

Abstract
Transgenic mice expressing prion protein (PrP) molecules with several different internal deletions display spontaneous neurodegenerative phenotypes that can be dose-dependently suppressed by coexpression of wild-type PrP. Each of these deletions, including the largest one (Δ32–134), retains 9 aa immediately following the signal peptide cleavage site (residues 23–31; KKRPKPGGW). These residues have been implicated in several biological functions of PrP, including endocytic trafficking and binding of glycosaminoglycans. We report here on our experiments to test the role of this domain in the toxicity of deleted forms of PrP. We find that transgenic mice expressing Δ23–134 PrP display no clinical symptoms or neuropathology, in contrast to mice expressing Δ32–134 PrP, suggesting that residues 23–31 are essential for the toxic phenotype. Using a newly developed cell culture assay, we narrow the essential region to amino acids 23–26, and we show that mutant PrP toxicity is not related to the role of the N-terminal residues in endocytosis or binding to endogenous glycosaminoglycans. However, we find that mutant PrP toxicity is potently inhibited by application of exogenous glycosaminoglycans, suggesting that the latter molecules block an essential interaction between the N terminus of PrP and a membrane-associated target site. Our results demonstrate that a short segment containing positively charged amino acids at the N terminus of PrP plays an essential role in mediating PrP-related neurotoxicity. This finding identifies a protein domain that may serve as a drug target for amelioration of prion neurotoxicity.
Introduction
Transmissible spongiform encephalopathies are progressive neurodegenerative conditions including Creutzfeldt–Jakob disease in humans and bovine spongiform encephalopathy in cattle (Prusiner, 2004). These diseases are caused by conformational conversion of the endogenous cell surface protein, the cellular isoform of prion protein (PrPC), into an insoluble, β-rich isoform, the scrapie isoform of prion protein (PrPSc), that is self-propagating and infectious (Prusiner, 1998; Aguzzi and Polymenidou, 2004). Historically, research in the prion field has focused on the chemical nature of the infectious agent, with relatively little attention being paid to the mechanism by which PrPSc and other abnormal forms of prion protein (PrP) cause neurodegeneration.
Loss of a normal functional activity of PrPC is unlikely, by itself, to explain prion neurotoxicity, since mice carrying targeted deletion of the PrP gene do not display a prion disease phenotype (Büeler et al., 1992; Manson et al., 1994; Mallucci et al., 2002). Nevertheless, several lines of evidence suggest that PrPC plays a role in neurotoxic signaling pathways that in some way contribute to prion-induced pathology (Harris and True, 2006). For example, genetic elimination of neuronal PrPC expression in adult mice greatly prolongs survival and causes reversal of scrapie-associated neuropathology, despite massive accumulation of PrPSc (Mallucci et al., 2003, 2007).
Transgenic mice expressing PrP molecules that harbor internal deletions in the unstructured, N-terminal half of the protein provide potentially important clues to the neurotoxic and neuroprotective signaling mechanisms underlying PrP-associated neurodegeneration (Solomon et al., 2010a). These deletions, including Δ32–121 and Δ32–134 (Shmerling et al., 1998), Δ94–134 (Baumann et al., 2007), and Δ105–125 (Li et al., 2007), induce a spontaneous neurological illness characterized by neuronal loss and/or white matter abnormalities. Significantly, the disease phenotype in each of these lines is suppressed in a dose-dependent fashion by coexpression of wild-type (WT) PrP, suggesting that WT and mutant molecules physically interact with each other and/or compete for binding to a common partner.
Interestingly, each of these neurotoxic PrP deletions, including the largest one (Δ32–134), retains 9 aa (residues 23–31, KKRPKPGGW) immediately following the signal peptide cleavage site. These residues have been implicated in several properties of PrPC, including endocytic trafficking (Sunyach et al., 2003; Taylor et al., 2005) and binding of glycosaminoglycans (GAGs) (Pan et al., 2002; Warner et al., 2002; Taubner et al., 2010). We therefore wondered whether the 23–31 domain might play some role in the neurotoxic properties of deleted forms of PrP.
The present study was undertaken to investigate this idea using a combination of transgenic and cell culture approaches. We report here that removal of residues 23–31 eliminates the toxicity of deleted forms of PrP in vivo as well as in a cell culture assay. We also demonstrate that PrP toxicity is not dependent on endocytosis or binding to endogenous cell surface GAGs, but is potently inhibited by exogenous GAGs. Our results have implications for the molecular mechanisms underlying PrP-associated toxicity, and they suggest that the 23–31 region of PrP may represent a novel target for therapeutic intervention in prion diseases.
Materials and Methods
Plasmid construction.
cDNAs encoding murine Δ23–134 and Δ32–134 PrPs were generated by PCR amplification using the following primers: Δ23–134, 5′-GTCCGAAAGCTTCTCGAGGCCGCCACCATGGCGAACCTTGGCTACTGGCTGCTGGCCCTCTTTGTGACTATGTGGACTGATGTCGGCCTCTGCAGGCCCATGATCCATTTTGGC-3′ (upstream) and 5′-TCGGACTCTAGACTCGAGTCATCATCCCACGATCAGGAAGAT (downstream); Δ32–134, 5′-TTGTACAAGCTTCTCGAGGCCGCCACCATGGCGAACCTTGGCTACTGG-3′ (upstream) and 5′-TCGGACTCTAGACTCGAGTCATCATCCCACGATCAGGAAGAT-3′ (downstream). WT and Δ105–125 PrP cDNAs were prepared as described previously (Li et al., 2007). The upstream primers contain HindIII and XhoI restriction sites, along with a Kozak consensus sequence. The downstream primers contained XhoI and XbaI sites. The resulting PCR products were digested with HindIII and XbaI and cloned into pcDNA 3.1 (+) Hygro (Invitrogen).
The QuikChange Site-Directed Mutagenesis kit (Stratagene) was used to introduce additional mutations into plasmids encoding Δ105–125 and Δ32–134 PrP. To mutate amino acids 23–27 from KKRPK to KRHPS, templates were subjected to PCR amplification using the following primers: 5′-GTCGGCCTCTGCAAACGACACCCATCGCCTGGAGGGTGGAACACCG-3′ (upstream) and 5′-CCGGTGTTCCACCCTCCAGGCGATGGGTGTCGTTTGCAGAGGCCGAC-3′ (downstream). To delete residues 23–26, the PCR amplification was performed using the following primers: 5′-TCGGCCTCTGCAAGCCTGGAGGGTGG-3′ (upstream) and 5′-CCACCCTCCAGGCTTGCAGAGGCCGAC-3′ (downstream).
Generation of Tg(Δ23–134) mice.
A fragment encoding Δ23–134 PrP was released from the pcDNA3.1 (+) Hygro plasmid described above by digestion with XhoI and was ligated into XhoI-digested expression vector MoPrP.Xho (Borchelt et al., 1996). The transgene was released from the recombinant plasmid by NotI restriction digest and purified on GFX PCR DNA purification columns (GE Healthcare). The purified DNA was then injected into the pronuclei of fertilized eggs from C57BL/6J × CBA F1 hybrid mice. Transgenic founders were bred initially to Tga20+/+ mice and then to Zurich I Prn-p0/0 mice on a pure C57BL/6 background (EMMA). Genotyping of transgenic mice was performed by PCR analysis of tail DNA prepared using the Puregene DNA Isolation kit (Gentra Systems). Primers P1 and P4 (Chiesa et al., 1998) were used for genotyping. These primers will amplify both Δ23–134 PrP and Tga20 transgenes, which can be distinguished from each other by size. P2 and P4 primers (Chiesa et al., 1998) were used to amplify the Prn-p+ and Prn-p0 alleles.
Histology.
Animals were perfused with 4% paraformaldehyde, after which brains were isolated and postfixed in the same solution. Paraffin sections were stained with hematoxylin and eosin as described previously (Li et al., 2007).
Immunofluorescence localization.
BHK, N2a, and HEK cells were maintained as previously described (Christensen and Harris, 2009). Cells were plated on glass coverslips at ∼50% confluence. The day after plating, cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
For surface staining, cells were placed on ice for 15 min, washed twice with cold PBS, and incubated in PrP antibody 6H4 (Prionics) in Opti-MEM (Invitrogen). Cells were then washed twice with PBS and fixed for 10 min at room temperature in 4% paraformaldehyde in PBS. After washing, cells were stained with Alexa Fluor 488 goat anti-mouse IgG (Invitrogen).
For internal staining, cells were fixed as above, permeabilized with 0.2% Triton X-100 for 5 min at room temperature, and then stained with antibody 6H4 along with rabbit polyclonal antibodies for the Golgi marker, giantin (Covance), or the ER marker, TRAP (Millipore). Cells were then stained with Alexa Fluor 488 goat anti-mouse IgG and Alexa Fluor 594 goat anti-rabbit secondary antibodies (Invitrogen).
After staining, coverslips were mounted on glass slides and viewed in Nikon TE-2000E inverted fluorescence microscope. Images captured with MetaMorph software (Molecular Devices).
Endocytosis assays.
N2a cells were plated on glass coverslips 1 d before transfection with Lipofectamine 2000 according to the manufacturer's instructions. Cells were removed from incubator, placed on ice for 15 min, and then surface-labeled on ice with PrP antibody 6H4 in Opti-MEM. Cells were then returned to 37°C for 60 min to induce PrP internalization. Next, cells were washed twice with PBS, treated with 0.5% trypsin/0.2% EDTA on ice to remove surface proteins, and fixed for 10 min at room temperature in 4% paraformaldehyde in PBS. Fixative was removed and cells were washed with PBS. Cells were permeabilized with 0.2% Triton X-100 for 5 min at room temperature, blocked for 1 h in 2% goat serum in PBS, and stained with fluorescently conjugated secondary antibodies in goat serum/PBS for 1 h. Finally, coverslips were mounted on glass slides and visualized by fluorescent microscopy as described above.
