

Abstract
Background
ASK1-interacting protein-1 (AIP1), a Ras GTPase-activating protein family member, is highly expressed in endothelial cells (EC) and vascular smooth muscle cells (VSMC). The role of AIP1 in VSMC and VSMC-proliferative disease is not known. We employed mouse graft arteriosclerosis models characterized by VSMC accumulation and intimal expansion to determine the function of AIP1.
Methods and Results
In a single minor histocompatibility antigen (male to female)-dependent aorta transplantation model, AIP1 deletion in the graft augmented neointima formation, an effect reversed in AIP1/ interferon-γ receptor (IFN-γR) doubly deficient aorta donors. In a syngeneic aortic transplantation model in which WT or AIP1-KO mouse aortas were transplanted into IFN-γ receptor deficient recipient and neointima formation induced by intravenous administration of adenovirus encoding a mouse IFN-γ transgene, donor grafts from AIP1-KO enhanced IFN-γ -induced VSMC proliferation and neointima formation. Mechanistically, knockout or knockdown of AIP1 in VSMC significantly enhanced IFN-γ-induced JAK-STAT signaling and IFN-γ-dependent VSMC migration and proliferation, two critical steps in neointima formation. Furthermore, AIP1 specifically binds to JAK2 and inhibits its activity.
Conclusion
AIP1 functions as a negative regulator in IFN-γ-induced intimal formation, in part, by downregulating IFN-γ-JAK2-STAT1/3-dependent migratory and proliferative signaling in VSMC.
Keywords: Arteriosclerosis, Atherosclerosis Vascular grafts, AIP1/DAB2IP
INTRODUCTION
Graft arteriosclerosis, the major cause of late cardiac allograft failure, Is characterized by arterial intimal hyperplasia due to recruitment and proliferation of VSMC, resulting in luminal obstruction and allograft ischemia. Although the mechanism underlying graft arteriosclerosis is not well understood, host T cell-dependent alloantigen recognition is critical1, 2. The evidence for a pathogenetic role of IFN-γ, a proinflammatory cytokine produced by effector T cells, in graft arteriosclerosis is particularly compelling. In mouse heart transplantation models, genetic absence of IFN-γ ameliorates alloimmune-mediated intimal hyperplasia of intramyocardial coronary arteries3–5. IFN-γ has also been implicated as a contributor to arteriosclerosis in other mouse models, e.g., genetic hyperlipidemias due to deficiency of ApoE or of low density lipoprotein receptors6–8. Moreover, microarray analysis of atherectomy specimens from sites of human post-angioplasty restenosis has shown a signature of IFN-γ-induced genes9. In a model in which human arterial segments are interposed into the infrarenal aorta of SCID/beige mice10, 11, human T cell-derived IFN-γ exacerbates graft intimal hyperplasia and lumen loss and its neutralization can prevent T cell-mediated EC and SMC dysfunction10–12. In addition, human IFN-γ (which does not act on mouse cells) can directly promote VSMC proliferation in and arteriosclerosis of the graft in the absence of leukocytes13, 14.
The canonical IFN-γ signaling pathways have been well established. Binding of IFN-γ to its receptor IFN-γR induces receptor dimerization, trans-activation of receptor-associated JAK1 and JAK2 and subsequent tyrosine phosphorylation of the IFN-γR. Phosphorylation of IFN-γR results in recruitment, JAK-mediated tyrosine phosphorylation, dimerization and nuclear translocation of STAT1 which in turn induces expression of IFN-γ responsive genes15, 16. IFN-γ may also activate STAT3 in some cell types including VSMC17–20, and STAT3 signaling is primarily involved in cellular growth and survival17–20. In VSMC, IFN-γ also activates STAT-independent but phosphatidylinositol 3-kinase (PI3K)-Akt-mammalian target of rapamycin (mTOR)-dependent proliferation21. Cellular mechanisms regulating these IFN-γ pathways are less well understood. In the present study, we have identified AIP1 as an endogenous cellular inhibitor of IFN-γ-dependent arteriosclerosis. ASK1-interacting protein 1 (AIP1, also known as DAB2-interacting protein DAB2IP), is a novel member of the Ras-GAP (Ras-GTPase-activating protein) protein family. In addition to the GAP domain, AIP1 also contains other structural domains including a plekstrin homology (PH) and protein kinase C conserved domain (C2) in its N-terminal half and a period-like domain, a proline-rich region, a leucine-zipper, phospho-serines for 14-3-3 binding and an Akt binding domain in its C-terminal half22–26. In EC, AIP1 functions as an adaptor molecule that activates pro-apoptotic ASK1 while inhibiting prosurvival IκBα kinase and PI3K22–29. AIP1 is also highly expressed in VSMC24, 27, 30. In the present study, we investigate the role of AIP1 in VSMC proliferation in vitro and intima formation in vivo.
MATERIALS AND METHODS
Expanded materials and methods are provided in the online supplement, including mouse graft transplantation model, graft analyses, aorta organ and cell culture, immunoprecipitation/immunoblotting, in vitro VSMC migration and proliferation assays, and statistical analyses.
RESULTS
AIP1 deletion enhances IFN-γ-dependent neointima formation in a mouse graft arteriosclerosis (GA) model
AIP1-KO mice appear developmentally normal with no obvious defects in blood vessel formation and maturation27. To determine the function of AIP1 in VSMC in pathological settings, we employed mouse graft arteriosclerosis models characterized by VSMC proliferation and intimal expansion. In our allograft model, a segment of a male donor thoracic aorta is interposed into the abdominal aorta of a female recipient. The host then mounts a T cell-mediated alloimmune responses against the male-specific minor histocompatibility antigen H-Y expressed by the graft31 in which T cell-produced IFN-γ drives graft VSMC proliferation1–4, 32, 33 (Supplemental Fig.I). WT male to WT female transplantation induced GA, characterized by infiltration of leukocytes and neointima formation with accumulation of VSMC (Fig.1A). AIP1-KO male donor grafts to WT female recipients generated significantly more neointima containing more SMA positive cells but similar numbers of leukocytes compared to the WT donor group (Fig.1A with quantification in Fig.1B). Expression of signature genes for Th1 (IFN-γ, TNF-α and T-bet), and Treg (TGF-β and Foxp3) but not Th2 (IL-4) or Th17 (IL-17) were detected by qRT-PCR, suggesting that Th1 and Treg are the major T cells involved in this mouse GA model. No significant differences between WT and AIP1-KO in gene expression of Th1 and Treg signature genes were detected (Supplemental Fig.ID). However, induction of IFN-γ-induced genes (IP-10 and Mig) was higher in the grafts of AIP1-KO than the WT donor groups as shown for both mRNA (Fig.1C) and protein (Fig.1D–E). These data suggest that AIP1 deletion does not alter the T subtypes in GA, but instead affects IFN-γ responses of VSMC to T cells. Of note, IFN-γ-induced chemokines, such as IP-10 and Mig, not only serve as IFN-γ signature genes, but also play active roles in GA progression. Specifically, IP-10 has been shown to be involved in VSMC migration and proliferation34, 35.
