entry into afferent lymphatics. Nature Immunology. 2005;6(9):895–901. doi: 10.1038/ni1240. [DOI] [PubMed] [Google Scholar]
- 77.Viola A, Luster AD. Chemokines and their receptors: drug targets in immunity and inflammation. Annual Review of Pharmacology and Toxicology. 2008;48:171–197. doi: 10.1146/annurev.pharmtox.48.121806.154841. [DOI] [PubMed] [Google Scholar]
- 78.Banks C, Bateman A, Payne R, Johnson P, Sheron N. Chemokine expression in IBD. Mucosal chemokine expression is unselectively increased in both ulcerative colitis and Crohn’s disease. Journal of Pathology. 2003;199(1):28–35. doi: 10.1002/path.1245. [DOI] [PubMed] [Google Scholar]
- 79.Puleston J, Cooper M, Murch S, et al. A distinct subset of chemokines dominates the mucosal chemokine response in inflammatory bowel disease. Alimentary Pharmacology and Therapeutics. 2005;21(2):109–120. doi: 10.1111/j.1365-2036.2004.02262.x. [DOI] [PubMed] [Google Scholar]
- 80.Kawashima D, Oshitani N, Jinno Y, et al. Augmented expression of secondary lymphoid tissue chemokine and EBI1 ligand chemokine in Crohn’s disease. Journal of Clinical Pathology. 2005;58(10):1057–1063. doi: 10.1136/jcp.2004.024828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mora JR, Bono MR, Manjunath N, et al. Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature. 2003;424(6944):88–93. doi: 10.1038/nature01726. [DOI] [PubMed] [Google Scholar]
- 82.Stagg AJ, Kamm MA, Knight SC. Intestinal dendritic cells increase T-cell expression of α4β7 integrin. European Journal of Immunology. 2002;32(5):1445–1454. doi: 10.1002/1521-4141(200205)32:5<1445::AID-IMMU1445>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 83.Johansson-Lindbom B, Svensson M, Wurbel MA, Malissen B, Márquez G, Agace W. Selective generation of gut tropic T cells in gut-associated lymphoid tissue (GALT): requirement for GALT dendritic cells and adjuvant. Journal of Experimental Medicine. 2003;198(6):963–969. doi: 10.1084/jem.20031244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mora JR, Cheng G, Picarella D, Briskin M, Buchanan N, Von Andrian UH. Reciprocal and dynamic control of CD8 T-cell homing by dendritic cells from skin- and gut-associated lymphoid tissues. Journal of Experimental Medicine. 2005;201(2):303–316. doi: 10.1084/jem.20041645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Johansson-Lindbom B, Svensson M, Pabst O, et al. Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T-cell homing. Journal of Experimental Medicine. 2005;202(8):1063–1073. doi: 10.1084/jem.20051100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jaensson E, Uronen-Hansson H, Pabst O, et al. Small intestinal CD103+ dendritic cells display unique functional properties that are conserved between mice and humans. Journal of Experimental Medicine. 2008;205(9):2139–2149. doi: 10.1084/jem.20080414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity. 2004;21(4):527–538. doi: 10.1016/j.immuni.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 88.Manicassamy S, Pulendran B. Retinoic acid-dependent regulation of immune responses by dendritic cells and macrophages. Seminars in Immunology. 2009;21(1):22–27. doi: 10.1016/j.smim.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Svensson M, Johansson-Lindbom B, Zapata F, et al. Retinoic acid receptor signaling levels and antigen dose regulate gut homing receptor expression on CD8+ T cells. Mucosal Immunology. 2008;1(1):38–48. doi: 10.1038/mi.2007.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stagg AJ, Norin KE, Midtvedt T, Kamm MA, Knight SC, Björkstén B. Mesenteric dendritic cells from germ-free mice cause less T-cell stimulation but still induce α4β7 integrin. Microbial Ecology in Health and Disease. 2007;19(3):171–183. [Google Scholar]
- 91.Ng SC, Kamm MA, Stagg AJ, Knight SC. Intestinal dendritic cells: their role in bacterial recognition, lymphocyte homing, and intestinal inflammation. Inflammatory Bowel Diseases. 2010;16(10):1787–1807. doi: 10.1002/ibd.21247. [DOI] [PubMed] [Google Scholar]