HEK cells stably expressing WT and mutant PrPs were plated on glass coverslips. The following day, cells were placed on ice for 10 min, and surface PrP was labeled with PrP antibody 6H4 in Opti-MEM on ice for 15 min. Cells were then washed, warm medium was added, and cells were incubated at 37°C for 30 min to allow endocytosis to occur. At the end of this period, cells were fixed without permeabilization as described above. All cells were labeled with Alexa 488-coupled anti-mouse IgG (Invitrogen). Coverslips were then mounted on glass slides and visualized as described above.
Western blotting and PrP insolubility assay.
Brain homogenates (10% w/v) were prepared by mechanical dissociation of single hemispheres using plastic pestles (South Jersey Precision Tool and Mold) in PBS containing Protease Inhibitor Cocktail Tablet (Roche). The suspension was clarified for 5 min at 2300 × g in a microcentrifuge. Total protein in the supernatant was quantitated using the BCA kit (Pierce). After separation by SDS-PAGE, proteins were transferred to PVDF membranes (Millipore) and probed overnight with PrP antibodies (6H4, 6D11, 32.1.1, or P45-66) in 5% nonfat milk, washed with TBS/0.1% Tween, and incubated in HRP-conjugated secondary antibodies (Sigma-Aldrich; or Pierce). Blots were developed with ECL detection system (GE Healthcare).
Detergent-resistant membrane localization.
Isolation of detergent-resistant membranes was conducted as previously described (Christensen and Harris, 2009). Briefly, HEK cells stably expressing WT or mutant Δ23–134 PrPs were lysed in 25 mm Tris-HCl, pH 7.4, 150 mm NaCl, 5 mm EDTA, and 1% Triton X-100, on ice. Lysates were mixed with a 60% OptiPrep solution (Sigma-Aldrich) to achieve at final concentration of 40% OptiPrep. An OptiPrep gradient ranging from 0% (top) to 30% (bottom) was layered atop the lysate/OptiPrep mixture. After 4°C ultracentrifugation for 4 h at 172,000 × g, a 200 μl aliquot of each gradient fraction was methanol precipitated and analyzed by Western blotting with anti-PrP antibody (6H4) and anti-flotillin antibody (BD Biosciences).
Cell culture assays for mutant PrP toxicity.
Cell viability and H2AX phosphorylation assays were performed as described previously (Massignan et al., 2010). In the first assay, HEK cells stably expressing WT or mutant PrPs were treated with Zeocin (500 μg/ml) or (2R,3S,4R,5R,6S)-5-amino-6-[(1R,2S,3S,4R,6S)-4,6-diamino-3-[(2R,3R,4R,5R)-3,5-dihydroxy-5-methyl-4-methylamino-oxan-2-yl]oxy-2-hydroxycyclohexyl] oxy-2-(1-hydroxyethyl)oxane-3,4-diol (G418) (400 μg/ml) for 48 h, after which cell viability was assayed by MTT reduction. In the second assay, HEK cells were treated for 30 min with Zeocin, after which cells were lysed, and phosphorylated H2AX analyzed by Western blotting using the Odyssey fluorescent imaging system (LI-COR). Actin levels were analyzed as a loading control.
GAG experiments.
To test the effect of exogenous GAGs on mutant PrP toxicity, heparin (100 μg/ml) (Sigma-Aldrich) or pentosan sulfate (100 μg/ml) were included during incubation with G418 or Zeocin. Three different molecular size forms of PS were tested: 3000 Da (Sigma-Aldrich); 4500 or 5000 Da (both from Biopharm). Cell viability or phosphorylated H2AX was then measured as described above.
To test the role of endogenous GAGs, cells were either pretreated for 1 h with 5 mU/ml heparinase II (Sigma-Aldrich) or were incubated for 24 h with 30 mm sodium chlorate in Opti-MEM before addition of Zeocin. Cell viability or phosphorylated H2AX were then measured as described above.
To measure the effect of chlorate on GAG sulfation, cells were pretreated with 30 mm sodium chlorate in Opti-MEM for 24 h, after which 25 μCi/ml 35SO4 (PerkinElmer) was added, and cells were incubated for an additional 48 h. Cells were then lysed, and radioactive counts in cell lysates were measured in a scintillation counter.
To measure the effect of heparinase digestion on cell surface GAGs, cells were incubated for 48 h in Opti-MEM containing 25 μCi/ml 35SO4, after which the labeling medium was removed and cells were incubated for 30 min in fresh medium containing heparinase II. Cells were then lysed, and radioactivity in cell lysates was measured in a scintillation counter.
Results
Construction of transgenic mice expressing Δ23–134 PrP
Tg(F35) mice expressing PrP deleted for residues 32–134 exhibit cerebellar ataxia and neurodegeneration, and succumb to illness by 100 d in the absence of full-length PrP (Shmerling et al., 1998). The Δ32–134 deletion preserves residues 23–31 (KKRPKPGGW) immediately following the signal sequence cleavage site (Fig. 1A). This region, which includes a cluster of four positively charged residues, is highly conserved in PrP from several mammalian species (Fig. 1B). Multiple positively charged residues are also present in the region following the signal peptide cleavage site in nonmammalian PrPs and in the PrP paralogs, Doppel and Shadoo (Fig. 1B,C).
Figure 1.

Schematic of PrP constructs and comparison of sequences in the 23–31 region. A, Schematic of wild-type PrP and three different deletion constructs. Structural domains of PrP are indicated by the colored blocks: SS (blue), signal sequence; OR (pink), octapeptide repeats; CC (red), charge cluster; HD (green), hydrophobic domain; GPI (purple), GPI attachment signal. The amino acid sequence of PrP in the 23–31 region is shown above the block diagram; the + symbols above the sequence indicate positively charged amino acids. The dotted lines indicated deleted regions in each construct. B, Consensus alignment for the conserved region immediately following the signal sequence cleavage site in PrP from different species. Sequences were aligned using VectorNTI software. Color coding is as follows: red foreground on yellow background indicates consensus residues that are completely conserved in all sequences; blue foreground on cyan background indicates consensus residues derived from a block of similar residues at that position; black foreground on green background indicates consensus residues derived from occurrence of >50% of a single residue at that position; green foreground on white background indicates residues that are weakly similar to the consensus residue at that position; black foreground on white background indicates nonsimilar residues. C, Consensus alignment for the conserved region immediately following the signal sequence cleavage site in PrP, Doppel, and Shadoo from different species. The color key is the same as in B.
To investigate the role of residues 23–31 in the neurotoxicity of Δ32–134 PrP, we generated transgenic mice expressing Δ23–134 PrP under control of a modified version of the endogenous Prn-p promoter. Δ23–134 represents a further deletion that abolishes the entire N terminus of PrP, including residues 23–31 (Fig. 1A). Seven transgene positive founders were recovered. In case Δ23–134 PrP proved to be highly toxic in the absence of WT PrP, we first bred Tg(Δ23–134) founders to Tga20+/+ mice (Fischer et al., 1996) that overexpress WT PrP. Tg(Δ23–134)/Tga20+/0 progeny were subsequently bred to Prn-p0/0 mice on the C57BL/6 background to obtain Tg(Δ23–134)/Prn-p0/0 offspring lacking endogenous or transgenically encoded WT PrP.
Two Tg(Δ23–134) lines, designated H (high) and L (low), were found to be positive for transgene expression by RT-PCR (data not shown) and Western blotting (Fig. 2A). The H and L lines express Δ23–134 PrP at ∼2 and 0.5 times the level of endogenous PrP, respectively. Thus, the Tg(Δ23–134H) line has a PrP expression level comparable with that of Tg(F35) mice expressing Δ32–134 PrP (Shmerling et al., 1998). On Western blots, the majority of Δ23–134 protein migrates as a single species of ∼30 kDa. After enzymatic removal of the N-linked oligosaccharides using PNGase F, the protein migrates as a single band of the expected size (∼16 kDa), slightly faster than the endogenous C1 fragment, which is missing residues 23–111 (Chen et al., 1995). Thus, similar to Δ32–134 PrP (Shmerling et al., 1998), the Δ23–134 protein exists primarily in a glycosylated form. Western blot analysis with a panel of PrP-specific antibodies demonstrates that, as expected, Δ23–134 PrP is recognized by a C-terminal specific antibody (6H4, residues 144–152), but not N-terminally directed antibodies (M32.1.1, residues 23–50; P45-66, residues 45–66; 6D11, residues 95–100) (Fig. 2B).
Figure 2.

Analysis of PrP expression in Tg(Δ23–134) mice. A, Brain homogenates containing equal amounts of total protein from mice expressing the indicated forms of PrP were either untreated (− symbols) or were treated with PNGase F to remove N-linked oligosaccharides (+ symbols). Samples were then subjected to Western blotting with anti-PrP antibody 6H4. The arrowhead points to the C1 cleavage product of full-length PrP (Chen et al., 1995) in lane 2. B, Brain homogenates containing equal amounts of total protein from mice expressing the indicated forms of PrP were subjected to Western blotting with the indicated anti-PrP antibodies. The schematic below the blot indicates the locations of the epitopes recognized by each of the antibodies. Molecular size markers are given in kilodaltons.