Fig.1. Effects of AIP1 deletion on the minor antigen-driven allograft arteriosclerosis.
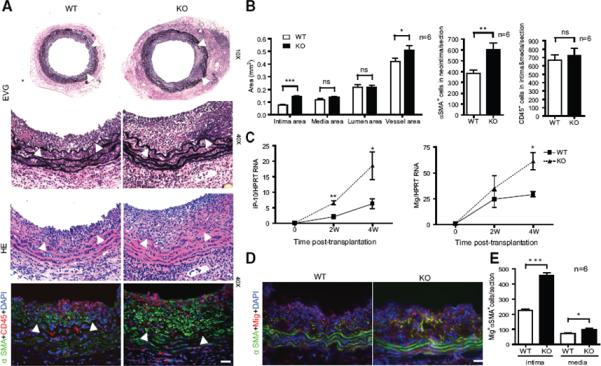
Aorta from WT or AIP1-KO mice was transplanted to female WT recipient (so that the host induces an alloimmune response against the minor Y antigen expressed on the graft EC). Aortas were harvested at 4 weeks post-transplantation. A. Histological analysis of artery grafts by Elastica-van Gieson (EVG), H&E and immunostaining with anti-α-SMA and anti-CD45. Representative photomicrographs are shown. Arrowheads mark the internal elastic lamina to delineate the intima from media. Scale bar: 50 μm. B. Morphometric assessment of artery graft intima, media, lumen and vessel areas by computer-assisted microscopy and quantification of α-SMA+ cells in intima and CD45+ cells in intima and media for each artery graft. C. Transcripts for IFN-γ signature genes Mig and IP-10 were quantified by qRT-PCR and normalized to hypoxanthine guanine phosphoribosyltransferase (HPRT) from WT and AIP1-KO grafts. D–E: Immunostaining with anti-Mig1 in grafts. Representative images are shown in D with quantification in E. All data in B, C and E are mean ± SEM from 6 mice per group. *P<0.05, **P<0.001, ***P<0.0001 comparing WT and AIP1-KO groups. ns: no significance.
To determine the role of IFN-γ signaling in AIP1 deletion-augmented GA, we bred AIP1-KO into IFN-γ receptor-deficient (IFN-γR-KO) background mice to generate AIP1-KO/IFN-γR-KO mice (DKO). Aortas from WT, IFN-γR-KO, AIP1-KO or DKO mice were transplanted to WT female recipients. Deletion of IFN-γR in donor grafts caused a reduced infiltration of leukocytes and neointima formation with decreases of SMA-positive cells compared to WT grafts (Supplemental Fig.IIA with quantification in Fig.IIC). Importantly, AIP1 deletion-augmented GA progression was diminished in the absence of IFN-γR in donor grafts (DKO vs AIP1-KO, Supplemental Fig.IIB with quantification in Fig.IID). These results support a critical role for IFN-γ signaling in GA progression in our mouse model as previously observed in a humanized mouse xenograft transplantation model10, 11, 13, 14.
AIP1 deletion augments IFN-γ-induced graft arteriosclerosis
Human IFN-γ alone is sufficient to induce neointima formation in human artery xenografts13, 14. To directly determine the role of AIP1 in IFN-γ-mediated GA progression, we established an IFN-γ-mediated mouse syngeneic graft arteriosclerosis model. WT or AIP1-KO male aortas were transplanted into IFN-γR-deficient recipient mice followed by intravenous injection of replication-deficient adenovirus encoding the mouse IFN-γ transgene (Ad-IFN-γ) or the control LacZ gene (Ad-LacZ) (Supplemental Fig.IIIA for the illustration and scheme of the protocol). Hepatic infection and liver transgene expression of Ad-LacZ was verified by X-gal staining in the Ad-LacZ group but not the Ad-IFN-γ groups as described previously14. Systemic expression of IFN-γ in serum was detected at a level of 120–150 ng/ml on day 3 and retained up to 5 weeks in the Ad-IFN-γ but not in the LacZ group (Supplemental Fig.IIIB). This extended duration of IFN-γ expression in IFN-γR-KO recipient is similar to that observed in the SCID/beige mice14. Aortas were harvested at 5 weeks post-injection of adenovirus for histological analysis and morphometric assessment of artery graft intima, media, lumen and vessel area. Expression of IFN-γ, but not LacZ control, induced neointimal formation (Fig.2A with quantification in Fig.2B). Unlike the allograft model, no obvious infiltration of leukocytes was detected (Supplemental Fig.IV). Importantly, AIP1-KO donor grafts generated significantly more neointima containing more SMA positive cells compared to the WT donor group (Fig.2C with quantification in Fig.2D).
Fig.2. AIP1 deletion enhances IFN-γ-induced intimal expansion in a syngeneic mouse artery transplantation model.

Thoracic artery from male donor WT or AIP1-KO littermates at 8 week of age were dissected and transplanted into abdominal aorta in male recipient IFN-γR KO mice. One week post-transplant, 1×109 pfu Ad-mIFN-γ or Ad-LacZ was injected via jugular vein into recipient mice. Grafts were harvested 6 weeks after transplantation for further analysis. A. Histological analysis of artery grafts by Elastica–van Gieson (EVG) staining. Representative photomicrographs are shown. Arrowheads mark the internal elastic lamina to delineate the intima from media. Scale bar: 50 μm. B. Morphometric assessment of artery graft intima, media, lumen and vessel areas was performed by computer-assisted microscopy. Data are mean ± SEM from 6 mice per group, *P<0.05 comparing WT and AIP1-KO groups with IFN-γ treatment. C–D. VSMC accumulation was measured by immunostaining with anti-α-SMA. Representative images from WT and KO groups are shown in C with quantification in D. Arrowheads mark the internal elastic lamina to delineate the intima from media. Scale bar: 50 μm. Data are mean ± SEM from 6 mice per group, *P<0.05 and **P<0.001, comparing WT vs KO groups.
