

Abstract
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma in children under 15 years of age and rare among persons older than 45 years of age. It is considered to result from malignant transformation of primitive mesenchymal cells. Although it has a relative predominance for head and neck region, it is found less often in oral cavity. Here we report a case of RMS of mandible in an adult patient, which was initially diagnosed as carcinosarcoma. Clinical and pathologic findings are described, which were confirmed by histochemical and immunohistochemical stains.
Keywords: Embryonal rhabdomyosarcoma, muscle tumor, rhabdomyosarcoma, sarcoma
INTRODUCTION
Rhabdomyosarcoma (RMS) is the most common malignant soft tissue tumor. This pathology was first delineated as a distinct entity by Arthur Purdy Stout. RMS primarily involves the head and neck region and shows a characteristic age distribution, most of the patients being within the second decade of life.[1]
No clear etiologic factors have been identified to account for the occurrence of this malignant neoplasm. There are however increasing evidences which suggest that gene abnormalities may play a role in the development of RMS.[2,3]
RMS exhibits a spectrum of histologic appearance which includes embryonal, botryoid, pleomorphic and alveolar variants.[4] Among these, botryoid RMS has been considered as a variant of embryonal form. The embryonal and botryoid variants occur in children between birth and 15 years age group (with mean age of occurrence being 8 years), while the alveolar type occurs in young adults between age group of 10 to 25 years (with mean age of occurrence being 16 years) and the pleomorphic variant occur in older people between the age group of 50–56 years.[1]
RMS is an example of group of tumors that often pose difficulty in diagnosis. Here we report a case which on incisional biopsy appeared to be poorly differentiated soft tissue sarcoma/carcinosarcoma, but after excisional biopsy and following immunohistochemical staining, a definitive diagnosis of RMS was established.
CASE REPORT
A 28-year-old male patient reported to the Dental Clinic with a chief complaint of pain in the right lower posterior tooth of 5-month duration. Patient gave a history of 2–3 episodes of pain during this period. There was no fever or pus discharge. There was a history of slight weight loss during this period.
On examination a diffuse extraoral swelling was seen in the right mandibular region extending superio-inferiorly from zygomatic process to the lower border of the mandible and anterior-posteriorly from the corner of the lip to the anterior border of tragus of the ear [Figure 1].
Figure 1.
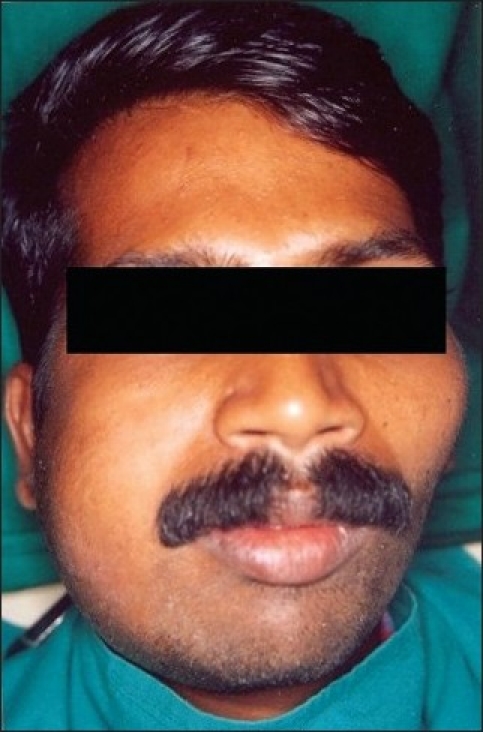
Swelling in the right mandibular region
Intraoral examination revealed an exophytic ulceroproliferative growth on buccal gingiva of about 5 cm × 3 cm size extending right lower second premolar to retromolar region [Figure 2]. The growth was fixed to underlying tissues. The teeth in the affected region exhibited grade II mobility and were displaced. The submandibular lymph nodes were bilaterally palpable, non-tender and fixed.
Figure 2.