Tg(Δ23–134) mice show no clinical illness or histopathology
Both the H and L lines of Tg(Δ23–134) mice on the Prn-p0/0 background remained clinically normal for >400 d (Fig. 3). Conversely, 100% of Tg(F35) mice on the Prn-p0/0 background exhibited tremor and ataxia by 32 d, which progressed to a total hindlimb paralysis and finally death by 100 d (Fig. 3). In addition, Tg(Δ23–134)/Prn-p0/0 mice of both lines demonstrated no histopathological abnormalities for at least 1 year (as long as they were observed) (Fig. 4) (data not shown). In contrast, Tg(F35)/Prn-p0/0 mice showed significant degeneration of cerebellar granule neurons by 10 weeks of age (Fig. 4). We conclude that Δ23–134 PrP, in contrast to Δ32–134 PrP expressed at an equivalent level, does not cause significant neurotoxicity in transgenic mice.
Figure 3.

Tg(Δ23–134) mice remain healthy, while Tg(F35) mice develop a fatal neurological illness. The plot shows the percentage of mice of the indicated genotypes remaining alive at each age. The number of mice, of either sex, in each group was 12 for Tg(Δ23–134H)/Prn-p0/0, 12 for Tg(Δ23–134L)/Prn-p0/0, 14 for Tg(F35)/Prn-p0/0, 7 for Tg(F35)/Prn-p+/0, 6 for Prn-p0/0, and 7 for Prn-p+/0.
Figure 4.
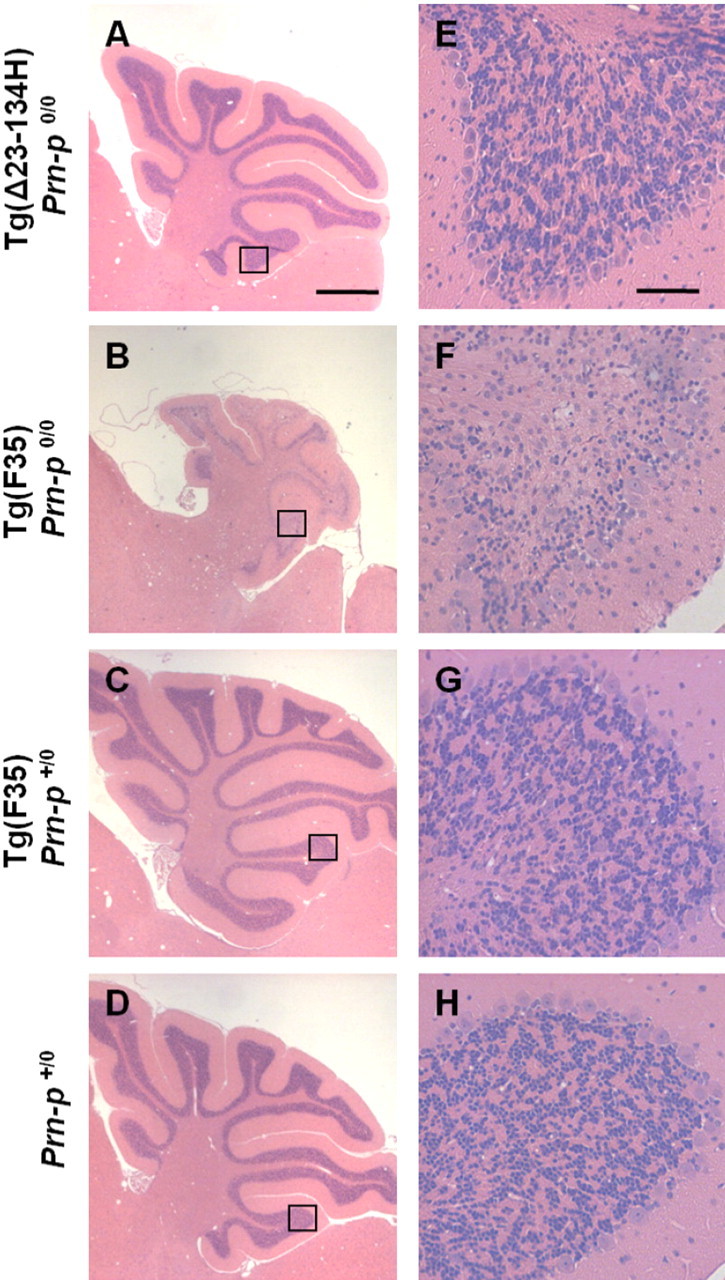
Tg(Δ23–134) mice display normal cerebellar histology, while Tg(F35) mice show cerebellar atrophy and massive loss of granule neurons. A–D show hematoxylin/eosin-stained cerebellar sections from 70-d-old mice of either sex of the indicated genotypes. The area outlined by the box in each panel is shown at higher magnification in E–H. Scale bars: (in A) A–D, 1 mm; (in E) E–H, 50 μm.
Cellular localization and biochemical properties of Δ23–134 PrP in cultured cells
We tested whether the lack of toxicity of Δ23–134 PrP was attributable to an alteration in its cellular localization compared with Δ32–134 PrP. In transfected BHK cells, Δ23–134 PrP, like WT and Δ32–134 PrPs, was localized to the cell surface when assayed by immunostaining of intact cells with PrP antibody 6H4 (Fig. 5A–C). Furthermore, the Δ23–134 protein, like the WT and Δ32–134 proteins, was attached to the cell surface by a glycosylphosphatidylinositol (GPI) anchor, as demonstrated by its release after treatment of cells with phosphatidylinositol-specific phospholipase C (PIPLC), a bacterial enzyme that cleaves the GPI anchor (Fig. 5D–F). After PIPLC treatment, both Δ23–134 and WT PrPs were detected in the cell culture medium by Western blotting of methanol-precipitated proteins (data not shown). Intracellularly, Δ23–134 PrP colocalized with the Golgi marker, giantin, in BHK cells, demonstrating trafficking through the secretory pathway, similar to WT and Δ32–134 PrPs (Fig. 5G–I). Similar results were obtained when localization and biochemical analyses were performed on Δ23–134 PrP expressed in transfected HEK and N2a cells (data not shown), as well as in cerebellar granule neurons cultured from Tg(Δ23–134) mice (Fig. 5J–M).
Figure 5.
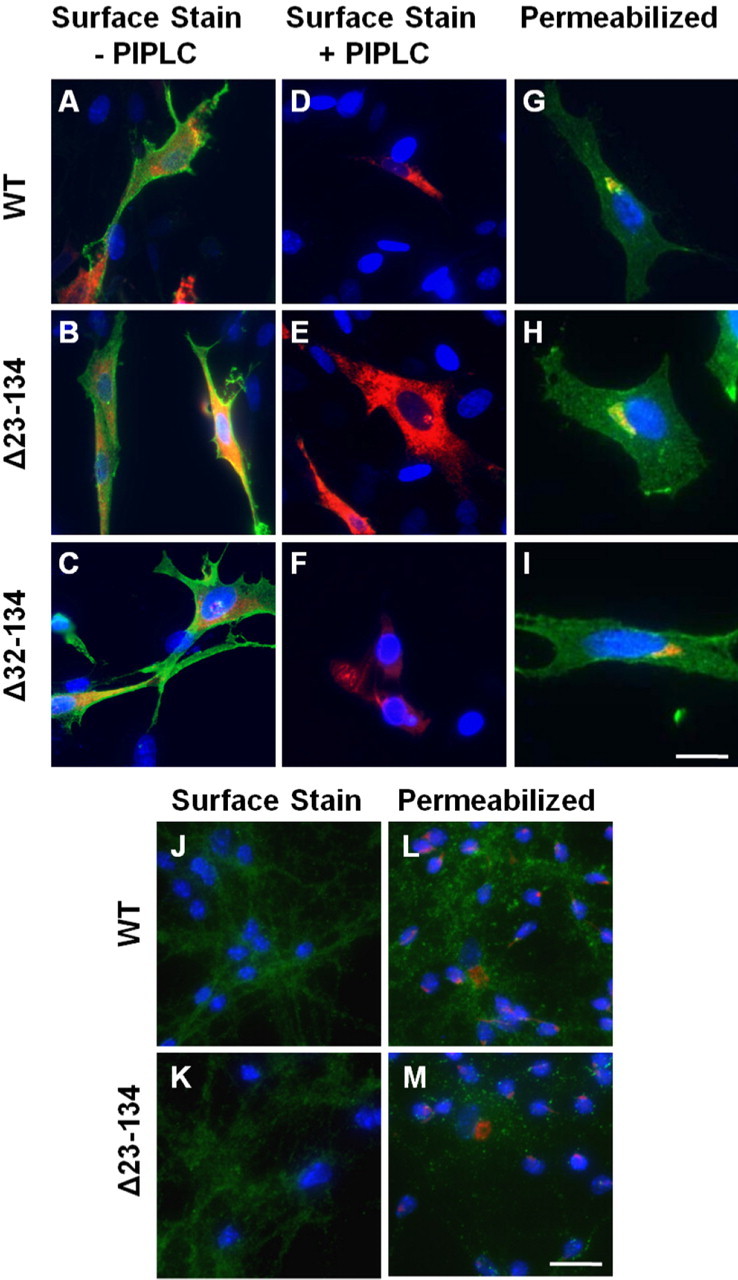
Δ23–134 PrP traffics through the Golgi apparatus and is GPI-anchored to the cell surface. A–I, BHK cells were transiently transfected with plasmids encoding the indicated forms of PrP, along with a plasmid encoding DsRed (A–F) targeted to the ER (to mark transfected cells). A–F, Cells were incubated either with (D–F) or without (A–C) PIPLC to cleave GPI-anchored cell surface proteins and were then stained live on ice with anti-PrP antibody 6H4. Cells were then fixed without permeabilization and stained with fluorescently tagged secondary antibody to reveal PrP. Nuclei were visualized by staining with DAPI. The panels show merged green (PrP), red (Ds-Red ER), and blue (DAPI) images. G–I, Fixed and permeabilized cells were stained with anti-PrP antibody 6H4 (green), along with an antibody to the Golgi marker, giantin (red), and DAPI (blue). J–M, Cerebellar granule neurons were isolated from either wild-type mice (J, L) or Tg(Δ23–134) mice (K, M). Cells were stained for PrP (green) either without permeabilization (J, K), or after fixation, permeabilization, and costaining for giantin (red) (L, M). Cell nuclei were stained with DAPI (blue). Scale bars: (in I) A–I, 25 μm; (in M) J–M, 40 μm.