To directly assess VSMC proliferation in the neointima, IFN-γR-KO mouse recipients with WT or AIP1-KO donor grafts were injected with BrdU at 3 weeks post-administration of adenovirus. Injection was every day for 2 weeks and grafts were harvested. VSMC proliferation was measured by co-staining with antibodies to α-SMA and BrdU. Proliferative VSMCs (SMA+ BrdU+ cells) were detected in the neointima of IFN-γ-treated but not LacZ group. AIP1-KO donor grafts showed an increased number of SMA+ BrdU+ cells in the neointima but not in the media (Fig.3A with quantification in Fig.3B). Augmented IFN-γ signaling in AIP1-KO was also observed by immunostaining with IFN-γ-activated phospho-JAK2, p-STAT1 and p-STAT3 (Fig.3C with quantification in Fig.3D) as well as the IFN-γ-responsive gene Mig (Fig.3E–G). Co-localizations of these IFN-γ-activated signaling molecules with α-SMA were observed, suggesting that the IFN-γ signaling pathway is activated in VSMC.
Fig.3. AIP1 deletion enhances IFN-γ-induced α-SMA positive cell accumulation and proliferation with augmented JAK2 activation in neointima in mouse artery transplantation model.

A–B. IFN-γR-KO mouse recipients with WT or AIP1-KO donor grafts were injected with BrdU at 3 weeks post-administration of adenoviruses. Injection was every day for 2 weeks and grafts were harvested. VSMC proliferation was measured by co-staining with antibodies to α-SMA and BrdU. Representative images are shown in A with quantifications of IFN-γ groups in B. SMA+BrdU+ cells were counted as proliferative VSMC. C–D. AIP1 deletion enhances IFN-γ-induced JAK2, STAT1 and STAT3 activation in syngeneic vessel grafts. Grafts from the syngeneic graft model were co-immunostaining with antibodies to α-SMA and pJAK2, p-STAT1 or p-STAT3. Representative images of IFN-γ groups are shown in C with quantifications in D. E. Transcripts for the IFN-γ signature gene Mig was quantified by qRT-PCR and normalized to HPRT from WT and AIP1-KO grafts. F–G: Immunostaining with anti-Mig1 in grafts. Representative images IFN-γ groups are shown in F with quantification in G. Arrowheads in A, C and F mark the internal elastic lamina to delineate the intima from media. Scale bar: 50 μm. All data are mean ± SEM from 6 mice per group. *P<0.05, **P<0.001, ***P<0.0001 comparing WT and AIP1-KO groups.
AIP1 deletion enhances IFN-γ responses in cultured aorta and isolated aortic VSMC
To determine if the effect of AIP1 is VSMC autonomous, we examined the IFN-γ responses in isolated aorta and cultured VSMC from WT and AIP1-KO mice. The organ culture of aortas and cell culture of VSMC were treated with mouse IFN-γ and activation of IFN-γ downstream signaling (phosphorylation of JAK2-STAT1/STAT3) was determined by Western blot with phospho-specific antibodies. AIP1 deletion in both whole aorta and isolated VSMC caused enhanced IFN-γ-induced activation of JAK2 and STAT1/3 (Fig.4A,B). Similarly, knockdown of AIP1 in human VSMC also augmented IFN-γ responses (Fig.4C). IFN-β-dependent signaling activates TYK2 instead of JAK2, and to test the specificity of AIP1 in VSMC for IFN-γ, we treated VSMC with IFN-β. In contrast to our results with IFN-γ, AIP1 deletion in mouse VSMC and AIP1 knockdown in human VSMC had no effects on the IFN-β-dependent signaling including activation of TYK2, STAT1 and STAT3 (Supplemental Fig.V). Taken together, these results suggest that AIP1 specifically inhibits IFN-γ responses in VSMC.
Fig.4. AIP1 deletion augmented the IFN-γ responses in cultured aorta and isolated aortic VSMC.
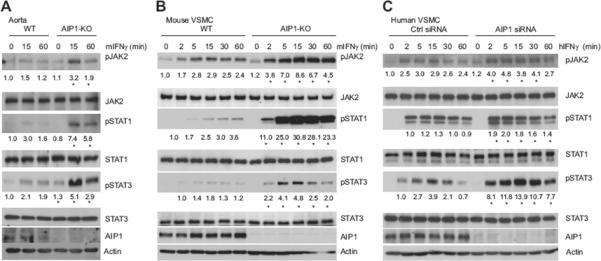
A. Aortas dissected from WT and AIP1-KO mice were cultured overnight in serum-free medium followed by a treatment with mouse IFN-γ (10 ng/ml) for indicated times. B. WT and AIP1-KO mouse VSMCs were cultured in serum-free medium for 24 hours, followed by treatment with mouse IFN-γ (10 ng/ml) for indicated times. C. Human VSMCs were transfected with AIP1 or control siRNA for 48 hours and cultured in serum-free medium for the other 24 hours, followed by treatment with human IFN-γ (10 ng/ml) for indicated times. In A–C, phosphorylation of JAK2, STAT1, and STAT3 and the total proteins were determined by Western blot with respective antibodies. AIP1 and tubulin were also determined. Relative activation of JAK2 (p-JAK2/JAK2), STAT1 (p-STAT1/STAT1) and STAT3 (p-STAT3/STAT3) is shown, with untreated WT or Ctrl siRNA as 1.0. For mouse VSMC and human VSMC, relative activation of STAT1/3 is normalized by taking the 2 min time point of IFN-γ treatment in WT or Ctrl siRNA group as 1.0 because the basal p-STAT1/3 was undetectable. All experiments were repeated at least three times, and data are mean ± SEM from three independent blots. *P<0.05 comparing WT vs AIP1-KO, and Ctrl siRNA vs AIP1 siRNA groups.
AIP1 binds JAK2 and inhibits JAK2 Activity
To define the mechanism by which AIP1 specifically inhibits IFN-γ signaling, we reasoned that AIP1 associates with IFN-γ-specific signaling complex (IFN-γR-JAK2) in VSMC. Our initial data co-immunoprecipitation assay using co-expression of AIP1 with IFN-γR, JAK1 or JAK2 in 293T indicated that AIP1 strongly bound to JAK2, but not to JAK1 or IFN-γR (the IFN-γRα subunit, not shown). Based on this observation, we further characterized the binding between AIP1 and JAK2. To this end, human VSMC were treated with human IFN-γ for various times and association of AIP1 with JAK2 was determined by a co-immunoprecipitation assay with anti-AIP1 followed by Western blot with anti-JAK2). Association of AIP1 with JAK2 was not detected in resting VSMC but was strongly induced by IFN-γ. Kinetics analysis indicated that the association of AIP1 with JAK2 peaked at 15 min and sustained upto 60 min (Fig.5A). IFN-γ-induced phosphorylation of JAK2 peaked at 5 min (see Fig.5C for human VSMC), suggesting that AIP1-JAK2 complex is formed at a late phase of IFN-γ signaling.
Fig.5. AIP1 binds JAK2 and inhibits JAK2 Activity.