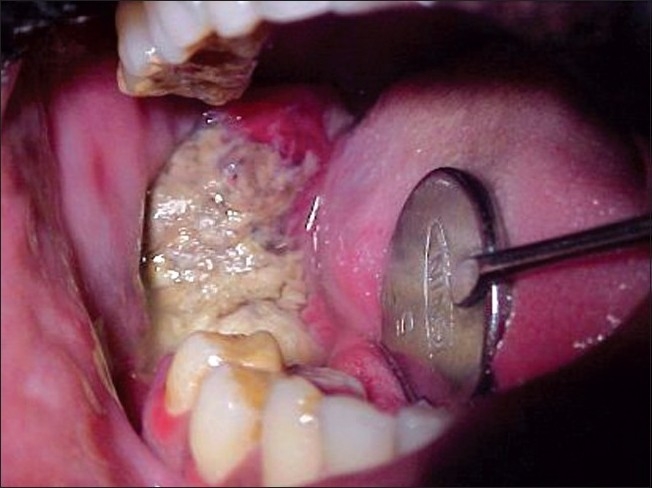
Ulceroproliferative growth in right mandibular region
Radiographic examination of the mandible revealed a destructive lesion involving 45 to 47 region with irregular borders. Erosion of the superior border of inferior alveolar canal was also noted [Figure 3].
Figure 3.
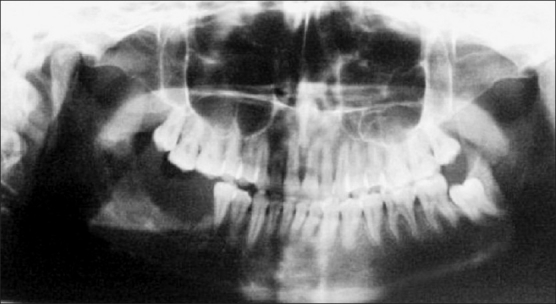
OPG showing destructive lesion
The tumor was subjected for histopathological examination via incisional biopsy and was diagnosed as spindle cell variant of squamous cell carcinoma (SCC). The patient underwent wide excision with marginal mandibular resection and supraomohyoid neck dissection. The excised tissue fragments were sent for histopathologic evaluation. Deviation of histological appearance from that of incisional biopsy prompted further immunohistochemistry which led to the diagnosis of RMS. The patient was advised chemotherapy, but he refused due to monetary reasons. Still, the hospital authorities provided him with 5 doses of Cobalt-60, after which he left the hospital. After 4 months patient returned with recurrence of swelling on same side, but denied any treatment after examination. The patient was finally lost for follow-up.
Pathology report
Incisional biopsy
The Hematoxylin and Eosin (H and E) stained sections revealed a highly cellular connective tissue stroma with many tumor cells lying beneath the epithelium. The tumor cells exhibited marked nuclear and cellular pleomorphism, nuclear hyperchromatism, prominent nucleoli, loss of cohesion between the cells, abundant abnormal mitotic figures and occasional spindle cell morphology of tumor cells. The intervening connective tissue stroma showed diffuse infiltration of chronic inflammatory cells and few erythrocytes filled blood vessel. The overlying epithelium was parakeratotic, stratified squamous epithelium with areas exhibiting surface ulceration [Figures 4, 5a, and 5b]. The decalcified sections also revealed similar features with new bone formation and few osteoclastic giant cells.
Figure 4.
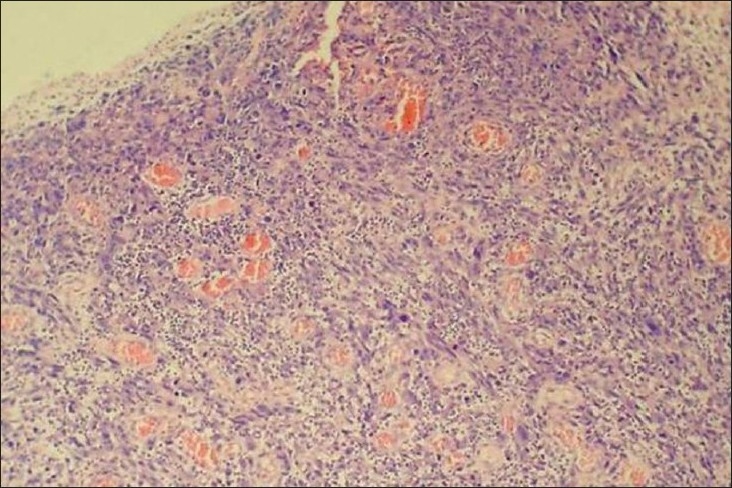
Tumor cells in the connective tissue stroma (H and E, 40×)
Figure 5.