After ultracentrifugation at 265,000 × g, Δ23–134 PrP in detergent lysates of transfected BHK cells was found in the supernatant fraction, similar to WT PrP and in contrast to PG14 PrP, an aggregation-prone mutant PrP (data not shown).
Additionally, when Δ23–134 PrP was analyzed by density gradient flotation, we found that, like WT PrP, Δ23–134 PrP was distributed in fractions that contained the lipid raft resident protein flotillin-1 (data not shown).
In conclusion, our analyses reveal that Δ23–134 PrP has a cellular localization and solubility similar to those of WT and Δ32–134 PrPs.
Δ23–134 PrP is not toxic in a cell culture assay
We previously reported a cell culture assay for the toxicity of certain mutant forms of PrP, including Δ32–134 and Δ105–125 (Massignan et al., 2010). The assay is based on the ability of these mutant PrP molecules to render cells hypersensitive to several cationic drugs, including G418, hygromycin, and Zeocin. The assay mimics several essential features of PrP toxicity seen in transgenic mice, including rescue by coexpression of wild-type PrP.
We assayed the toxic activity of Δ23–134 PrP and compared it with the activity of Δ32–134 and Δ105–125 PrPs. Stably transfected HEK cells expressing these mutant PrP molecules were incubated for 48 h in the presence of 400 μg/ml G418, and cell viability was assayed by enzymatic reduction of the MTT reagent. As reported previously (Massignan et al., 2010), we found that cells expressing Δ32–134 or Δ105–125 PrP showed significantly reduced viability compared with control cells expressing WT PrP or empty vector, with Δ105–125 being more potent than Δ32–134, reflective of the relative toxicities of these two molecules in transgenic mice (Fig. 6). In contrast, HEK cells expressing Δ23–134 did not show altered viability in this assay. Similar results were obtained for several clones expressing each construct at equivalent levels as determined by Western blotting (data not shown). These results demonstrate that the lack of toxicity of Δ23–134 in transgenic mice is reflected in a cell culture assay.
Figure 6.

Δ23–134 PrP is not toxic in a cell culture assay. HEK cells stably expressing the indicated constructs were treated for 48 h in the presence or absence of 400 μg/ml G418, after which cell viability was measured by MTT reduction. Cell viability is expressed as the value for MTT reduction (A570) of G418-treated cells as a percentage of the value for untreated cells. The bars show mean values ± SEM. The asterisks indicate values that are significantly different from those for WT PrP (p < 0.0001).
Mutant PrP toxicity is not correlated with endocytosis
The discrepancy between the toxic activities of Δ32–134 and Δ23–134 PrPs in both in vivo and in vitro settings suggests that residues 23–31 may represent an important toxicity-determining region. This positively charged region has been previously implicated in several properties of PrP, one of which is endocytic trafficking and targeting to clathrin-coated pits (Nunziante et al., 2003; Sunyach et al., 2003; Taylor et al., 2005). We therefore investigated whether endocytosis was necessary for the toxicity of deleted forms of PrP.
We used two complementary assays to measure the endocytosis of WT, Δ23–134, Δ32–134, and Δ105–125 PrPs. Our initial experiments used transiently transfected N2a neuroblastoma cells, which were previously shown to exhibit significant constitutive endocytosis of PrPC (Shyng et al., 1993, 1995a). After immunolabeling surface PrP at 4°C, cells were incubated at 37°C to initiate endocytosis. After 1 h, cells were treated with trypsin to remove residual surface PrP and were then fixed, permeabilized, and incubated with a fluorescently conjugated secondary antibody to selectively visualize internalized PrP. Cells expressing all constructs that were immunolabeled at 4°C but not warmed to 37°C demonstrated strong labeling of the cell surface, reflective of the initial distribution of the PrP before endocytosis (Fig. 7A–D). After incubation at 37°C, both WT and Δ105–125 PrPs exhibited punctate intracellular fluorescence staining corresponding to endosomal compartments to which antibody-labeled PrP had been delivered (Fig. 7E,H). In marked contrast, Δ23–134 and Δ32–134 PrPs were not significantly endocytosed, since no antibody-labeled PrP appeared inside the cells (Fig. 7F,G).
Figure 7.
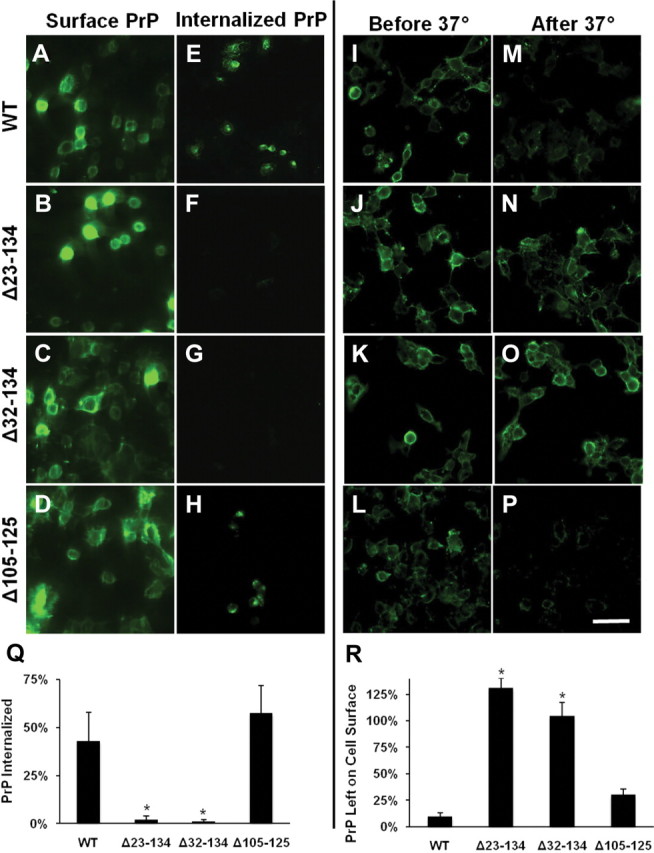
Δ105–125 PrP, but not Δ23–134 or Δ32–134 PrPs, are endocytosed in N2a and HEK cells. A–H, Living N2a cells expressing the indicated constructs were surface-labeled for PrP at 4°C. One set of cultures (A–D) was immediately fixed and stained with fluorescent secondary antibody to confirm the initial distribution of surface PrP. The other set of cultures (E–H) was warmed to 37°C for 1 h to allow endocytosis to occur, after which surface PrP was removed with trypsin, and then the cells were fixed, permeabilized, and stained with fluorescent secondary antibody to reveal the distribution of internalized PrP. Accumulations of PrP in endocytic structures are visible in E and H, but not in F and G. I–P, Living HEK cells expressing the indicated constructs were surface labeled for PrP at 4°C. One set of cultures (I–L) was immediately fixed and stained with fluorescent secondary antibody to confirm the initial distribution of surface PrP. The other set of cultures (M–P) was warmed to 37°C for 1 h to allow endocytosis to occur, after which cells were stained with fluorescent secondary antibody without permeabilization to reveal the amount of PrP remaining on the surface. Loss of surface PrP, indicative of endocytosis, is evident in M and P, but not in N and O. Scale bar: (in P) A–P, 50 μm. Quantification of PrP endocytosis using ImageJ software was performed by measuring the average pixel intensity of PrP immunofluorescence inside the cell (Q) or on the cell surface (R) after warming to 37°C compared with the initial surface immunofluorescence. PrP signals were normalized to fluorescence signal for DAPI, as a measure of cell number. The bars show mean values ± SEM. The asterisks indicate values that are significantly different from those for WT PrP (p < 0.001).
We confirmed these results in another cell type using a second kind of assay to measure PrP endocytosis. In this experiment, using stably transfected HEK cells, internalization of PrP was scored by observing the loss of antibody-bound PrP from the cell surface. Cells were labeled at 4°C with antibody 6H4 and were then incubated at 37°C to initiate endocytosis. Subsequently, cells were fixed without permeabilization and PrP remaining on the cell surface was visualized by incubation with a fluorescently labeled secondary antibody. In control cells that were not warmed to 37°C, surface staining for PrP was, as expected, observed for each of the constructs (Fig. 7I–L). After incubation at 37°C, surface staining remained visible for Δ23–134 and Δ32–134 PrPs (Fig. 7N,O), but not for WT or Δ105–125 PrPs (Fig. 7M,P), reflecting impaired endocytosis of the two former mutants, and paralleling the results obtained using the other assay in N2a cells.
Since neither Δ32–134 nor Δ23–134 PrP were efficiently endocytosed, we conclude that the lack of toxicity of the latter mutant cannot be attributed to its impaired endocytosis. In addition, Δ105–125 PrP, which elicits a neurodegenerative phenotype like Δ32–134 PrP (Li et al., 2007), is constitutively endocytosed. Together, these results demonstrate a lack of correlation between mutant PrP neurotoxicity and endocytic trafficking.
Lys23-Pro26 are necessary for the toxicity of Δ105–125 and Δ32–134 PrPs
To further examine the connection between PrP toxicity and endocytosis, we performed mutagenesis of the 23–31 region of Δ105–125 PrP. We constructed two versions of Δ105–125 PrP with alterations in the 23–31 region that have been shown previously to abolish endocytosis of WT PrP: the triple mutation K24R/R25H/K27S [similar to that in the study by Sunyach et al. (2003)] and deletion Δ23–26 (Taylor et al., 2005). As expected, both of these alterations abolished endocytosis of Δ105–125 PrP as assayed by loss of surface staining for PrP in HEK cells (Fig. 8A–F) and quantified in Figure 8G.