A. AIP1 associates with JAK2 in response to IFN-γ in VSMC. Human VSMCs were cultured in serum-free medium for 24 hours, followed by treatment with human IFN-γ (10 ng/ml) for indicated times. Association of AIP1 with IFN-γR-JAK2 was determined by a co-immunoprecipitation assay with anti-AIP1 followed by Western blot with anti-JAK2 or anti-IFN-γR. B. Schematic diagram for AIP1 structural domains and expression constructs. PH, PH domain; C2, PKC conserved domain in which K1 and K2 indicate the 2 lysine clusters; PER, period-like domain; LZ, leucine-zipper motif; ΔPR, deletion of the PR; N, AIP1-N; C, AIP1-C; C-PR, AIP1 containing PR motif at the C-terminal. C. The N-terminal domain, but not the C-terminal domain, of AIP1 binds to JAK2 and inhibits JAK2 activity. Various AIP1 constructs (AIP1-F, AIP1-N and AIP1-C) were co-expressed with JAK2 in 293T cells, and AIP1-JAK2 association was determined by co-immunoprecipitation assay with anti-JAK2, followed by Western blot with anti-FLAG. D. The N-terminal domain of AIP1 binds to and inhibits JAK2 activity. AIP1-N and JAK2 were expressed separately or co-expressed into 293T cells as indicated. Phospho-JAK2 and total JAK2 and AIP1-N were determined by Western blot with respective antibodies. E. Both PH and C2 domain of AIP1 are required for the JAK2 binding. Various AIP1 N-terminal truncates (AIP1-N, PHC2, PH and ΔPH) were co-expressed with JAK2 in 293T cells, and AIP1-JAK2 association was determined by co-immunoprecipitation assay with JAK2 followed by Western blot with anti-Flag. Phospho-JAK2 and total JAK2 in cell lysate was determined by Western blot with respective antibodies. All experiments were repeated at least three times.
AIP1 contains a PH domain, a C2 domain and a GAP domain in the N-terminal half while a period-like domain; a leucine-zipper motif and a proline-rich region in the C-terminal half (Fig.5B). We first mapped the critical domain of AIP1 for the JAK2 binding. Various AIP1 constructs (AIP1-F, AIP1-N and AIP1-C with N-terminal FLAG tag) were co-expressed with JAK2 in 293T cells and cells were untreated or treated with IFN-γ. AIP1-JAK2 association was determined by co-immunoprecipitation assay with anti-JAK2 followed by Western blot with anti-FLAG. Association of AIP1-F with JAK2 could be detected in the resting cells and was enhanced by IFN-γ. AIP1-N strongly associated with JAK2 in both resting and IFN-γ-treated cells. In contrast, AIP1-C did not bind to JAK2 under either condition (Fig.5C). These data suggest that the N-terminal domain of AIP1 is responsible for JAK2 binding. We also examined the effect of AIP1 co-expression on JAK2 activity. Overexpression of JAK2 induced an auto- or trans-phosphorylation of JAK2 which was further enhanced by IFN-γ treatment as determined a p-JAK2-specific antibody (Fig.5C). Interestingly, AIP1-F weakly and AIP1-N strongly inhibited JAK2 activity, correlated with the binding ability of AIP1-F and AIP1-N with JAK2 (Fig.5C). The specific inhibitory effect of AIP1-N on the JAK2 activity was further confirmed by comparing to the control vector (Fig.5D). We then defined which structural domain (the PH, C2 or GAP) at the N-terminal half is required for the JAK2 binding. Various AIP1 N-terminal truncates were generated- AIP1-N (containing all three domains), PHC2 (containing the PH and C2 domains), PH (the PH-only) and ΔPH (with a deletion of the PH) (Fig.5B). A deletion of the PH domain (AIP1-ΔPH) or the GAP domain (AIP1-PHC2) reduced the binding of AIP1 to JAK2. A further deletion of the C2 domain (AIP1-PH) ablated the ability to bind JAK2. These results suggest that an intact N-terminal half is required for the JAK2 binding. Importantly, the strength of the binding of AIP1 mutants to JAK2 correlated with their inhibitory effects on the JAK2 autophosphorylation (Fig.5E).
AIP1 deletion enhances IFN-γ-JAK2-dependent VSMC migration and proliferation
To correlate the role of AIP1 in IFN-γ signaling to its effect on the in vivo neointima formation, we determined the effects of AIP1 deletion and IFN-γ signaling inhibitors on VSMC migration and growth, two critical steps involved in neointima formation36, 37. Human VSMCs were transfected with a control or AIP1 siRNA followed by treatment with human IFN-γ in the absence or presence of a specific inhibitor to JAK2 (AG490), STAT1 (Fludarabine, a purine analog that has been shown to specifically inhibit STAT1 in VSMC)38 or STAT3 (Stattic, a nonpeptide small molecule inhibitor of STAT3 activation and dimerization)39. IFN-γ-induced VSMC migration was measured by monolayer “wound healing” assay. AIP1 knockdown in human VSMC had no effects on the migration of resting VSMC, but significantly augmented IFN-γ-induced VSMC proliferation (Supplemental Fig.VIA with quantification in Fig.VIB). This augmented VSMC migration by AIP1 deletion was blunted in the presence of a JAK2, STAT1 or STAT3-specific inhibitor (Supplemental Fig VIC with quantification in Fig VID). Similar effects of AIP1 on VSMC migration were observed in a transwell migration assay (Fig.6 A–B). In contrast, IFN-γ-induced VSMC migration was significantly inhibited by overexpression of AIP1 in a monolayer assay (Supplemental Fig.VIE–F). We have also performed migration assay using mouse VSMC isolated from WT, AIP1-KO and AIP1-KO/IFNγR-KO (DKO) mice, and AIP1 deletion-augmented cell migration was diminished in DKO VSMC, suggesting that IFNγR is required for AIP1-mediated responses (Supplemental Fig.VIG–H). IFN-γ induced VSMC proliferation was measured by a WST-1 assay. AIP1 knockdown in human VSMC significantly augmented IFN-γ-induced VSMC proliferation (Fig.6C), and enhanced VSMC proliferation caused by AIP1 deletion was blunted by the JAK2 and STAT3-specific inhibitors (Fig.6D). These data suggest that AIP1 regulates IFN-γ-induced VSMC migration and proliferation in a JAK2-STAT3-dependent manner. PDGF-β is a potent VSMC mitogen in vivo and in vitro. To exclude possible role of PDGF-β in AIP1-regulated VSMC growth, we first measured PDGF-β expression in the mouse syngeneic graft model. IFN-γ did not upregulate gene expression of PDGF-β (Supplemental Fig.VII). We then examined the effects of AIP1-KO on PDGF-β-induced VSMC growth and signaling. In contrast to the observations for IFN-γ responses, AIP1 deletion in mouse VSMC aorta and knockdown in human VSMC had no effects on PDGF-β-induced signaling as determined for phosphorylation of PDGFRβ, PLC-γ, Akt and JAK2. (Supplemental Fig.VII). Similarly, AIP1 deletion or knockdown had no effects on PDGFβ-induced VSMC proliferation (not shown). These data suggest that AIP1 specifically regulates IFN-γ-dependent functions in VSMC relevant for arteriosclerosis.