- 92.Coombes JL, Siddiqui KRR, Arancibia-Cárcamo CV, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β -and retinoic acid-dependent mechanism. Journal of Experimental Medicine. 2007;204(8):1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kang SG, Wang C, Matsumoto S, Kim CH. High and low vitamin A therapies induce distinct foxP3+ T-cell subsets and effectively control intestinal inflammation. Gastroenterology. 2009;137(4):1391–e6. doi: 10.1053/j.gastro.2009.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chi H. Sphingosine-1-phosphate and immune regulation: trafficking and beyond. Trends in Pharmacological Sciences. 2011;32(1):16–24. doi: 10.1016/j.tips.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Meyer zu Heringdorf D, Jakobs KH. Lysophospholipid receptors: signalling, pharmacology and regulation by lysophospholipid metabolism. Biochimica et Biophysica Acta. 2007;1768(4):923–940. doi: 10.1016/j.bbamem.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 96.Chae SS, Proia RL, Hla T. Constitutive expression of the S1P1 receptor in adult tissues. Prostaglandins and Other Lipid Mediators. 2004;73(1-2):141–150. doi: 10.1016/j.prostaglandins.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 97.Graeler M, Goetzl EJ. Activation-regulated expression and chemotactic function of sphingosine 1-phosphate receptors in mouse splenic T cells. FASEB Journal. 2002;16(14):1874–1878. doi: 10.1096/fj.02-0548com. [DOI] [PubMed] [Google Scholar]
- 98.Jaillard C, Harrison S, Stankoff B, et al. Edg8/S1P5: an oligodendroglial receptor with dual function on process retraction and cell survival. Journal of Neuroscience. 2005;25(6):1459–1469. doi: 10.1523/JNEUROSCI.4645-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Matloubian M, Lo CG, Cinamon G, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427(6972):355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 100.Shiow LR, Rosen DB, Brdičková N, et al. CD69 acts downstream of interferon-α/β to inhibit S1P 1 and lymphocyte egress from lymphoid organs. Nature. 2006;440(7083):540–544. doi: 10.1038/nature04606. [DOI] [PubMed] [Google Scholar]
- 101.Pham THM, Okada T, Matloubian M, Lo CG, Cyster JG. S1P1 receptor signaling overrides retention mediated by Gαi-coupled receptors to promote T-cell egress. Immunity. 2008;28(1):122–133. doi: 10.1016/j.immuni.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rathinasamy A, Czeloth N, Pabst O, Förster R, Bernhardt G. The origin and maturity of dendritic cells determine the pattern of sphingosine 1-phosphate receptors expressed and required for efficient migration. Journal of Immunology. 2010;185(7):4072–4081. doi: 10.4049/jimmunol.1000568. [DOI] [PubMed] [Google Scholar]
- 103.Allende ML, Bektas M, Lee BG, et al. Sphingosine-1-phosphate lyase deficiency produces a pro-inflammatory response while impairing neutrophil trafficking. Journal of Biological Chemistry. 2011;286(9):7348–7358. doi: 10.1074/jbc.M110.171819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Walzer T, Chiossone L, Chaix J, et al. Natural killer cell trafficking in vivo requires a dedicated sphingosine 1-phosphate receptor. Nature Immunology. 2007;8(12):1337–1344. doi: 10.1038/ni1523. [DOI] [PubMed] [Google Scholar]
- 105.Spiege S, Milstien S. The outs and the ins of sphingosine-1-phosphate in immunity. Nature Reviews Immunology. 2011;11:403–415. doi: 10.1038/nri2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ammit AJ, Hastie AT, Edsall LC, et al. Sphingosine 1-phosphate modulates human airway smooth muscle cell functions that promote inflammation and airway remodeling in asthma. The FASEB Journal. 2001;15(7):1212–1214. doi: 10.1096/fj.00-0742fje. [DOI] [PubMed] [Google Scholar]