Figure 8.
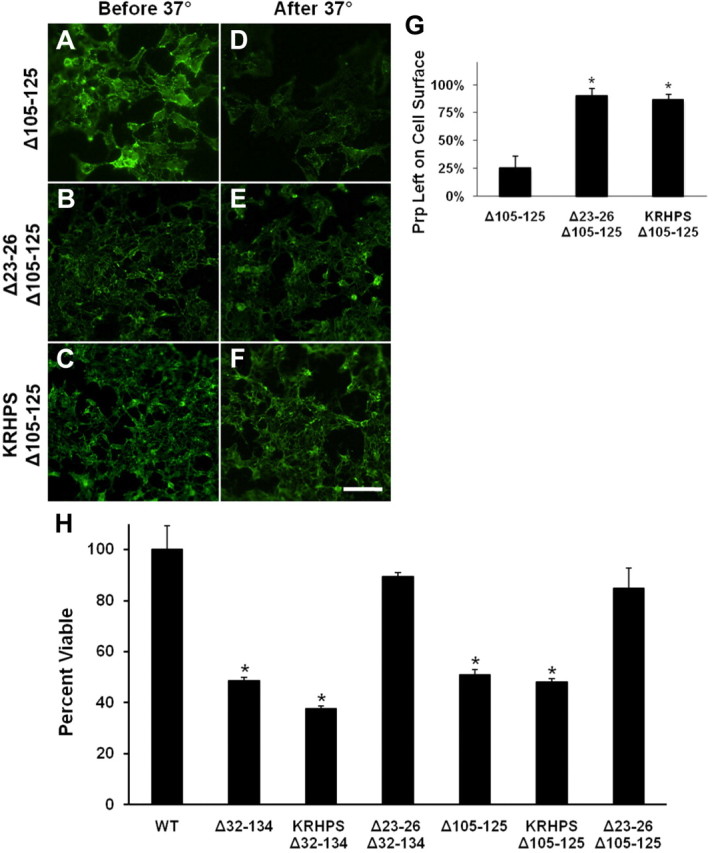
Residues 23–26 are essential for the endocytosis and toxicity of Δ32–134 and Δ105–125 PrPs. Transfected HEK cells expressing the indicated constructs were surface labeled for PrP at 4°C. (KRHPS indicates the triple mutation K24R/R25H/K27S.) One set of cultures (A–C) was immediately fixed and stained with fluorescent secondary antibody to confirm the initial distribution of surface PrP. The other set of cultures (D–F) was warmed to 37°C for 1 h to allow endocytosis to occur, after which cells were stained with fluorescent secondary antibody without permeabilization to reveal the amount of PrP remaining on the surface. Loss of surface PrP, indicative of endocytosis, is evident in D, but not in E and F. Scale bar: (in F) A–F, 50 μm. G, Quantification of PrP endocytosis using ImageJ software was performed by measuring the average pixel intensity of PrP immunofluorescence on the cell surface after warming to 37°C compared with the initial surface immunofluorescence. PrP signals were normalized to fluorescence signal for DAPI, as a measure of cell number. The bars show mean values ± SEM. The asterisks indicate values that are significantly different from those for WT PrP (p < 0.001). H, HEK cells expressing the indicated constructs were treated for 48 h in the presence or absence of 400 μg/ml G418, after which cell viability was measured by MTT reduction. (KRHPS indicates the triple mutation K24R/R25H/K27S.) Cell viability is expressed as the value for MTT reduction (A570) of G418-treated cells as a percentage of the value for untreated cells. The bars show mean values ± SEM. The asterisks indicate values that are significantly different from those for WT PrP (p < 0.0001).
We next tested how these alterations in the 23–31 region affected the toxicity of Δ105–125 PrP as measured by the drug hypersensitivity assay. Interestingly, introduction of the K24R/R25H/K27S triple mutation had no effect on the toxicity of Δ105–125 PrP, whereas the Δ23–26 deletion rendered Δ105–125 PrP nontoxic (Fig. 8H). A similar phenomenon was observed when these two alterations were introduced into the Δ32–134 PrP molecule (Fig. 8H). Together, these results demonstrate that the toxicity of Δ105–125 and Δ32–134 PrPs is independent of endocytosis, but requires the presence of 4 aa (Lys23-Pro26) at the N terminus.
Mutant PrP toxicity is abrogated by exogenous GAGs
Another well documented functional activity of the N-terminal region of PrP is binding to GAGs. Residues 23–35 have been shown to constitute one of several GAG binding sites in PrP (Pan et al., 2002; Warner et al., 2002; Taubner et al., 2010), and there is evidence that binding of GAGs alters several activities of PrPC, including its endocytic trafficking (Shyng et al., 1995b) and its conversion to PrPSc (Caughey and Raymond, 1993). We therefore investigated the role of GAG binding in the toxicity of mutant PrP using in vitro assays.
To explore the role of GAG binding in the toxicity of Δ32–134 and Δ105–125 PrPs, we tested the effect of heparin and pentosan sulfate (PS), two GAGs known to bind PrPC, in the drug hypersensitivity assay. We found that addition of both compounds to HEK cells expressing Δ105–125 PrP significantly diminished cell death in the presence of G418 (Fig. 9A) or Zeocin (data not shown) when assayed by MTT reduction. Pentosan sulfate had a more potent effect than heparin, corresponding to the higher affinity of the former for PrPC (Brimacombe et al., 1999). The rescuing effect of PS was seen with both low- and high-molecular-weight forms of PS, indicating that the effect was not dependent on the size of the molecule (data not shown). As an additional control, we treated untransfected HEK cells with heparin in the presence of G418 and did not observe any effect on cell viability even at very high concentrations of G418 that were sufficient to kill a significant proportion of the cells (data not shown). This result demonstrated that suppression of Δ105–125 toxicity by heparin is not due to an overall increase in cell viability, or inhibition of the action of G418. We observed a similar rescuing effect of heparin and PS from the toxicity of Δ32–134 PrP in the HEK cell viability assay with G418 (data not shown).
Figure 9.
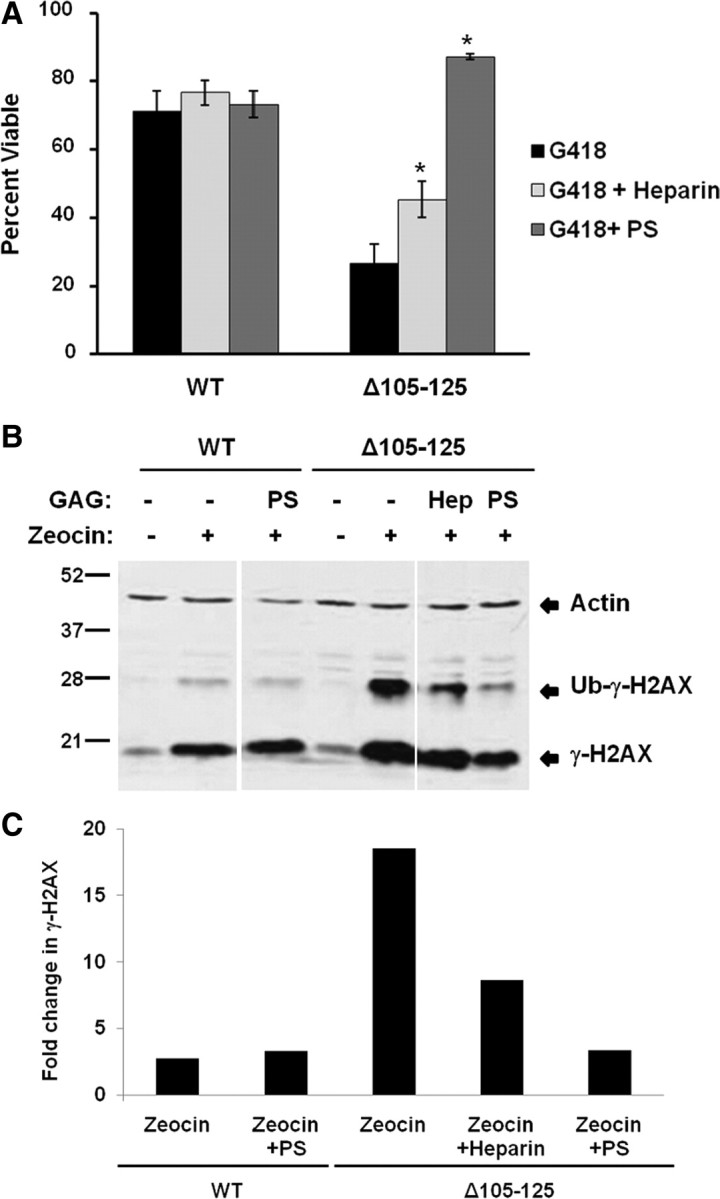
GAGs suppress the toxicity of Δ105–125 PrP in cell culture. A, HEK cells expressing WT or Δ105–125 PrP were treated for 48 h in the presence or absence of 400 μg/ml G418, after which cell viability was measured by MTT reduction. Cell viability is expressed as the value for MTT reduction (A570) of G418-treated cells as a percentage of the value for untreated cells. Where indicated, PS or heparin (each at 100 μg/ml) were present during the 48 h incubation period. The bars show mean values ± SEM. The asterisks indicate values that are significantly different between PS- or heparin-treated cells compared with cells not treated with GAGs (p < 0.0001). B, HEK cells expressing WT or Δ105–125 PrP were pretreated for 1 h in the presence of absence of heparin (“Hep”) (100 μg/ml) or PS (100 μg/ml), after which incubation was continued for an additional 30 min in the presence or absence of Zeocin (400 μg/ml). Cells were then lysed and analyzed for phosphorylated H2AX (γ-H2AX) and actin by Western blotting. The positions of γ-H2AX, ubiquitinated γ-H2AX (Ub-γ-H2AX), and actin are indicated. C, Western blot signals for γ-H2AX were quantitated and normalized to the amount of actin. γ-H2AX levels in the presence of Zeocin were expressed as a fold change relative to levels in the absence of Zeocin. A single experiment, representative of at least three similar ones, is shown.