Fig.6. AIP1 deletion enhances IFN-γ-induced VSMC migration and proliferation via the JAK2-STAT1/3-dependent pathways.

Human VSMCs were transfected with AIP1 or control siRNA for 48 hours and cultured in serum-free medium for the other 24 hours. A–B. AIP1 knockdown enhances VSMC migration in a transwell assay. Human VSMCs with siRNA knockdown were subjected to a transwell assay in response to human IFN-γ (10ng/ml). Migratory cell numbers/mm2 in the absence or presence of JAK2 or STAT3-specific inhibitor were quantified in A and B, respectively. Data are mean±SEM from duplicates (6 different areas in each well) from three independent experiments. **, P<0.001 comparing AIP1 siRNA vs Ctrl siRNA groups, ***, P<0.0001 comparing inhibitor vs DMSO groups. C–D. Human VSMCs with siRNA knockdown were subjected to cell proliferation assay response to human IFN-γ (10ng/ml) in the absence (C) or presence (D) of a JAK2 or STAT3-specific inhibitor. Cell proliferation was measured by WST1 assay. Data are mean±SEM from triplicates in each group from three independent experiments. *, P<0.05, **, P<0.001 and ***, P<0.0001 comparing IFN-γ-treated Ctrl siRNA vs AIP1 siRNA groups in A and C, DMSO vs inhibitor groups in B and D.
DISCUSSION
In the present study, we used two mouse graft arteriosclerosis models - the male-specific minor histocompatibility antigen H-Y-dependent aortic allograft arteriosclerosis model and IFN-γ transgene-mediated syngeneic graft arteriosclerosis model to investigate the role of AIP1 in GA. We demonstrate that AIP1-KO mice exhibit dramatically enhanced VSMC proliferation and intimal expansion. The donor grafts from AIP1-KO in both models exhibited enhanced IFN-γ signaling and pathological responses. Mechanistically, we show that knockout or knockdown of AIP1 in VSMC significantly augments IFN-γ-induced signaling, and JAK2-STAT1/3-dependent VSMC migration and proliferation, two critical steps in neointima formation. Furthermore, we show that AIP1 via its intact N-terminal domains associates with JAK2 in response to IFN-γ and inhibits its activity. We conclude that AIP1 inhibits intimal formation and graft arteriosclerosis by downregulating IFN-γ-JAK2-STAT1/3-dependent migratory and proliferative signaling in VSMC.
Vascular IFN-γ signaling in mouse GA models
In the present study, we have created two new mouse models to verify the role of IFN-γ in mouse GA. In the first model, a male donor aortic segment is transplanted into female recipient so that the host induces alloreactive T cell-mediated alloimmune responses against the male-specific minor histocompatibility antigen H-Y expressed on the grafts31. Deletion of IFN-γR in donor grafts blunted leukocyte infiltration and VSMC accumulation in the neointima. This experiment demonstrated a role of IFN-γ in mice consistent with previous findings from a humanized mouse xenograft transplantation model where IFN-γ signaling in graft is critical for GA progression10, 11, 13, 14. In the second model, we determined if IFN-γ alone is sufficient to induce neointima formation by transplanting a male aorta is transplanted into a male IFN-γR-KO recipient animal in which mouse IFN-γ is then systemically expressed by intravenously injection of replication-deficient adenovirus encoding the mouse IFN-γ transgene (Ad-IFN-γ). We observed that expression of IFN-γ, but not LacZ control, induced neointimal formation. Under these conditions, we observed intimal expansion of the graft in the absence of infiltrating leukocytes, once again consistent with the previous observations in a human artery-SCID mouse transplantation model in which vascular IFN-γ signaling alone is sufficient to induce neointima formation in the absence of leukocytes13, 14.
AIP1 on IFN-γ-dependent VSMC effects
The direct effect of IFN-γ on VSMC in these new mouse GA model is an intriguing finding11, 13, 14. We excluded a possible role of PDGF-β (a potent VSMC mitogen) in VSMC growth. However, the molecular mechanism by which IFN-γ induces VSMC migration and proliferation is not fully understood. Although STAT1-dependent signaling has been generally considered as anti-proliferative and apoptotic signaling, the observation that IFN-γ-activated PI3K mediates a serine phosphorylation and subsequent full activation of STAT1 may argue that STAT1 has pro-survival effects40. Moreover, recent studies suggest in some cell types including VSMC that IFN-γ also induces activation of STAT317–20 which is primarily responsible for cellular growth and survival17–20. The central finding of our current study is that we have identified AIP1 as a novel negative regulator of IFN-γ-JAK2-STAT1/3 signaling in VSMC. It has previously been shown that JAK-STAT signaling can be negatively regulated through three main mechanisms: the dephosphorylation of JAKs or STATs by various protein tyrosine phosphatases such as Src homology region 2 domain-containing phosphatases 1 and 2; the inactivation of JAKs by the suppressor of cytokine signaling (SOCS) family of proteins; and the inhibition of the transcriptional activity of STATs by protein inhibitor of STAT (PIAS) proteins41, 42. Here, we demonstrate that AIP1 directly binds to JAK2 and inhibits the JAK2 kinase activity. Therefore, AIP1 acts in a similar manner as the SOCS family (SOCS1). We do not yet know if AIP1 and SOCS1 function synergistically in inhibition of IFN-γ signaling.
An important finding in this study is that AIP1 associates with JAK2 in response to IFN-γ and inhibits JAK2 autokinase activity, thereby preventing JAK2-dependent STAT1/3 tyrosine phosphorylation. Consistent with a previous report14, we have observed that IFN-γ also activates PI3K-Akt-mTOR signaling in VSMC. AIP1 knockdown human VSMC exhibited an enhanced IFN-γ-induced phosphorylation of Akt-mTOR (Supplemental Fig.VIII). In contrast, AIP1 deletion had no effect on either PDGF-BB-induced JAK2 or Akt activation (Supplemental Fig.VII). It is plausible that AIP1 inhibits JAK2 and PI3K-Akt in a receptor complex-dependent manner. In supporting this model, our previous studies have demonstrated that AIP1 associates with VEGF-activated VEGFR2-PI3K complex in which AIP1 via its C2 domain and proline-rich motif binds to VEGFR2 and PI3K, respectively27, 43. A JAK2 inhibitor had a stronger inhibitory effect than an Akt inhibitor on AIP1 deletion-augmented VSMC migration in response to IFN-γ (Supplemental Fig.VIII). These data suggest that IFN-γ-induced JAK2-STAT1/3 is a primary target of AIP1 in VSMC.