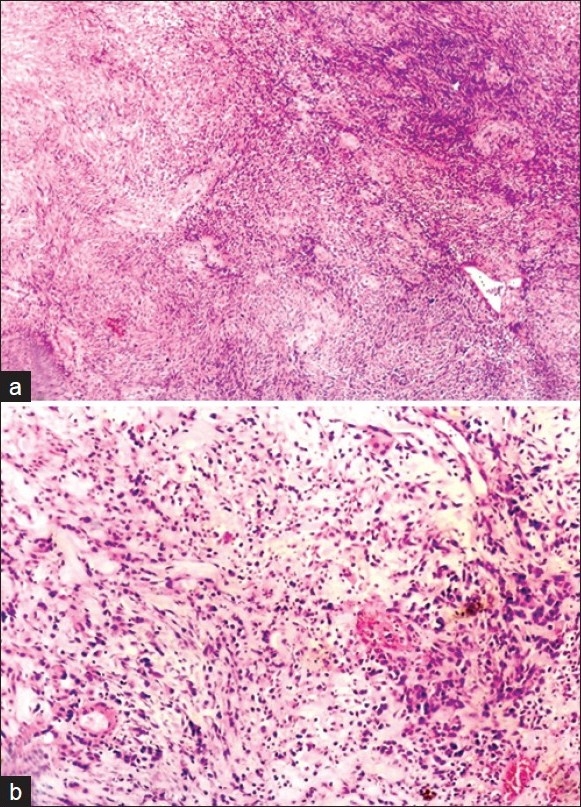
(a, b) Incisional biopsy showing sarcomatous features (H and E, 40×, 100×)
Special stains and immunohistochemistry
Histochemical staining showed scattered glycogen containing tumor cells, which were periodic acid Schiff (PAS) positive, but did not take up Masson trichrome stain. Immunohistochemically, the tumor cells stained positively for cytokeratin (CK-19) [Figure 6].
Figure 6.
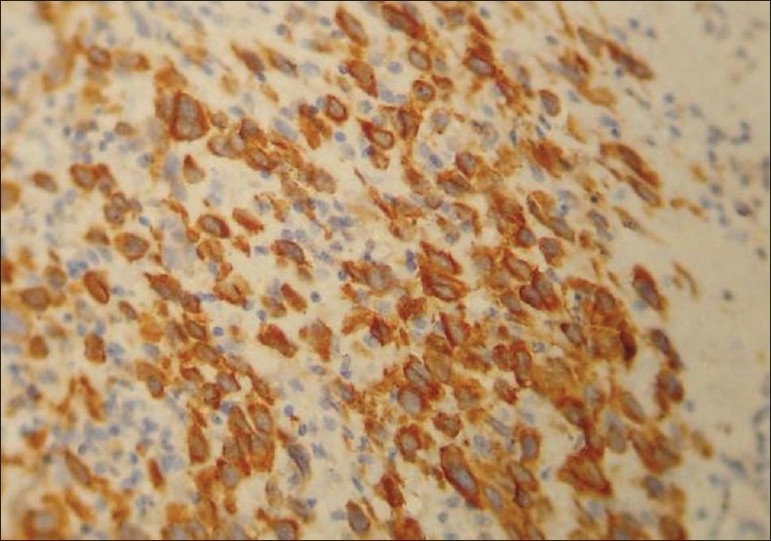
Tumor cells showing CK-19 positivity (400×)
Based on all the above findings, the tumor was categorized as carcinosarcoma or spindle cell variant of SCC. However, request was made to send the excisional biopsy specimen to rule out other spindle cell neoplasms.
Excisional biopsy
The H and E stained sections from excisional tissue revealed highly cellular connective tissue stroma containing round to ovoid eosinophilic and pleomorphic tumor cells with darkly stained vesicular nuclei, cytoplasm of these tumor cells was either granular or fibrillar resembling myofibrils of the muscle cells. Some cells lacked the cross striations and even cell outlines. These cellular features mimicked rhabdomyoblasts. In certain regions tumor cells exhibited spindle cell morphology with elongated, hyperchromatic nuclei and moderate amount of eosinophilic cytoplasm with a small number of peripherally placed myofibrils in some cells.
The stroma also exhibited moderate amount of mixed inflammatory cell infiltrate mainly containing lymphocytes, plasma cells, neutrophils and few eosinophils and mast cells [Figure 7]. The overlying epithelium was atrophic, hyperkeratotic stratified squamous epithelium exhibiting inter and intracellular oedema in the spinous layer with few areas of ulceration.
Figure 7.