To confirm these results using another assay, we tested the effect of heparin and PS on Zeocin-induced phosphorylation of histone H2AX in cells expressing Δ105–125 PrP. H2AX is a variant histone that is rapidly phosphorylated in response to double-strand breaks in DNA induced by genotoxic agents like Zeocin. We showed previously that HEK cells expressing Δ105–125 PrP exhibit enhanced phosphorylation of H2AX after treatment with Zeocin (Massignan et al., 2010), presumably reflecting increased entry of Zeocin into the cells. We observed here that both heparin and PS suppressed this Zeocin-induced increase in H2AX phosphorylation (Fig. 9B,C), consistent with the ability of these compounds to suppress Zeocin-induced cell death as measured by MTT reduction (data not shown). Again, PS was more potent than heparin.
Since GAGs have been shown to induce endocytosis of PrP (Shyng et al., 1995b), we tested the possibility that the suppressive effect of these compounds on mutant PrP toxicity was due to removal of PrP from the cell surface. PrP internalization was analyzed as described above, based on loss of surface staining for PrP. We found that treatment with PS or heparin at concentrations that suppressed Δ105–125 and Δ32–134 toxicity induced internalization of WT and Δ105–125 PrP, but not Δ32–134 PrP (data not shown). These results demonstrate that heparin and PS are likely to be rescuing mutant PrP toxicity by a mechanism that does not depend on internalization of PrP from the cell surface.
Mutant PrP toxicity is not dependent on endogenous GAGs
Given our observation that exogenous GAGs are capable of inhibiting the toxicity of mutant PrPs, we wondered whether endogenous, cell-derived GAGs might be required as part of the mechanism by which mutant PrPs exert their toxic activity. We therefore analyzed the effect of interfering with the synthesis of endogenous GAG chains, and of cleaving these chains enzymatically, on the toxicity of Δ105–125 PrP in the HEK cell drug hypersensitivity assay.
Chlorate inhibits GAG chain sulfation by acting as a competitive inhibitor of ATP-sulfurylase (Baeuerle and Huttner, 1986). We pretreated HEK cells expressing Δ105–125 PrP for 24 h with sodium chlorate, and then analyzed H2AX phosphorylation in response to Zeocin exposure. We observed that chlorate treatment had no significant effect on the level of H2AX phosphorylation (Fig. 10). To confirm that chlorate treatment was inhibiting GAG sulfation, we quantitated cellular incorporation of 35SO4. We found that incubation with chlorate reduced 35SO4 incorporation to ∼60% of the levels in untreated cells (data not shown). The residual level of cell-associated radioactivity after chlorate treatment most likely represents 35SO4 incorporation into other intracellular sulfated proteins and amino acids (data not shown).
Figure 10.

Inhibition of endogenous GAG sulfation does not affect the toxicity of Δ105–125 PrP. A, HEK cells expressing WT or Δ105–125 PrP were incubated for 24 h in the presence or absence of 30 mm sodium chlorate (ClO3) or 100 μg/ml PS, after which incubation was continued for an additional 30 min in the presence or absence of Zeocin (400 μg/ml). Cells were then lysed and analyzed for phosphorylated H2AX (γ-H2AX) and actin by Western blotting. The positions of γ-H2AX, ubiquitinated γ-H2AX (Ub-γ-H2AX), and actin are indicated. B, Western blot signals for Ub-γ-H2AX were quantitated and normalized to the amount of actin. Ub-γ-H2AX levels in the presence of Zeocin were expressed as a fold change relative to levels in the absence of Zeocin. A single experiment, representative of at least three similar ones, is shown.
We also investigated the effect on Δ105–125 PrP toxicity of enzymatically digesting cell surface GAG chains using heparinase II. This enzyme cleaves heparan sulfate chains at the one to four linkages between hexosamines and uronic acid residues. We pretreated HEK cells expressing Δ105–125 PrP for 30 min with heparinase II and then analyzed Zeocin-induced phosphorylation of H2AX. We found that heparinase II treatment had no effect on phosphorylated H2AX levels (Fig. 11). To confirm that heparinase II had cleaved cell surface GAG chains, we monitored release of radioactivity from cells that had been previously labeled with 35SO4. We found that cell-associated radioactivity decreased by ∼40% after heparinase II treatment for both wild-type (data not shown) and Δ105–125 PrPs (data not shown), similar to the reduction produced by treatment with chlorate.
Figure 11.

Cleavage of cell surface GAGs does not affect the toxicity of Δ105–125 PrP. A, HEK cells expressing WT or Δ105–125 PrP were incubated for 1 h in the presence or absence of 5 mU/ml heparinase II (“Hep”) or 100 μg/ml PS, after which incubation was continued for an additional 30 min in the presence or absence of Zeocin (400 μg/ml). Cells were then lysed and analyzed for phosphorylated H2AX (γ-H2AX) and actin by Western blotting. The positions of γ-H2AX, ubiquitinated γ-H2AX (Ub-γ-H2AX), and actin are indicated. B, Western blot signals for Ub-γ-H2AX were quantitated and normalized to the amount of actin. Ub-γ-H2AX levels in the presence of Zeocin were expressed as a fold change relative to levels in the absence of Zeocin. A single experiment, representative of at least three similar ones, is shown.
Together, these results indicate that cell surface heparan sulfate chains and sulfation of endogenous GAG chains are not necessary for the toxicity of Δ105–125 PrP in an in vitro assay.
Discussion
Several deletions in the N-terminal tail of PrP induce spontaneous neurodegeneration in transgenic mice (Shmerling et al., 1998; Baumann et al., 2007; Li et al., 2007) and display toxic activity in a cell culture assay (Massignan et al., 2010). Each of these deletions, including the largest one (Δ32–134), retains a highly conserved segment of 9 aa immediately following the signal peptide cleavage site (residues 23–31; KKRPKPGGW). This study was undertaken to investigate the role of residues 23–31 in the toxic activity of deleted forms of PrP. Our results demonstrate that these residues are essential for toxicity in both transgenic mice and cell culture. The contribution of these residues does not depend on their role in endocytic trafficking or binding of endogenous GAGs. However, we have discovered that addition of exogenous GAG molecules can dramatically suppress the toxicity of deleted PrP forms. These results suggest possible mechanisms underlying PrP toxicity, and they implicate the 23–31 region as a possible therapeutic target in treatment of prion diseases.
Importance of residues 23–31
We report here that, in contrast to Tg(F35) mice expressing Δ32–134 PrP, which develop a spontaneous neurodegenerative illness within 3 weeks of birth, Tg(Δ23–134) mice remain healthy and free of neuropathology for >1 year. Both kinds of mice express the transgene at comparable levels under control of the identical, modified PrP promoter. Since the only difference between the proteins encoded by these two transgenes is the presence (in Δ32–134) or absence (in Δ23–134) of residues 23–31, our results strongly implicate 23–31 as an essential neurotoxicity-determining region. These in vivo results were confirmed by a new cell culture assay we developed based on hypersensitivity to the toxic effects of several cationic drugs (Massignan et al., 2010). This assay demonstrates that the removal of as few as 4 aa from the N-terminal region (Lys23-Pro26) is sufficient to abrogate the toxicity of Δ32–134 and Δ105–125 PrPs.
Interestingly, there is evidence that the 23–31 region may also be essential for the ability of wild-type PrP to protect cells against the toxic effects of deleted forms of PrP or Doppel (Atarashi et al., 2003; Drisaldi et al., 2004; Yoshikawa et al., 2008). In this regard, we have found that Δ23–134 and Δ23–31 PrPs display greatly diminished ability to suppress the neurodegenerative phenotype of mice coexpressing Δ32–134 PrP (J.A. Turnbaugh, L. Westergard, D.A. Harris, unpublished observations).
How might the 23–31 region play a role in the toxic activity of deleted forms of PrP? These nine residues have been implicated previously in several biological functions of PrPC, including the following: endocytic trafficking and localization to clathrin-coated pits (Sunyach et al., 2003; Taylor et al., 2005), binding of GAGs (Pan et al., 2002; Warner et al., 2002; Taubner et al., 2010), nuclear localization (Gu et al., 2003), retrograde axonal transport (Hachiya et al., 2004a,b), antimicrobial activity (Pasupuleti et al., 2009), binding of the Alzheimer's Aβ peptide (Chen et al., 2010), regulation of β-secretase activity (Parkin et al., 2007), and transduction of polypeptide segments across the lipid bilayer (Wadia et al., 2008). Of these putative functions, the most extensively characterized are endocytic trafficking and GAG binding. The relationship between these two functions and the toxicity of PrP mutants was therefore the focus of the studies reported here.
Endocytosis is not required for toxicity
PrPC is a GPI-anchored membrane protein that is constitutively endocytosed via clathrin-coated pits and recycled back to the cell surface (Shyng et al., 1993, 1994). Previous studies indicated that the N-terminal region of PrPC, including residues 23–31, plays an important role in targeting the protein to clathrin-coated pits and promoting its subsequent endocytosis. Deletion of Lys23-Pro26 (Taylor et al., 2005) or mutation of three of the four positively charged residues (Sunyach et al., 2003) has been shown to significantly impair endocytosis of mammalian PrPC. In addition, several N-terminal deletions reduce endocytosis of chicken PrPC (Shyng et al., 1995a).