- 107.Kitano M, Hla T, Sekiguchi M, et al. Sphingosine 1-phosphate/sphingosine 1-phosphate receptor 1 signaling in rheumatoid synovium: regulation of synovial proliferation and inflammatory gene expression. Arthritis and Rheumatism. 2006;54(3):742–753. doi: 10.1002/art.21668. [DOI] [PubMed] [Google Scholar]
- 108.Brinkmann V, Billich A, Baumruker T, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nature Reviews Drug Discovery. 2010;9(11):883–897. doi: 10.1038/nrd3248. [DOI] [PubMed] [Google Scholar]
- 109.Chiba K. A new therapeutic approach for autoimmune diseases by the sphingosine 1-phosphate receptor modulator, fingolimod (FTY720) Yakugaku Zasshi. 2009;129(6):655–665. doi: 10.1248/yakushi.129.655. [DOI] [PubMed] [Google Scholar]
- 110.Lan YY, De Creus A, Colvin BL, et al. The sphingosine-1-phosphate receptor agonist FTY720 modulates dendritic cell trafficking in vivo. American Journal of Transplantation. 2005;5(11):2649–2659. doi: 10.1111/j.1600-6143.2005.01085.x. [DOI] [PubMed] [Google Scholar]
- 111.Budde K, Schmouder RL, Nashan B, et al. Pharmacodynamics of single doses of the novel immunosuppressant FTY720 in stable renal transplant patients. American Journal of Transplantation. 2003;3(7):846–854. doi: 10.1034/j.1600-6143.2003.00130.x. [DOI] [PubMed] [Google Scholar]
- 112.Budde K, Schmouder RL, Brunkhorst R, et al. First human trial of FTY720, a novel immunomodulator, in stable renal transplant patients. Journal of the American Society of Nephrology. 2002;13(4):1073–1083. doi: 10.1681/ASN.V1341073. [DOI] [PubMed] [Google Scholar]
- 113.Kahan BD, Karlix JL, Ferguson RM, et al. Pharmacodynamics, pharmacokinetics, and safety of multiple doses of FTY720 in stable renal transplant patients: a multicenter, randomized, placebo-controlled, phase I study. Transplantation. 2003;76(7):1079–1084. doi: 10.1097/01.TP.0000084822.01372.AC. [DOI] [PubMed] [Google Scholar]
- 114.Tedesco-Silva H, Mourad G, Kahan BD, et al. FTY720, a novel immunomodulator: efficacy and safety results from the first phase 2A study in de novo renal transplantation. Transplantation. 2005;79(11):1553–1560. [PubMed] [Google Scholar]
- 115.Daniel C, Sartory N, Zahn N, Geisslinger G, Radeke HH, Stein JM. FTY720 ameliorates Th1-mediated colitis in mice by directly affecting the functional activity of CD4+CD25+ regulatory T cells. Journal of Immunology. 2007;178(4):2458–2468. doi: 10.4049/jimmunol.178.4.2458. [DOI] [PubMed] [Google Scholar]
- 116.Deguchi Y, Andoh A, Yagi Y, et al. The S1P receptor modulator FTY720 prevents the development of experimental colitis in mice. Oncology Reports. 2006;16(4):699–703. [PubMed] [Google Scholar]
- 117.Daniel C, Sartory NA, Zahn N, et al. FTY720 ameliorates oxazolone colitis in mice by directly affecting T helper type 2 functions. Molecular Immunology. 2007;44(13):3305–3316. doi: 10.1016/j.molimm.2007.02.026. [DOI] [PubMed] [Google Scholar]
- 118.Fujii R, Kanai T, Nemoto Y, et al. FTY720 suppresses CD4+CD44highCD62L- effector memory T-cell-mediated colitis. American Journal of Physiology. 2006;291(2):G267–G274. doi: 10.1152/ajpgi.00496.2005. [DOI] [PubMed] [Google Scholar]
- 119.Mizushima T, Ito T, Kishi D, et al. Therapeutic effects of a new lymphocyte homing reagent FTY720 in interleukin-10 gene-deficient mice with colitis. Inflammatory Bowel Diseases. 2004;10(3):182–192. doi: 10.1097/00054725-200405000-00002. [DOI] [PubMed] [Google Scholar]
- 120.Awad AS, Ye H, Huang L, et al. Selective sphingosine 1-phosphate 1 receptor activation reduces ischemia-reperfusion injury in mouse kidney. American Journal of Physiology. 2006;290(6):F1516–F1524. doi: 10.1152/ajprenal.00311.2005. [DOI] [PubMed] [Google Scholar]
- 121.Bolick DT, Srinivasan S, Kim KW, et al. Sphingosine-1-phosphate prevents tumor necrosis factor-α-mediated monocyte adhesion to aortic endothelium in mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(5):976–981. doi: 10.1161/01.ATV.0000162171.30089.f6. [DOI] [PubMed] [Google Scholar]