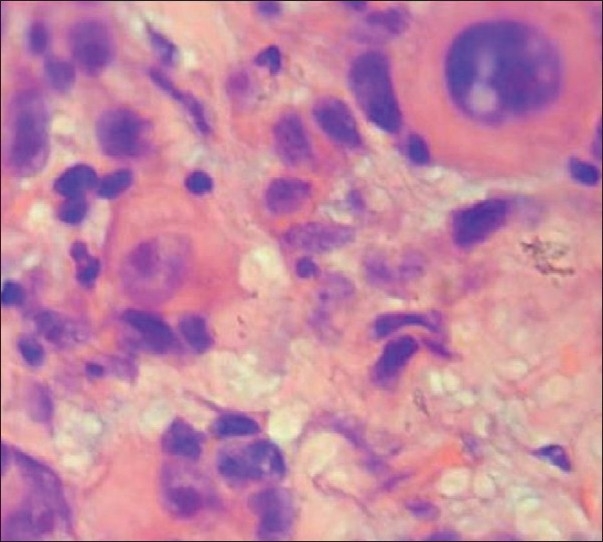
Connective tissue stroma showing rhabdomyoblast (H and E, 400×)
Special stains and immunohistochemistry
The van-Gieson stain showed bundles of collagen fibers in the connective tissue stroma whereas reticulin stain was weakly positive [Figure 8]. PAS positive and diastase digestable intracytoplasmic granules were seen in some of the tumor cells [Figure 9]. Longitudinal fibrils and striations were noticed with phosphotungstic acid hematoxylin (PTAH) stain in the cytoplasm of tumor cells.
Figure 8.
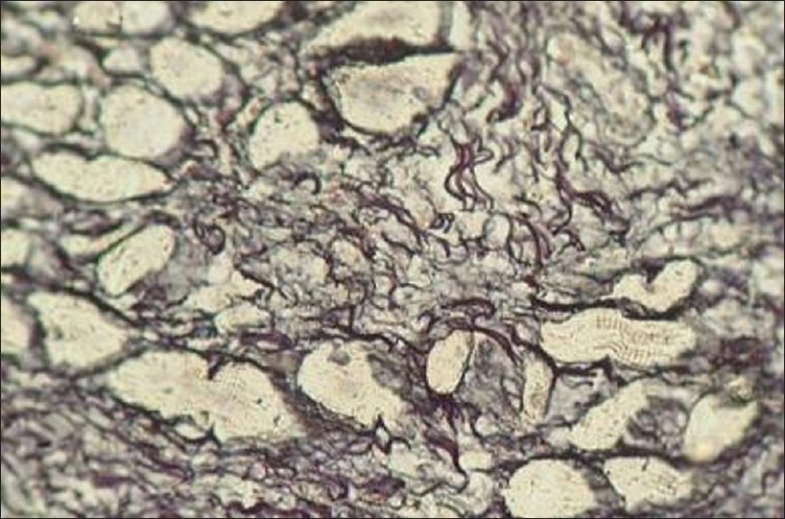
Bundles of collagen fibers showing weak reticulin positivity (400×)
Figure 9.
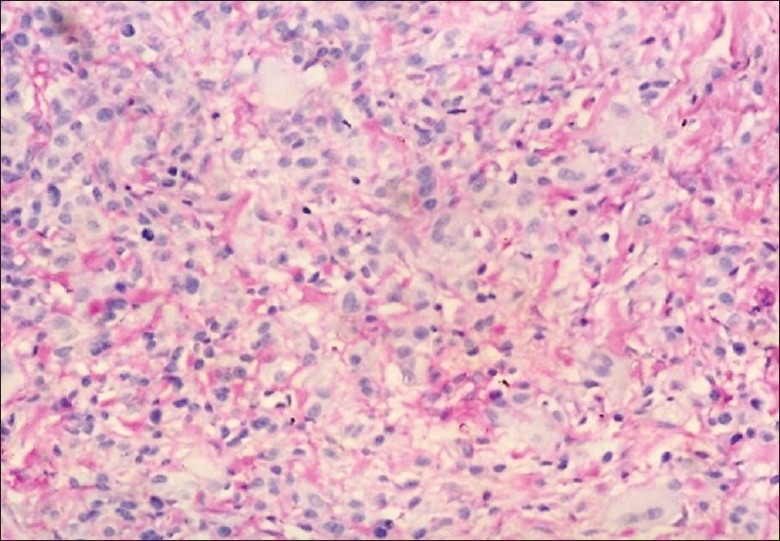
PAS positive and diastase digestable intracytoplasmic granules in tumor cells (400×)
Immunohistochemical staining was performed on the sections for smooth muscle actin, desmin and MyoD1. A positive staining was demonstrated for all these antibodies [Figures 10–12]. On the basis of all the above findings, the tumor was diagnosed as embryonal RMS.