Our results indicate that endocytosis is not required for the toxicity of deleted forms of PrP. Neither Δ23–134 nor Δ32–134 PrP were endocytosed, yet the latter mutant was toxic in our in vitro assay and in transgenic mice. Moreover, the triple mutation K24R/R25H/K27S abolished endocytosis of Δ105–125 PrP but had no effect on its toxicity. Thus, we conclude that the lack of toxicity of Δ23–134 PrP both in transgenic mice and in cell culture is not caused by impaired endocytosis.
Exogenous GAGs inhibit mutant PrP toxicity, but endogenous GAGs are not required for toxicity
Previous studies have identified GAG binding sites in PrP using recombinant protein and synthetic peptides. Three regions of PrP were identified as sufficient for binding of heparin and heparan sulfate (HS), including residues 23–52, 53–93, and 110–128 (Pan et al., 2002; Warner et al., 2002). The first region contains the sequence KKRPKP (residues 23–28), which is similar to a previously identified HS binding consensus sequence XBBBXXBX, where B is a basic residue and X is an uncharged or hydrophobic residue (Cardin and Weintraub, 1989). There is evidence that the GAG-binding domains of PrP may interact with glypican-1, an endogenous heparan sulfate proteoglycan that mediates lipid raft association of PrPC (Taylor et al., 2009). Moreover, exogenous GAGs stimulate endocytosis of PrPC (Shyng et al., 1995b). In addition to their interaction with PrPC, GAGs have also been shown to inhibit formation of PrPSc, and to delay disease in animal models (Caughey and Raymond, 1993; Doh-ura et al., 2004).
Our data demonstrate that two GAGs, heparin and PS, when applied exogenously, significantly suppress the toxicity elicited by PrP mutants in our cell culture assay. The strength of this effect correlates with the binding affinity of these molecules for PrP (Brimacombe et al., 1999), with PS being more potent than heparin. In contrast, we report that the toxicity of Δ105–125 and Δ32–134 PrPs is not dependent on endogenous GAGs, since inhibition of GAG sulfation using chlorate or enzymatic release of surface GAG chains with heparinase does not affect toxic activity in the cell culture assay. Together, these results suggest that exogenously applied GAGs do not act by interrupting a functional interaction between PrP and endogenous GAGs. Rather, mutant PrP toxicity may depend on binding to other, non-GAG molecules on the cell surface through the 23–31 region, perhaps via electrostatic interactions involving positively charged residues. This interaction may be competitively inhibited by supraphysiological levels of exogenous, negatively charged GAGs. A prediction from the results presented here is that intracerebral administration of GAGs such as PS will ameliorate neurodegeneration in Tg(Δ105–125) or Tg(F35) mice. We are currently testing this possibility.
Possible mechanisms for involvement of residues 23–31 in the toxicity of mutant PrP and PrPSc
How does the 23–31 region of PrP function in a cellular context to influence PrP toxicity? We have recently found that expression in transfected cells of Δ105–125 PrP induces large, spontaneous ionic currents that can be detected by patch-clamping techniques (Solomon et al., 2010b). These currents, which are produced by relatively nonselective, cation-permeable channels or pores in the cell membrane, can be silenced by overexpression of wild-type PrP, paralleling the ability of wild-type PrP to suppress the neurodegenerative phenotype of Tg(Δ105–125) mice. Similar currents are induced by PrP molecules carrying several different point mutations in the central region that cause familial prion diseases in humans. These results indicate that the neurotoxicity of some mutant forms of PrP is attributable to enhanced ion channel activity.
Interestingly, the ionic currents associated with Δ105–125 PrP can be rapidly inhibited by treatment with PS (Solomon et al., 2010b). In addition, we have found that deletion of residues 23–31 abolishes the current-inducing ability of Δ105–125 PrP (Solomon et al., 2011). These results suggest that exogenous GAGs block interactions of the 23–31 region of PrP with the membrane or with a cell surface receptor, and this interaction is essential for inducing current activity.
It has been reported that residues 23–29 of PrP can function as an autonomous “protein transduction domain” (PTD) that is capable of translocating polypeptides (either PrP itself or reporter proteins) across the lipid bilayer into the cytoplasm (Wadia et al., 2008). This process, which is thought to involve lipid raft-mediated macropinocytosis, depends on an interaction of the PTD with cell surface GAGs, and can be inhibited by exogenous GAGs. Thus, it is possible that the toxicity of Δ105–125, Δ32–134, and other deleted forms of PrP involves the PTD activity of the N terminus, perhaps via formation of channels or pores in the lipid bilayer.
The results reported here may have relevance to the mechanisms by which PrPSc elicits toxic effects during natural prion diseases. We have postulated that PrPSc may activate some of the same neurotoxic signaling pathways as Δ105–125 and other deleted forms of PrP (Li et al., 2007). If so, then residues 23–31 may play an important role in mediating scrapie-induced neurodegeneration. To test this idea, we are currently investigating whether PrP molecules deleted for residues 23–31, which convert to PrPSc (J. A. Turnbaugh and D. A. Harris, unpublished data), are impaired in their ability to induce neuropathology in scrapie-infected mice. Interestingly, GAGs have been reported to significantly prolong incubation times in mouse models of scrapie, an effect that has generally been attributed to the ability of GAGs to inhibit conversion of PrPC into PrPSc (Caughey and Raymond, 1993; Doh-ura et al., 2004). Our results raise the possibility that GAGs also mitigate scrapie symptoms by blocking a PrPSc-related toxic signaling pathway. N-terminal residues of PrP, including the 23–31 region, have been reported to play a role in several aspects of PrPSc formation and processing (Zulianello et al., 2000; Supattapone et al., 2001; Solforosi et al., 2007; Ostapchenko et al., 2008; Ashok and Hegde, 2009), suggesting that this region may affect both toxicity and PrPSc conversion phenomena. Given these considerations, the 9 aa domain we have studied here represents an attractive target for therapeutic intervention. Small molecules that bind to this positively charged region may prove to be effective in reducing the toxic effects of PrPSc as well as inhibiting its formation.
Footnotes
This work was supported by NIH Grants NS40975 and NS052526 (D.A.H.). Tg(F35) mice were obtained from Adriano Aguzzi (Institute of Neuropathology at the University of Zurich, Zurich, Switzerland), and Prn-p0/0 and Tga20 mice from Charles Weissmann (The Scripps Research Institute, Jupiter, FL). We thank Cheryl Adles and Su Deng for mouse maintenance and genotyping. We acknowledge Aimin Li for help with primer design and cloning, and advice on the course of the project. We are grateful to Michael Green and Richard Stewart for critically reading this manuscript. We appreciate help from Emiliano Biasini for work on the PIPLC release experiments.