AIP1 regulation in GA
We have previously demonstrated that AIP1 in vascular EC functions as an endogenous inhibitor of inflammatory responses in mouse models27. It has been proposed that that vascular EC may selectively recruit or activate IFN-γ-secreting T cells to graft vessels, and IFN-γ in turn contributes to pathogenesis of GA by modulating functions of VSMC. Therefore, the functional effect of IFN-γ on EC may be an early event which occurs prior to and is causally linked to SMC accumulation1, 12. We cannot exclude the role of EC-expressed AIP1 in the allograft model where AIP1 in EC may control infiltration of T cells which in turn regulate VSMC proliferation12. However, our studies using in vitro VSMC culture and the IFN-γ-induced syngeneic graft model support a critical role of VSMC-expressed AIP1 in regulation of GA. Further investigation will be warranted to use EC and VSMC-specific AIP1 knockout mice to determine the relative importance of these effects. Nevertheless, our results in both EC and VSMC suggest that AIP1 is an important regulator in maintaining normal function of the vasculature. A recent human genome-wide association study (GWAS) has identified AIP1 (DAB2IP) as a susceptibility gene for abdominal aortic aneurysm, peripheral vascular disease, early onset of myocardial infarction and pulmonary embolism30. AIP1 expression is primarily regulated by epigenetic modification in its promoter region in tumor29, 44. A future analysis of the regulation of AIP1 expression during GA progression should help to define if AIP1 is a potential therapeutic target for the prevention of GA and other vascular diseases.
Supplementary Material
Novelty and Significance.
What Is Known?
A critical role of the proinflamamtory cytokine IFN-γ in mediating graft arteriosclerosis, characterized by vascular smooth muscle cell accumulation and intimal expansion, has been demonstrated in a humanized graft transplantation model.
AIP1, a Ras GTPase-activating protein family member, is involved in inflammatory responses in vascular endothelial cells.
What New Information Does This Article Contribute?
We have established and characterized two mouse allograft and syngeneic artery transplant models. We have shown that IFN-γ is necessary and sufficient to induced graft arteriosclerosis in these models.
AIP1 prevents arteriosclerosis by inhibiting IFN-γ-dependent smooth muscle cell proliferation and intimal expansion.
AIP1 functions as a new regulator in IFN-γ signaling by inhibiting JAK2 kinase.
Graft arteriosclerosis, the major cause of late cardiac allograft failure, is characterized by arterial intimal hyperplasia due to recruitment and proliferation of vascular smooth muscle cell, resulting in luminal obstruction and allograft ischemia. Although the role of IFN-γ in graft arteriosclerosis has been linked to human allografts and demonstrated in a humanized graft model, mouse models for IFN-γ-dependent graft arteriosclerosis have not been established. We have established and characterized two mouse artery transplant models - a single minor histocompatibility antigen (male to female)-dependent aorta transplantation model and IFN-γ-induced syngeneic graft transplantation model. We have demonstrated, for the first time, that IFN-γ is a critical mediator in mouse graft arteriosclerosis models. These two models will offer us tools to define role of a specific gene involved in graft arteriosclerosis. Using these models we have demonstrated that AIP1 prevents arteriosclerosis by inhibiting IFN-γ-dependent smooth muscle cell proliferation and intimal expansion. Mechanistically, AIP1 specifically inhibits IFN-γ-induced JAK-STAT signaling and IFN-γ-dependent vascular smooth muscle cell migration and proliferation, two critical steps in neointima formation. Our study has provided AIP1 as a potential therapeutic target for the prevention of graft arteriosclerosis and other vascular diseases.
ACKNOWLEDGEMENTS
None
Source of funding This work was supported by NIH grants P01HL070295 and R01 HL065978.
Non-standard abbreviations and acronyms
- AIP1
ASK1-interacting protein-1
- GA
graft arteriosclerosis
- EC
endothelial cells
- SMA
smooth muscle cell actin
- VSMC
vascular smooth muscle cells
Footnotes
Disclosure None declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Mitchell RN. Allograft arteriopathy: pathogenesis update. Cardiovasc Pathol. 2004;13:33–40. doi: 10.1016/S1054-8807(03)00108-X. [DOI] [PubMed] [Google Scholar]
- 2.Tellides G, Pober JS. Interferon-gamma axis in graft arteriosclerosis. Circ Res. 2007;100:622–632. doi: 10.1161/01.RES.0000258861.72279.29. [DOI] [PubMed] [Google Scholar]
- 3.Nagano H, Libby P, Taylor MK, Hasegawa S, Stinn JL, Becker G, Tilney NL, Mitchell RN. Coronary arteriosclerosis after T-cell-mediated injury in transplanted mouse hearts: role of interferon-gamma. Am J Pathol. 1998;152:1187–1197. [PMC free article] [PubMed] [Google Scholar]
- 4.Nagano H, Mitchell RN, Taylor MK, Hasegawa S, Tilney NL, Libby P. Interferon-gamma deficiency prevents coronary arteriosclerosis but not myocardial rejection in transplanted mouse hearts. J Clin Invest. 1997;100:550–557. doi: 10.1172/JCI119564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Furukawa Y, Cole SE, Shah RV, Fukumoto Y, Libby P, Mitchell RN. Wild-type but not interferon-gamma-deficient T cells induce graft arterial disease in the absence of B cells. Cardiovasc Res. 2004;63:347–356. doi: 10.1016/j.cardiores.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 6.Gupta S, Leatham EW, Carrington D, Mendall MA, Kaski JC, Camm AJ. Elevated Chlamydia pneumoniae antibodies, cardiovascular events, and azithromycin in male survivors of myocardial infarction [see comments] Circulation. 1997;96:404–407. doi: 10.1161/01.cir.96.2.404. [DOI] [PubMed] [Google Scholar]