Figure 10.
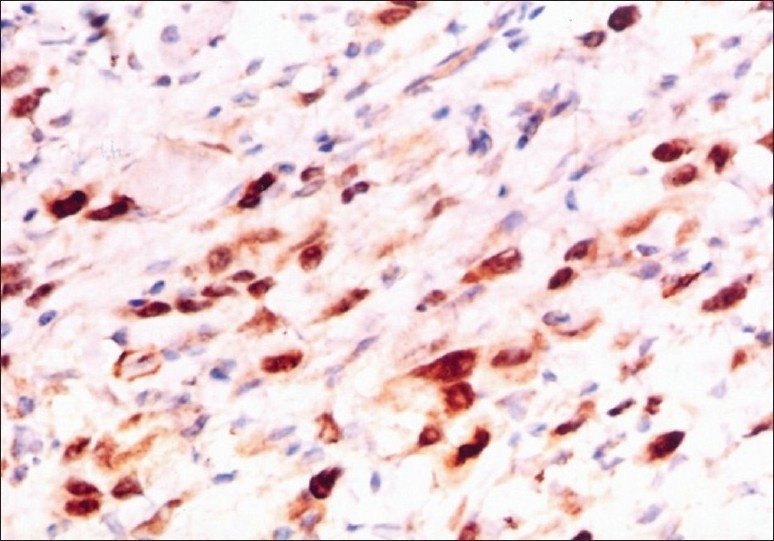
Tumor cells showing positive staining with Myo D1 (100×)
Figure 12.
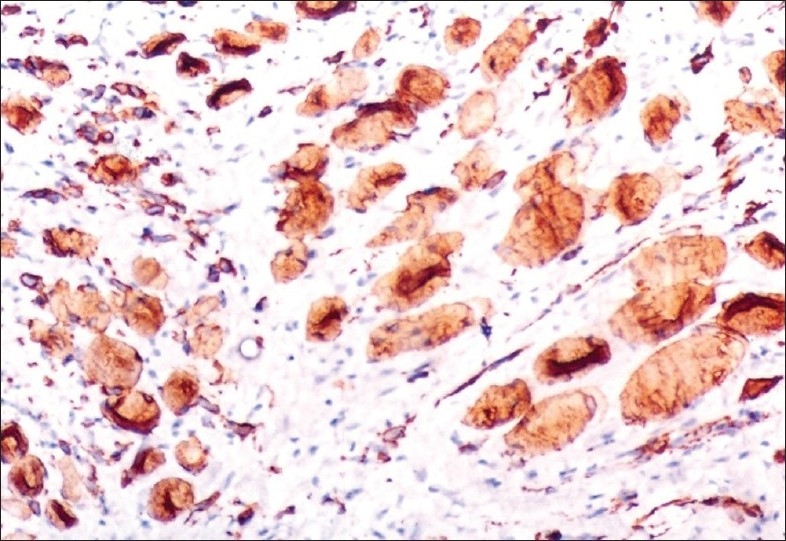
Tumor cells showing positive staining with Desmin (400×)
Figure 11.