- 122.Song J, Matsuda C, Kai Y, et al. A novel sphingosine 1-phosphate receptor agonist, 2-amino-2-propanediol hydrochloride (KRP-203), regulates chronic colitis in interleukin-10 gene-deficient mice. Journal of Pharmacology and Experimental Therapeutics. 2008;324(1):276–283. doi: 10.1124/jpet.106.119172. [DOI] [PubMed] [Google Scholar]
- 123.Miller DH, Khan OA, Sheremata WA, et al. A controlled trial of natalizumab for relapsing multiple sclerosis. The New England Journal of Medicine. 2003;348(1):15–23. doi: 10.1056/NEJMoa020696. [DOI] [PubMed] [Google Scholar]
- 124.Rudick RA, Stuart WH, Calabresi PA, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. The New England Journal of Medicine. 2006;354(9):911–923. doi: 10.1056/NEJMoa044396. [DOI] [PubMed] [Google Scholar]
- 125.Stuve O, Gold R, Chan A, Mix E, Zettl U, Kieseier BC. α4-Integrin antagonism with natalizumab : effects and adverse effects. Journal of Neurology. 2008;255(supplement 6):58–65. doi: 10.1007/s00415-008-6011-0. [DOI] [PubMed] [Google Scholar]
- 126.Hesterberg PE, Winsor-Hines D, Briskin MJ, et al. Rapid resolution of chronic colitis in the cotton-top tamarin with an antibody to a gut-homing integrin α4β7. Gastroenterology. 1996;111(5):1373–1380. doi: 10.1053/gast.1996.v111.pm8898653. [DOI] [PubMed] [Google Scholar]
- 127.Barish CF. Alicaforsen therapy in inflammatory bowel disease. Expert Opinion on Biological Therapy. 2005;5(10):1387–1391. doi: 10.1517/14712598.5.10.1387. [DOI] [PubMed] [Google Scholar]
- 128.Yacyshyn B, Chey WY, Wedel MK, Yu RZ, Paul D, Chuang E. A randomized, double-masked, placebo-controlled study of alicaforsen, an antisense inhibitor of intercellular adhesion molecule 1, for the treatment of subjects with active Crohn's disease. Clinical Gastroenterology and Hepatology. 2007;5(2):215–220. doi: 10.1016/j.cgh.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 129.Yacyshyn BR, Chey WY, Goff J, et al. Double blind, placebo controlled trial of the remission inducing and steroid sparing properties of an ICAM-1 antisense oligodeoxynucleotide, alicaforsen (ISIS 2302), in active steroid dependent Crohn’s disease. Gut. 2002;51(1):30–36. doi: 10.1136/gut.51.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Schreiber S, Nikolaus S, Malchow H, et al. Absence of efficacy of subcutaneous antisense ICAM-1 treatment of chronic active Crohn’s disease. Gastroenterology. 2001;120(6):1339–1346. doi: 10.1053/gast.2001.24015. [DOI] [PubMed] [Google Scholar]
- 131.Miner P, Jr., Wedel M, Bane B, Bradley J. An enema formulation of alicaforsen, an antisense inhibitor of intercellular adhesion molecule-1, in the treatment of chronic, unremitting pouchitis. Alimentary Pharmacology and Therapeutics. 2004;19(3):281–286. doi: 10.1111/j.1365-2036.2004.01863.x. [DOI] [PubMed] [Google Scholar]
- 132.Bommakanti S, Patil A, Eshoa C, Chitambar CR. Case reports: efalizumab-associated lymphoproliferative disease. Journal of Drugs in Dermatology. 2007;6(6):646–648. [PubMed] [Google Scholar]
- 133.Okigami H, Takeshita K, Tajimi M, et al. Inhibition of eosinophilia in vivo by a small molecule inhibitor of very late antigen (VLA)-4. European Journal of Pharmacology. 2007;559(2-3):202–209. doi: 10.1016/j.ejphar.2006.11.065. [DOI] [PubMed] [Google Scholar]
- 134.Cortijo J, Sanz MJ, Iranzo A, et al. A small molecule, orally active, α4β1/ α4β7 dual antagonist reduces leukocyte infiltration and airway hyper-responsiveness in an experimental model of allergic asthma in Brown Norway rats. British Journal of Pharmacology. 2006;147(6):661–670. doi: 10.1038/sj.bjp.0706658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Johansson-Lindbom B, Agace WW. Generation of gut-homing T cells and their localization to the small intestinal mucosa. Immunological Reviews. 2007;215(1):226–242. doi: 10.1111/j.1600-065X.2006.00482.x. [DOI] [PubMed] [Google Scholar]
- 136.Koenecke C, Förster R. CCR9 and inflammatory bowel disease. Expert Opinion on Therapeutic Targets. 2009;13(3):297–306. doi: 10.1517/14728220902762928. [DOI] [PubMed] [Google Scholar]