References
- Aguzzi A, Polymenidou M. Mammalian prion biology: one century of evolving concepts. Cell. 2004;116:313–327. doi: 10.1016/s0092-8674(03)01031-6. [DOI] [PubMed] [Google Scholar]
- Ashok A, Hegde RS. Selective processing and metabolism of disease-causing mutant prion proteins. PLoS Pathog. 2009;5:e1000479. doi: 10.1371/journal.ppat.1000479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi R, Nishida N, Shingematsu K, Goto S, Kondo T, Sakaguchi S, Katamine S. Deletion of N-terminal residues 23-88 from prion prion protein (PrP) abrogates the potential to rescue PrP-deficient mice from PrP-like protein/doppel-induced neurodegeneration. J Biol Chem. 2003;278:28944–28949. doi: 10.1074/jbc.M303655200. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, Huttner WB. Chlorate—a potent inhibitor of protein sulfation in intact cells. Biochem Biophys Res Commun. 1986;141:870–877. doi: 10.1016/s0006-291x(86)80253-4. [DOI] [PubMed] [Google Scholar]
- Baumann F, Tolnay M, Brabeck C, Pahnke J, Kloz U, Niemann HH, Heikenwalder M, Rülicke T, Bürkle A, Aguzzi A. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 2007;26:538–547. doi: 10.1038/sj.emboj.7601510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13:159–163. doi: 10.1016/s1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- Brimacombe DB, Bennett AD, Wusteman FS, Gill AC, Dann JC, Bostock CJ. Characterization and polyanion-binding properties of purified recombinant prion protein. Biochem J. 1999;342:605–613. [PMC free article] [PubMed] [Google Scholar]
- Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behavior of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- Cardin AD, Weintraub HJ. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis. 1989;9:21–32. doi: 10.1161/01.atv.9.1.21. [DOI] [PubMed] [Google Scholar]
- Caughey B, Raymond GJ. Sulfated polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J Virol. 1993;67:643–650. doi: 10.1128/jvi.67.2.643-650.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Yadav SP, Surewicz WK. Interaction between human prion protein and Aβ oligomers: the role of N-terminal residues. J Biol Chem. 2010;285:26377–26383. doi: 10.1074/jbc.M110.145516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SG, Teplow DB, Parchi P, Teller JK, Gambetti P, Autilio-Gambetti L. Truncated forms of the human prion protein in normal brain and in prion diseases. J Biol Chem. 1995;270:19173–19180. doi: 10.1074/jbc.270.32.19173. [DOI] [PubMed] [Google Scholar]
- Chiesa R, Piccardo P, Ghetti B, Harris DA. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron. 1998;21:1339–1351. doi: 10.1016/s0896-6273(00)80653-4. [DOI] [PubMed] [Google Scholar]
- Christensen HM, Harris DA. A deleted prion protein that is neurotoxic in vivo is localized normally in cultured cells. J Neurochem. 2009;108:44–56. doi: 10.1111/j.1471-4159.2008.05719.x. [DOI] [PubMed] [Google Scholar]
- Doh-ura K, Ishikawa K, Murakami-Kubo I, Sasaki K, Mohri S, Race R, Iwaki T. Treatment of transmissible spongiform encephalopathy by intraventricular drug infusion in animal models. J Virol. 2004;78:4999–5006. doi: 10.1128/JVI.78.10.4999-5006.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drisaldi B, Coomaraswamy J, Mastrangelo P, Strome B, Yang J, Watts JC, Chishti MA, Marvi M, Windl O, Ahrens R, Major F, Sy MS, Kretzschmar H, Fraser PE, Mount HT, Westaway D. Genetic mapping of activity determinants within cellular prion proteins: N-terminal modules in PrPC offset pro-apoptotic activity of the Doppel helix B/B' region. J Biol Chem. 2004;279:55443–55454. doi: 10.1074/jbc.M404794200. [DOI] [PubMed] [Google Scholar]
- Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996;15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Hinnerwisch J, Fredricks R, Kalepu S, Mishra RS, Singh N. Identification of cryptic nuclear localization signals in the prion protein. Neurobiol Dis. 2003;12:133–149. doi: 10.1016/s0969-9961(02)00014-1. [DOI] [PubMed] [Google Scholar]
- Hachiya NS, Watanabe K, Sakasegawa Y, Kaneko K. Microtubules-associated intracellular localization of the NH2-terminal cellular prion protein fragment. Biochem Biophys Res Commun. 2004a;313:818–823. doi: 10.1016/j.bbrc.2003.11.167. [DOI] [PubMed] [Google Scholar]
- Hachiya NS, Watanabe K, Yamada M, Sakasegawa Y, Kaneko K. Anterograde and retrograde intracellular trafficking of fluorescent cellular prion protein. Biochem Biophys Res Commun. 2004b;315:802–807. doi: 10.1016/j.bbrc.2004.01.126. [DOI] [PubMed] [Google Scholar]
- Harris DA, True HL. New insights into prion structure and toxicity. Neuron. 2006;50:353–357. doi: 10.1016/j.neuron.2006.04.020. [DOI] [PubMed] [Google Scholar]
- Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105–125. EMBO J. 2007;26:548–558. doi: 10.1038/sj.emboj.7601507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallucci G, Dickinson A, Linehan J, Klöhn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;30:871–874. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- Mallucci GR, Ratté S, Asante EA, Linehan J, Gowland I, Jefferys JG, Collinge J. Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. EMBO J. 2002;21:202–210. doi: 10.1093/emboj/21.3.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallucci GR, White MD, Farmer M, Dickinson A, Khatun H, Powell AD, Brandner S, Jefferys JG, Collinge J. Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice. Neuron. 2007;53:325–335. doi: 10.1016/j.neuron.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol. 1994;8:121–127. doi: 10.1007/BF02780662. [DOI] [PubMed] [Google Scholar]
- Massignan T, Stewart RS, Biasini E, Solomon IH, Bonetto V, Chiesa R, Harris DA. A novel, drug-based, cellular assay for the activity of neurotoxic mutants of the prion protein. J Biol Chem. 2010;285:7752–7765. doi: 10.1074/jbc.M109.064949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunziante M, Gilch S, Schatzl HM. Essential role of the prion protein N terminus in subcellular trafficking and half-life of the cellular prion protein. J Biol Chem. 2003;278:3726–3734. doi: 10.1074/jbc.M206313200. [DOI] [PubMed] [Google Scholar]
- Ostapchenko VG, Makarava N, Savtchenko R, Baskakov IV. The polybasic N-terminal region of the prion protein controls the physical properties of both the cellular and fibrillar forms of PrP. J Mol Biol. 2008;383:1210–1224. doi: 10.1016/j.jmb.2008.08.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan T, Wong BS, Liu T, Li R, Petersen RB, Sy MS. Cell-surface prion protein interacts with glycosaminoglycans. Biochem J. 2002;368:81–90. doi: 10.1042/BJ20020773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin ET, Watt NT, Hussain I, Eckman EA, Eckman CB, Manson JC, Baybutt HN, Turner AJ, Hooper NM. Cellular prion protein regulates β-secretase cleavage of the Alzheimer's amyloid precursor protein. Proc Natl Acad Sci U S A. 2007;104:11062–11067. doi: 10.1073/pnas.0609621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasupuleti M, Roupe M, Rydengård V, Surewicz K, Sunewicz WK, Chalupka A, Malmsten M, Särensen OE, Schmidtchen A. Antimicrobial activity of human prion protein is mediated by its N-terminal region. PLoS One. 2009;4:e7358. doi: 10.1371/journal.pone.0007358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB, editor. Prion biology and diseases. Ed 2. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 2004. [Google Scholar]
- Shmerling D, Hegyi I, Fischer M, Blättler T, Brandner S, Götz J, Rülicke T, Flechsig E, Cozzio A, von Mering C, Hangartner C, Aguzzi A, Weissmann C. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell. 1998;93:203–214. doi: 10.1016/s0092-8674(00)81572-x. [DOI] [PubMed] [Google Scholar]
- Shyng SL, Huber MT, Harris DA. A prion protein cycles between the cell surface and an endocytic compartment in cultured neuroblastoma cells. J Biol Chem. 1993;268:15922–15928. [PubMed] [Google Scholar]
- Shyng SL, Heuser JE, Harris DA. A glycolipid-anchored prion protein is endocytosed via clathrin-coated pits. J Cell Biol. 1994;125:1239–1250. doi: 10.1083/jcb.125.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng SL, Moulder KL, Lesko A, Harris DA. The N-terminal domain of a glycolipid-anchored prion protein is essential for its endocytosis via clathrin-coated pits. J Biol Chem. 1995a;270:14793–14800. doi: 10.1074/jbc.270.24.14793. [DOI] [PubMed] [Google Scholar]
- Shyng SL, Lehmann S, Moulder KL, Harris DA. Sulfated glycans stimulate endocytosis of the cellular isoform of the prion protein, PrPC, in cultured cells. J Biol Chem. 1995b;270:30221–30229. doi: 10.1074/jbc.270.50.30221. [DOI] [PubMed] [Google Scholar]
- Solforosi L, Bellon A, Schaller M, Cruite JT, Abalos GC, Williamson RA. Toward molecular dissection of PrPC-PrPSc interactions. J Biol Chem. 2007;282:7465–7471. doi: 10.1074/jbc.M610051200. [DOI] [PubMed] [Google Scholar]
- Solomon IH, Schepker JA, Harris DA. Prion neurotoxicity: insights from prion protein mutants. Curr Issues Mol Biol. 2010a;12:51–61. [PMC free article] [PubMed] [Google Scholar]
- Solomon IH, Huettner JE, Harris DA. Neurotoxic mutants of the prion protein induce spontaneous ionic currents in cultured cells. J Biol Chem. 2010a;285:26719–26726. doi: 10.1074/jbc.M110.134619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon IH, Khatri N, Biasini E, Massignan T, Huettner JE, Harris DA. An N-terminal polybasic domain and cell surface localization are required for mutant prion protein toxicity. J Biol Chem. 2011;286:14724–14736. doi: 10.1074/jbc.M110.214973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunyach C, Jen A, Deng J, Fitzgerald KT, Frobert Y, Grassi J, McCaffrey MW, Morris R. The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J. 2003;22:3591–3601. doi: 10.1093/emboj/cdg344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supattapone S, Muramoto T, Legname G, Mehlhorn I, Cohen FE, DeArmond SJ, Prusiner SB, Scott MR. Identification of two prion protein regions that modify scrapie incubation time. J Virol. 2001;75:1408–1413. doi: 10.1128/JVI.75.3.1408-1413.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubner LM, Bienkiewicz EA, Copié V, Caughey B. Structure of the flexible amino-terminal domain of prion protein bound to a sulfated glycan. J Mol Biol. 2010;395:475–490. doi: 10.1016/j.jmb.2009.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DR, Watt NT, Perera WS, Hooper NM. Assigning functions to distinct regions of the N-terminus of the prion protein that are involved in its copper-stimulated, clathrin-dependent endocytosis. J Cell Sci. 2005;118:5141–5153. doi: 10.1242/jcs.02627. [DOI] [PubMed] [Google Scholar]
- Taylor DR, Whitehouse IJ, Hooper NM. Glypican-1 mediates both prion protein lipid raft association and disease isoform formation. PLoS Pathog. 2009;5:e1000666. doi: 10.1371/journal.ppat.1000666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadia JS, Schaller M, Williamson RA, Dowdy SF. Pathologic prion protein infects cells by lipid-raft dependent macropinocytosis. PLoS One. 2008;3:e3314. doi: 10.1371/journal.pone.0003314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner RG, Hundt C, Weiss S, Turnbull JE. Identification of the heparan sulfate binding sites in the cellular prion protein. J Biol Chem. 2002;277:18421–18430. doi: 10.1074/jbc.M110406200. [DOI] [PubMed] [Google Scholar]
- Yoshikawa D, Yamaguchi N, Ishibashi D, Yamanaka H, Okimura N, Yamaguchi Y, Mori T, Miyata H, Shigematsu K, Katamine S, Sakaguchi S. Dominant-negative effects of the N-terminal half of prion protein on neurotoxicity of prion protein-like protein/doppel in mice. J Biol Chem. 2008;283:24202–24211. doi: 10.1074/jbc.M804212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zulianello L, Kaneko K, Scott M, Erpel S, Han D, Cohen FE, Prusiner SB. Dominant-negative inhibition of prion formation diminished by deletion mutagenesis of the prion protein. J Virol. 2000;74:4351–4360. doi: 10.1128/jvi.74.9.4351-4360.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]