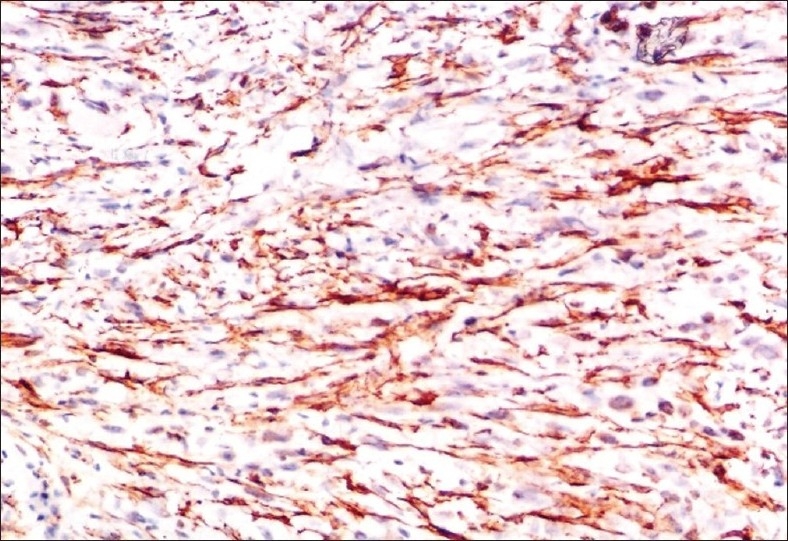
Tumor cells showing positive staining with Actin (100×)
DISCUSSION
RMS is one of the most common mesenchymal malignancies of childhood and adolescence.[1,2,5] It represents 5–15% of all malignant solid tumors and 4 to 8% of all malignant diseases in children under 15 years of age.[5] It is rare in individuals older than 45 years of age.[1]
Head and neck is the principal location of RMS and accounts for 36% of these tumors. Based upon the primary site of involvement of tumor, RMS of head and neck has been subdivided into orbital and non-orbital types. The non-orbital RMSs are further subdivided into the parameningeal and non-parameningeal types.[1,5] The tumors of oral cavity are included under non-orbital and non-parameningeal group, which accounts for 28% of head and neck RMSs.[2,6]
Oral RMS occurs more commonly in males and majority are seen in first two decades of life (mean age 19.6 years). Even the case on hand was in a male patient. A site predilection within the oral region has not been well established.[7] The soft palate appears to be the most common site for oral RMS as stated by some authors.[3] The frequent presentation in the posterior mandibular region including apparently, several intrabony cases, is noteworthy; as it is not recognized as a common site for occurrence within the mouth.[7] The present case also involves the mandibular posterior region. The patients with oral RMSs often have a rapidly enlarging painless mass, usually larger than 1 cm in diameter at time of presentation[2,3,8] with local infiltration, pain, ankyloglossia, paresthesia, and trismus. Primary RMS of head and neck are rarely associated with lymphatic spread.[2,5] Pain was the main presenting symptom in the case reported in this paper.
RMS has been classified by Horn and Enterline on the basis of clinical and histopathologic features into four subtypes: Pleomorphic, alveolar, embryonal, and botryoid. However, the botryoid RMS is a descriptive term for a RMS with polypoid or grape like gross appearance and is considered a variant of embryonal form.[1,3]
Tsokos et al.,[9] developed a new classification scheme related to prognosis. They classified RMS into two groups: Those with favourable histologic feature and prognosis and those with unfavourable histologic feature and prognosis. Prognostically, the embryonal type is more favourable followed by alveolar type. Histologically, the most common type of head and neck RMSs are of embryonal type, so was the presently seen case.[1,7] Further more in the IRS I, II trials, 71% of tumors were of embryonal, and 13% were of alveolar type.[2]
The cytogenetic abnormalities of embryonal and alveolar RMS are distinct. Embryonal rhabdomyosarcoma is characterized by a consistent loss of heterozygosity (LOH) for multiple closely linked loci at chromosome 11p15.5. This loss of heterozygosity may result in activation of tumor suppressor gene or genes, including the human tyrosine hydroxylase gene. Others have reported trisomy 8 as a consistent finding in embryonal RMS.[1]
RMS poses the biggest challenge in routine histopathology. The evidence of skeletal muscle differentiation may not be discernable in sections stained with H and E.[10] The absence of typical strap shaped rhabdomyocytes along with the lack of cytoplasmic cross-banding explains the limitation of standard light microscopy.[4] Moreover, the embryonal subtype of RMS can be exceedingly difficult to distinguish from other poorly differentiated round and spindle cell sarcomas like Ewing's sarcoma, neuroblastoma, peripheral primitive neuroectodermal tumors, and malignant lymphomas.[1,10]
Many special stains can be used to make an accurate and rapid diagnosis of RMS using light microscopy. Massons trichrome stains the cytoplasm of differentiated rhabdomyoblasts in deep red and PTAH stains these myoblasts in deep blue. Among these stains, the former stain is valuable in scanning the sections for the presence of differentiated rhabdomyoblasts and also in identification of “ribbon” and “strap” cells, whereas, the latter stain assists in studying myofibrils and cross-striations. However, surprisingly in the present case, the incisional biopsy was negative for Masson trichrome and in addition the skeletal muscle differentiation was not clearly evident in H and E-stained sections. Hence, a diagnosis in favour of sarcomatous tumor was made. Further, the sections from the excisional tissue showed spindle shaped cells with discernable cross-striations suggesting the presence of myofibrils in the cell cytoplasm. The sections were subjected for PTAH staining, which revealed longitudinal myofibrils and striations in cytoplasm of tumor cells, confirming the skeletal muscle differentiation. PAS stain with or without diastase digestion is considered to be useful in identifying the glycogen content in the cells. This further helps to differentiate rhabdomyosarcoma from other tumors which do not contain glycogen, such as neuroblastoma.[11] A positive staining for PAS was obtained in present case also. Reticulin preparations usually reveal little interstitial collagen and thus help in ruling out sarcomas.[1] In our case, the incisional biopsy specimen was weakly positive for reticulin staining and also, the sections were positive for van Geison staining, thus confirming the presence of bundles of collagen fibres in the connective tissue. Keeping in mind all the above findings, markers were the only choice to reach a definitive diagnosis.