- 137.Wermers JD, McNamee EN, Wurbel MA, Jedlicka P, Rivera-Nieves J. The Chemokine receptor CCR9 is required for the T-cell-mediated regulation of chronic ileitis in mice. Gastroenterology. 2011;140(5):1526–1535.e3. doi: 10.1053/j.gastro.2011.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wurbel MA, McIntire MG, Dwyer P, Fiebiger E. CCL25/CCR9 interactions regulate large intestinal inflammation in a murine model of acute colitis. PLoS ONE. 2011;6(1, article e16442) doi: 10.1371/journal.pone.0016442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kunkel EJ, Campbell JJ, Haraldsen G, et al. Lymphocyte CC chemokine receptor 9 and epithelial thymus-expressed chemokine (TECK) expression distinguish the small intestinal immune compartment: epithelial expression of tissue-specific chemokines as an organizing principle in regional immunity. Journal of Experimental Medicine. 2000;192(5):761–767. doi: 10.1084/jem.192.5.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Piali L, Weber C, LaRosa G, et al. The chemokine receptor CXCR3 mediates rapid and shear-resistant adhesion-induction of effector T lymphocytes by the chemokines IP10 and Mig. European Journal of Immunology. 1998;28(3):961–972. doi: 10.1002/(SICI)1521-4141(199803)28:03<961::AID-IMMU961>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 141.Bonecchi R, Bianchi G, Bordignon PP, et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. Journal of Experimental Medicine. 1998;187(1):129–134. doi: 10.1084/jem.187.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Singh UP, Venkataraman C, Singh R, Lillard JW., Jr. CXCR3 axis: role in inflammatory bowel disease and its therapeutic implication. Endocrine, Metabolic and Immune Disorders Drug Targets. 2007;7(2):111–123. doi: 10.2174/187153007780832109. [DOI] [PubMed] [Google Scholar]
- 143.Scarpini E, Galimberti D, Baron P, et al. IP-10 and MCP-1 levels in CSF and serum from multiple sclerosis patients with different clinical subtypes of the disease. Journal of the Neurological Sciences. 2002;195(1):41–46. doi: 10.1016/s0022-510x(01)00680-3. [DOI] [PubMed] [Google Scholar]
- 144.Hanaoka R, Kasama T, Muramatsu M, et al. A novel mechanism for the regulation of IFN-gamma inducible protein-10 expression in rheumatoid arthritis. Arthritis Research and Therapy. 2003;5(2):R74–81. doi: 10.1186/ar616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Aggarwal A, Agarwal S, Misra R. Chemokine and chemokine receptor analysis reveals elevated interferon-inducible protein-10 (IP)-10/CXCL10 levels and increased number of CCR5+ and CXCR3+ CD4 T cells in synovial fluid of patients with enthesitis-related arthritis (ERA) Clinical and Experimental Immunology. 2007;148(3):515–519. doi: 10.1111/j.1365-2249.2007.03377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Loetscher P, Pellegrino A, Gong JH, et al. The Ligands of CXC Chemokine Receptor 3, I-TAC, Mig, and IP10, Are Natural Antagonists for CCR3. Journal of Biological Chemistry. 2001;276(5):2986–2991. doi: 10.1074/jbc.M005652200. [DOI] [PubMed] [Google Scholar]
- 147.Byrne FR, Winters A, Brankow D, et al. An antibody to IP-10 is a potent antagonist of cell migration in vitro and in vivo and does not affect disease in several animal models of inflammation. Autoimmunity. 2009;42(3):171–182. doi: 10.1080/08916930802629547. [DOI] [PubMed] [Google Scholar]
- 148.Palmen MJHJ, Dijkstra CD, Van Der Ende MB, Pena AS, Van Rees EP. Anti-CD11b/CD18 antibodies reduce inflammation in acute colitis in rats. Clinical and Experimental Immunology. 1995;101(2):351–356. doi: 10.1111/j.1365-2249.1995.tb08363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kobayashi Y. Neutrophil infiltration and chemokines. Critical Reviews in Immunology. 2006;26(4):307–315. doi: 10.1615/critrevimmunol.v26.i4.20. [DOI] [PubMed] [Google Scholar]