- 7.Whitman SC, Ravisankar P, Elam H, Daugherty A. Exogenous interferon-gamma enhances atherosclerosis in apolipoprotein E−/− mice. Am J Pathol. 2000;157:1819–1824. doi: 10.1016/s0002-9440(10)64820-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buono C, Come CE, Stavrakis G, Maguire GF, Connelly PW, Lichtman AH. Influence of interferon-gamma on the extent and phenotype of diet-induced atherosclerosis in the LDLR-deficient mouse. Arterioscler Thromb Vasc Biol. 2003;23:454–460. doi: 10.1161/01.ATV.0000059419.11002.6E. [DOI] [PubMed] [Google Scholar]
- 9.Zohlnhofer D, Richter T, Neumann F, Nuhrenberg T, Wessely R, Brandl R, Murr A, Klein CA, Baeuerle PA. Transcriptome analysis reveals a role of interferon-gamma in human neointima formation. Mol Cell. 2001;7:1059–1069. doi: 10.1016/s1097-2765(01)00239-8. [DOI] [PubMed] [Google Scholar]
- 10.Lorber MI, Wilson JH, Robert ME, Schechner JS, Kirkiles N, Qian HY, Askenase PW, Tellides G, Pober JS. Human allogeneic vascular rejection after arterial transplantation and peripheral lymphoid reconstitution in severe combined immunodeficient mice. Transplantation. 1999;67:897–903. doi: 10.1097/00007890-199903270-00018. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Burns WR, Tang PC, Yi T, Schechner JS, Zerwes HG, Sessa WC, Lorber MI, Pober JS, Tellides G. Interferon-gamma plays a nonredundant role in mediating T cell-dependent outward vascular remodeling of allogeneic human coronary arteries. Faseb J. 2004;18:606–608. doi: 10.1096/fj.03-0840fje. [DOI] [PubMed] [Google Scholar]
- 12.Koh KP, Wang Y, Yi T, Shiao SL, Lorber MI, Sessa WC, Tellides G, Pober JS. T cell-mediated vascular dysfunction of human allografts results from IFN-gamma dysregulation of NO synthase. J Clin Invest. 2004;114:846–856. doi: 10.1172/JCI21767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tellides G, Tereb DA, Kirkiles-Smith NC, Kim RW, Wilson JH, Schechner JS, Lorber MI, Pober JS. Interferon-gamma elicits arteriosclerosis in the absence of leukocytes. Nature. 2000;403:207–211. doi: 10.1038/35003221. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Bai Y, Qin L, Zhang P, Yi T, Teesdale SA, Zhao L, Pober JS, Tellides G. Interferon-gamma induces human vascular smooth muscle cell proliferation and intimal expansion by phosphatidylinositol 3-kinase dependent mammalian target of rapamycin raptor complex 1 activation. Circ Res. 2007;101:560–569. doi: 10.1161/CIRCRESAHA.107.151068. [DOI] [PubMed] [Google Scholar]
- 15.Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 16.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 17.Peilot H, Rosengren B, Bondjers G, Hurt-Camejo E. Interferon-gamma induces secretory group IIA phospholipase A2 in human arterial smooth muscle cells. Involvement of cell differentiation, STAT-3 activation, and modulation by other cytokines. J Biol Chem. 2000;275:22895–22904. doi: 10.1074/jbc.M002783200. [DOI] [PubMed] [Google Scholar]
- 18.Qing Y, Stark GR. Alternative activation of STAT1 and STAT3 in response to interferon-gamma. J Biol Chem. 2004;279:41679–41685. doi: 10.1074/jbc.M406413200. [DOI] [PubMed] [Google Scholar]
- 19.Ramana CV, Kumar A, Enelow R. Stat1-independent induction of SOCS-3 by interferon-gamma is mediated by sustained activation of Stat3 in mouse embryonic fibroblasts. Biochem Biophys Res Commun. 2005;327:727–733. doi: 10.1016/j.bbrc.2004.12.074. [DOI] [PubMed] [Google Scholar]
- 20.Shen Y, Devgan G, Darnell JE, Jr., Bromberg JF. Constitutively activated Stat3 protects fibroblasts from serum withdrawal and UV-induced apoptosis and antagonizes the proapoptotic effects of activated Stat1. Proc Natl Acad Sci U S A. 2001;98:1543–1548. doi: 10.1073/pnas.041588198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bai Y, Ahmad U, Wang Y, Li JH, Choy JC, Kim RW, Kirkiles-Smith N, Maher SE, Karras JG, Bennett CF, Bothwell AL, Pober JS, Tellides G. Interferon-gamma induces X-linked inhibitor of apoptosis-associated factor-1 and Noxa expression and potentiates human vascular smooth muscle cell apoptosis by STAT3 activation. J Biol Chem. 2008;283:6832–6842. doi: 10.1074/jbc.M706021200. [DOI] [PubMed] [Google Scholar]
- 22.Zhang H, Zhang H, Lin Y, Li J, Pober JS, Min W. RIP1-mediated AIP1 phosphorylation at a 14-3-3-binding site is critical for tumor necrosis factor-induced ASK1-JNK/p38 activation. J Biol Chem. 2007;282:14788–14796. doi: 10.1074/jbc.M701148200. [DOI] [PubMed] [Google Scholar]
- 23.Zhang H, Zhang R, Luo Y, D'Alessio A, Pober JS, Min W. AIP1/DAB2IP, a novel member of the Ras-GAP family, transduces TRAF2-induced ASK1-JNK activation. J Biol Chem. 2004;279:44955–44965. doi: 10.1074/jbc.M407617200. [DOI] [PubMed] [Google Scholar]
- 24.Zhang R, He X, Liu W, Lu M, Hsieh JT, Min W. AIP1 mediates TNF-alpha-induced ASK1 activation by facilitating dissociation of ASK1 from its inhibitor 14-3-3. J Clin Invest. 2003;111:1933–1943. doi: 10.1172/JCI17790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie D, Gore C, Liu J, Pong RC, Mason R, Hao G, Long M, Kabbani W, Yu L, Zhang H, Chen H, Sun X, Boothman DA, Min W, Hsieh JT. Role of DAB2IP in modulating epithelial-to-mesenchymal transition and prostate cancer metastasis. Proc Natl Acad Sci U S A. 2010;107:2485–2490. doi: 10.1073/pnas.0908133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie D, Gore C, Zhou J, Pong RC, Zhang H, Yu L, Vessella RL, Min W, Hsieh JT. DAB2IP coordinates both PI3K-Akt and ASK1 pathways for cell survival and apoptosis. Proc Natl Acad Sci U S A. 2009;106:19878–19883. doi: 10.1073/pnas.0908458106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang H, He Y, Dai S, Xu Z, Luo Y, Wan T, Luo D, Jones D, Tang S, Chen H, Sessa WC, Min W. AIP1 functions as an endogenous inhibitor of VEGFR2-mediated signaling and inflammatory angiogenesis in mice. J Clin Invest. 