Immunohistochemistry is of great help in this context and several markers have been applied to diagnose rhabdomyosarcoma, but their diagnostic value, sensitivity and specifity vary substantially. The markers that are positive in RMS are desmin, myoglobin, myosin, vimentin, muscle specific actin (HHF 35), sarcomeric actin, smooth muscle actin, and Troponin-T.[1] Occasionally, S100 protein and cytokeratin may also be positive.[12]
A positive reaction for vimentin confirms a mesenchymal derived tumor[4] and desmin indicates muscular differentiation.[2] Desmin is among the earliest muscle structural gene to be expressed in myotome of embryo. It has been regarded as the best single marker for the diagnosis of poorly differentiated RMS.[13] Desmin is very sensitive and is identified in as much as 75–100% of RMSs. However, it is not very specific for skeletal muscle,[12] i.e., although desmin can recognize the myogenic phenotype, they do not distinguish between skeletal and smooth muscle differentiation. Myoglobin on the other hand appears to be specific for skeletal muscle, but it has average sensitivity.[12] Furthermore, with this staining is restricted to more differentiated cells, and this antigen may also be detected in non-muscle cells as a result of diffusion.[1] Other markers which are somewhat less sensitive and specific are S100 protein and cytokeratin. These have also been demonstrated in occasional undifferentiated tumor cells and rhabdomyoblasts, respectively. In a study done by Jean et al., 12% tumors showed staining for S100 proteins and 5% tumors stained strongly for cytokeratin. Pathologists should be aware of such unexpected staining to avoid any erroneous diagnosis[12] as it happened in our case.
The recent recognition of the role of myogenic regulatory proteins in skeletal muscle commitment and differentiation has led to the use of two proteins among the four of the MyoD family, as an excellent marker for all RMS subtypes showing high sensitivity and specificity. The MyoD family comprises of four proteins Myogenin, MyoD1, myf-5, and MRF-4. Antibodies to myogenin and MyoD1 exhibit both high sensitivity and specificity not only in normal tissues but also in neoplasms. Nuclear expression of both proteins was found solely in developing fetal muscle cell; no nuclear expression being found in normal adult tissue. Myogenin and MyoD1 nuclear expression was noted in 91% RMS by Gang-Hong Lee et al., whereas neither was it detected in any of the neuroblastoma or Ewing sarcoma/peripheral primitive neuroectodermal tumor (pPNET) cases examined. Thus, both these proteins offer advantage of high sensitivity and specificity in identifying RMS.[13] In the present case, the incisional biopsy sections were stained for cytokeratin (CK 19). A positive staining made an erroneous diagnosis of an epithelial malignancy. This along with the vague picture from routine and special staining led to the diagnosis of tumor as carcinosarcoma. But later, sections from excisional tissue gave a better histologic picture of the tumor suggestive of various stages in embryogenesis and differentiation of skeletal muscle. This was further confirmed by staining the sections for desmin, smooth muscle actin and MyoD1. A positive staining was demonstrated, thus confirming the diagnosis of RMS of embryonal type.
The management of rhabdomyosarcoma is multidisciplinary approach with optimal integration of surgery, radiotherapy and chemotherapy. Dramatic survival rates have been achieved with multimodal treatment approach.