- 150.Melgar S, Drmotova M, Rehnström E, Jansson L, Michaëlsson E. Local production of chemokines and prostaglandin E2 in the acute, chronic and recovery phase of murine experimental colitis. Cytokine. 2006;35(5-6):275–283. doi: 10.1016/j.cyto.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 151.Hanai H, Takeuchi K, Iida T, et al. Relationship between fecal calprotectin, intestinal inflammation, and peripheral blood neutrophils in patients with active ulcerative colitis. Digestive Diseases and Sciences. 2004;49(9):1438–1443. doi: 10.1023/b:ddas.0000042243.47279.87. [DOI] [PubMed] [Google Scholar]
- 152.Podolin PL, Bolognese BJ, Foley JJ, et al. A potent and selective nonpeptide antagonist of CXCR2 inhibits acute and chronic models of arthritis in the rabbit. Journal of Immunology. 2002;169(11):6435–6444. doi: 10.4049/jimmunol.169.11.6435. [DOI] [PubMed] [Google Scholar]
- 153.Schuh JM, Blease K, Hogaboam CM. CXCR2 is necessary for the development and persistence of chronic fungal asthma in mice. Journal of Immunology. 2002;168(3):1447–1456. doi: 10.4049/jimmunol.168.3.1447. [DOI] [PubMed] [Google Scholar]
- 154.Buanne P, Di Carlo E, Caputi L, et al. Crucial pathophysiological role of CXCR2 in experimental ulcerative colitis in mice. Journal of Leukocyte Biology. 2007;82(5):1239–1246. doi: 10.1189/jlb.0207118. [DOI] [PubMed] [Google Scholar]
- 155.Bento AF, Leite DFP, Claudino RF, Hara DB, Leal PC, Calixto JB. The selective nonpeptide CXCR2 antagonist SB225002 ameliorates acute experimental colitis in mice. Journal of Leukocyte Biology. 2008;84(4):1213–1221. doi: 10.1189/jlb.0408231. [DOI] [PubMed] [Google Scholar]
- 156.Schall TJ, Proudfoot AE. Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nature Reviews Immunology. 2011;11(5):355–363. doi: 10.1038/nri2972. [DOI] [PubMed] [Google Scholar]
- 157.Keshav S, Johnson D, Bekker P, Schall TJ. PROTECT-1 study demonstrated efficacy of the intestine-specific chemokine receptor antagonist CCX282-B (Traficet-EN) in treatment of patients with moderate-to-severe Crohn's disease. Gastroenterology. 2009;136:p. A-65. [Google Scholar]
- 158.Mattes J, Collison A, Plank M, Phipps S, Foster PS. Antagonism of microRNA-126 suppresses the effector function of T H2 cells and the development of allergic airways disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(44):18704–18709. doi: 10.1073/pnas.0905063106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Suárez Y, Fernández-Hernando C, Pober JS, Sessa WC. Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circulation Research. 2007;100(8):1164–1173. doi: 10.1161/01.RES.0000265065.26744.17. [DOI] [PubMed] [Google Scholar]
- 160.Poliseno L, Tuccoli A, Mariani L, et al. MicroRNAs modulate the angiogenic properties of HUVECs. Blood. 2006;108(9):3068–3071. doi: 10.1182/blood-2006-01-012369. [DOI] [PubMed] [Google Scholar]
- 161.Harris TA, Yamakuchi M, Ferlito M, Mendell JT, Lowenstein CJ. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(5):1516–1521. doi: 10.1073/pnas.0707493105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Suárez Y, Wang C, Manes TD, Pober JS. Cutting edge: TNF-induced microRNAs regulate TNF-induced expression of E-selectin and intercellular adhesion molecule-1 on human endothelial cells: feedback control of inflammation. Journal of Immunology. 2010;184(1):21–25. doi: 10.4049/jimmunol.0902369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nguyen HTT, Dalmasso G, Yan Y, et al. MicroRNA-7 modulates CD98 expression during intestinal epithelial cell differentiation. Journal of Biological Chemistry. 2010;285(2):1479–1489. doi: 10.1074/jbc.M109.057141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Glauben R, Siegmund B. Inhibition of histone deacetylases in inflammatory bowel diseases. Molecular Medicine. 2011;17(5-6):426–433. doi: 10.2119/molmed.2011.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Nicodeme E, Jeffrey KL, Schaefer U, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468(7327):1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]