2008;118:3904–3916. doi: 10.1172/JCI36168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wan T, Liu T, Zhang H, Tang S, Min W. AIP1 functions as Arf6-GAP to negatively regulate TLR4 signaling. J Biol Chem. 2010;285:3750–3757. doi: 10.1074/jbc.M109.069385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Min J, Zaslavsky A, Fedele G, McLaughlin SK, Reczek EE, De Raedt T, Guney I, Strochlic DE, Macconaill LE, Beroukhim R, Bronson RT, Ryeom S, Hahn WC, Loda M, Cichowski K. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat Med. 2010;16:286–294. doi: 10.1038/nm.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gretarsdottir S, Baas AF, Thorleifsson G, Holm H, den Heijer M, de Vries JP, Kranendonk SE, Zeebregts CJ, van Sterkenburg SM, Geelkerken RH, van Rij AM, Williams MJ, Boll AP, Kostic JP, Jonasdottir A, Jonasdottir A, Walters GB, Masson G, Sulem P, Saemundsdottir J, Mouy M, Magnusson KP, Tromp G, Elmore JR, Sakalihasan N, Limet R, Defraigne JO, Ferrell RE, Ronkainen A, Ruigrok YM, Wijmenga C, Grobbee DE, Shah SH, Granger CB, Quyyumi AA, Vaccarino V, Patel RS, Zafari AM, Levey AI, Austin H, Girelli D, Pignatti PF, Olivieri O, Martinelli N, Malerba G, Trabetti E, Becker LC, Becker DM, Reilly MP, Rader DJ, Mueller T, Dieplinger B, Haltmayer M, Urbonavicius S, Lindblad B, Gottsater A, Gaetani E, Pola R, Wells P, Rodger M, Forgie M, Langlois N, Corral J, Vicente V, Fontcuberta J, Espana F, Grarup N, Jorgensen T, Witte DR, Hansen T, Pedersen O, Aben KK, de Graaf J, Holewijn S, Folkersen L, Franco-Cereceda A, Eriksson P, Collier DA, Stefansson H, Steinthorsdottir V, Rafnar T, Valdimarsson EM, Magnadottir HB, Sveinbjornsdottir S, Olafsson I, Magnusson MK, Palmason R, Haraldsdottir V, Andersen K, Onundarson PT, Thorgeirsson G, Kiemeney LA, Powell JT, Carey DJ, Kuivaniemi H, Lindholt JS, Jones GT, Kong A, Blankensteijn JD, Matthiasson SE, Thorsteinsdottir U, Stefansson K. Genome-wide association study identifies a sequence variant within the DAB2IP gene conferring susceptibility to abdominal aortic aneurysm. Nat Genet. 2010;42:692–697. doi: 10.1038/ng.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scott DM, Ehrmann IE, Ellis PS, Chandler PR, Simpson E. Why do some females reject males? The molecular basis for male-specific graft rejection. J Mol Med. 1997;75:103–114. doi: 10.1007/s001090050095. [DOI] [PubMed] [Google Scholar]
- 32.Koulack J, McAlister VC, MacAulay MA, Bitter-Suermann H, MacDonald AS, Lee TD. Importance of minor histocompatibility antigens in the development of allograft arteriosclerosis. Clin Immunol Immunopathol. 1996;80:273–277. doi: 10.1006/clin.1996.0123. [DOI] [PubMed] [Google Scholar]
- 33.Raisanen-Sokolowski A, Glysing-Jensen T, Koglin J, Russell ME. Reduced transplant arteriosclerosis in murine cardiac allografts placed in interferon-gamma knockout recipients. Am J Pathol. 1998;152:359–365. [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Yue T, Ohlstein E, Sung C, Feuerstein G. Interferon-inducible Protein-10 Involves Vascular Smooth Muscle Cell Migration, Proliferation, and Inflammatory Response. J Biol Chem. 1996;271:24286–24293. doi: 10.1074/jbc.271.39.24286. [DOI] [PubMed] [Google Scholar]
- 35.Zhao D, Hu Y, Miller G, Luster A, Mitchell R, Libby P. Differential Expression of the IFN-γ -Inducible CXCR3-Binding Chemokines, IFN-Inducible Protein 10, Monokine Induced by IFN, and IFN-Inducible T Cell α Chemoattractant in Human Cardiac Allografts: Association with Cardiac Allograft Vasculopathy and Acute Rejection. J Immunol. 2002;169:1556–1560. doi: 10.4049/jimmunol.169.3.1556. [DOI] [PubMed] [Google Scholar]
- 36.Dzau VJ, Braun-Dullaeus RC, Sedding DG. Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat Med. 2002;8:1249–1256. doi: 10.1038/nm1102-1249. [DOI] [PubMed] [Google Scholar]
- 37.Newby AC, George SJ. Proliferation, migration, matrix turnover, and death of smooth muscle cells in native coronary and vein graft atherosclerosis. Curr Opin Cardiol. 1996;11:574–582. doi: 10.1097/00001573-199611000-00004. [DOI] [PubMed] [Google Scholar]
- 38.Torella D, Curcio A, Gasparri C, Galuppo V, De Serio D, Surace F, Cavaliere A, Leone A, Coppola C, Ellison G, Indolfi C. Fludarabine prevents smooth muscle proliferation in vitro and neointimal hyperplasia in vivo through specific inhibition of STAT-1 activation. Am J Physiol - Heart and Circulatory Physiology. 2007;292:H2933–2943. doi: 10.1152/ajpheart.00887.2006. [DOI] [PubMed] [Google Scholar]
- 39.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13:1235–1242. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 40.Nguyen H, Ramana CV, Bayes J, Stark GR. Roles of phosphatidylinositol 3-kinase in interferon-gamma-dependent phosphorylation of STAT1 on serine 727 and activation of gene expression. J Biol Chem. 2001;276:33361–33368. doi: 10.1074/jbc.M105070200. [DOI] [PubMed] [Google Scholar]
- 41.Krebs DL, Hilton DJ. SOCS: physiological suppressors of cytokine signaling. J Cell Sci. 2000;113:2813–2819. doi: 10.1242/jcs.113.16.2813. [DOI] [PubMed] [Google Scholar]
- 42.Shuai K, Liu B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol. 2005;5:593–605. doi: 10.1038/nri1667. [DOI] [PubMed] [Google Scholar]
- 43.Luo D, Luo Y, He Y, Zhang H, Zhang R, Li X, Dobrucki WL, Sinusas AJ, Sessa WC, Min W. Differential functions of tumor necrosis factor receptor 1 and 2 signaling in ischemia-mediated arteriogenesis and angiogenesis. Am J Pathol. 2006;169:1886–1898. doi: 10.2353/ajpath.2006.060603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen H, Toyooka S, Gazdar AF, Hsieh JT. Epigenetic regulation of a novel tumor suppressor gene (hDAB2IP) in prostate cancer cell lines. J Biol Chem. 2003;278:3121–3130. doi: 10.1074/jbc.M208230200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.