CONCLUSION
Our case was embryonal RMS of oral cavity (mandible) in an adult patient. The ubiquitous distribution and diverse histologic pattern of RMS posed a challenge to the diagnosis but with aid of special stains, suggestion for skeletal muscle neoplasm was obtained. Finally, the judicious use of immunohistochemical markers such as desmin, smooth muscle actin and MyoD1 ruled out other differential diagnosis and the tumor was categorized as embryonal RMS. It is extremely important that pathologists should be aware of unexpected staining of RMS by cytokeratins and sometimes S100 to avoid any erroneous diagnosis.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- 1.Enzinger FM, Weiss SW, Goldblum JR. Soft tissue tumors. 5th ed. St Louis, Mo, USA: Mosby; 2008. Rhabdomyosarcoma; pp. 595–631. [Google Scholar]
- 2.Doval DC, Kannan V, Acharya RS, Mukherjee G, Shenoy AM, Bapsy PP. Rhabdomyosarcoma of the tongue. Br J Oral Maxillofac Surg. 1994;32:183–6. doi: 10.1016/0266-4356(94)90107-4. [DOI] [PubMed] [Google Scholar]
- 3.Chen SY, Thakur A, Miller AS, Harwick RD. Rhabdomyosarcoma of the oral cavity: Report of four cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1995;80:192–201. doi: 10.1016/s1079-2104(05)80202-1. [DOI] [PubMed] [Google Scholar]
- 4.Bergamini JA, Nadimi H, Kuo PC. Rhabdomyosarcoma of the parapharyngeal space in an adult patient: An immunohistochemical study. J Oral Maxillofac Surg. 1989;47:414–7. doi: 10.1016/0278-2391(89)90349-2. [DOI] [PubMed] [Google Scholar]
- 5.Maurer HM, Moon T, Donaldson M, Fernandez C, Gehan EA, Hammond D, et al. The intergroup rhabdomyosarcoma study: A preliminary report. Cancer. 1977;40:2015–26. doi: 10.1002/1097-0142(197711)40:5<2015::aid-cncr2820400505>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 6.Sutow WW, Lindberg RD, Gehan EA, Ragab AH, Raney RB, Jr, Ruymann F, et al. Three year relapse-free survival rates in childhood. Rhabdomyosarcoma of the head and neck: Report from the Intergroup Rhabdomyosarcoma Study. Cancer. 1982;49:2217–21. doi: 10.1002/1097-0142(19820601)49:11<2217::aid-cncr2820491102>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 7.Peters E, Cohen M, Altini M, Murray J. Rhabdomyosarcoma of the oral and paraoral region. Cancer. 1989;63:963–6. doi: 10.1002/1097-0142(19890301)63:5<963::aid-cncr2820630529>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 8.Sippel HW, Nyberg CD. Embryonal Rhabdomyosarcoma of mandible and maxilla: Report of case. J Oral Surg. 1969;27:731–4. [PubMed] [Google Scholar]
- 9.Tsokos M, Webber BL, Parham DM, Wesley RA, Miser A, Miser JS, et al. Rhabdomyosarcoma. A new classification scheme related to prognosis. Arch Pathol Lab Med. 1992;116:847–55. [PubMed] [Google Scholar]
- 10.Kahn HJ, Yeger H, Kassim O, Jorgensen AO, MacLennan DH, Baumal R, et al. Immunohistochemical and electron microscopic assessment of childhood Rhabdomyosarcoma: Increased frequency of diagnosis over routine histologic methods. Cancer. 1983;51:1897–903. doi: 10.1002/1097-0142(19830515)51:10<1897::aid-cncr2820511023>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 11.Rubin BP, Hasserjian RP, Singer S, Janecka I, Fletcher JA, Fletcher CD. Spindle Cell Rhabdomyosarcoma (so-called) in adults: Report of two cases with emphasis on differential diagnosis. Am J Surg Pathol. 1998;22:459–64. doi: 10.1097/00000478-199804000-00011. [DOI] [PubMed] [Google Scholar]
- 12.Coindre JM, de Mascarel A, Trojani M, de Mascarel I, Pages A. Immunohistochemical study of rhabdomyosarcoma.unexpected staining with S100 protein and cytokeratin. J Pathol. 1988;155:127–32. doi: 10.1002/path.1711550209. [DOI] [PubMed] [Google Scholar]
- 13.Wang NP, Marx J, McNutt MA, Rutledge JC, Gown AN. Expression of myogenic regulatory proteins (myogenin and MyoD1) in small blue round cell tumors of childhood. Am J Pathol. 1995;147:1799–810. [PMC free article] [PubMed] [Google Scholar]