

Abstract
Vesicular Zn2+ regulates postsynaptic neuronal excitability upon its corelease with glutamate. We previously demonstrated that synaptic Zn2+ acts via a distinct metabotropic zinc-sensing receptor (mZnR) in neurons to trigger Ca2+ responses in the hippocampus. Here, we show that physiological activation of mZnR signaling induces enhanced K+/Cl− cotransporter 2 (KCC2) activity and surface expression. As KCC2 is the major Cl− outward transporter in neurons, Zn2+ also triggers a pronounced hyperpolarizing shift in the GABAA reversal potential. Mossy fiber stimulation-dependent upregulation of KCC2 activity is eliminated in slices from Zn2+ transporter 3-deficient animals, which lack synaptic Zn2+. Importantly, activity-dependent ZnR signaling and subsequent enhancement of KCC2 activity are also absent in slices from mice lacking the G-protein-coupled receptor GPR39, identifying this protein as the functional neuronal mZnR. Our work elucidates a fundamentally important role for synaptically released Zn2+ acting as a neurotransmitter signal via activation of a mZnR to increase Cl− transport, thereby enhancing inhibitory tone in postsynaptic cells.
Introduction
Neurons contain two major pools of Zn2+. One pool is composed of Zn2+ bound to intracellular proteins, such as enzymes, transcription factors and metal-binding proteins. This bound Zn2+ can be liberated into the cytoplasm during oxidative or nitrosative neuronal injury, leading to cell death (Aizenman et al., 2000; Zhang et al., 2004, 2006; Redman et al., 2009). The second pool is synaptic Zn2+, packaged into a subpopulation of glutamate-containing synaptic vesicles by the Zn2+ transporter 3 (ZnT3), and released into the synaptic cleft during neuronal activity in a Ca2+-dependent manner (Qian and Noebels, 2005, 2006; Paoletti et al., 2009). Vesicular Zn2+ regulates neuronal excitability and can strongly influence seizure activity (Vogt et al., 2000; Smart et al., 2004; Sensi et al., 2009). Indeed, removal of synaptic Zn2+ by dietary means, chemical chelation, or via genetic deletion of ZnT3, leads to enhanced susceptibility to epileptic seizures (Cole et al., 2000; Blasco-Ibáñez et al., 2004; Takeda et al., 2005), a phenomenon that may be reflective of some forms of human epilepsy (Goldberg and Sheehy, 1982; Ganesh and Janakiraman, 2008). By comparison, elevation of Zn2+ levels either by dietary means or by direct infusion into the brain can delay seizures in kindled animals (Fukahori and Itoh, 1990; Elsas et al., 2009).
Synaptic Zn2+ is known to allosterically modulate glutamate, GABA and glycine ionotropic receptors (Smart et al., 2004; Madry et al., 2008; Paoletti et al., 2009), and has heretofore been classified as a neuromodulator (Laube et al., 1995; Vogt et al., 2000; Hosie et al., 2003; Smart et al., 2004; Paoletti et al., 2009). However, the recent identification of a metabotropic Zn2+-sensing receptor (mZnR) in hippocampal neurons suggests that synaptically released Zn2+ can profoundly alter postsynaptic cell function via a distinct Gq-linked pathway that triggers the release of Ca2+ from intracellular stores (Besser et al., 2009). GPR39, a previously considered orphan Gq-coupled receptor, was shown to mediate Zn2+-dependent signaling in a recombinant expression system (Yasuda et al., 2007), and we have recently suggested that this receptor is linked to mZnR activity in the CA3 region of the hippocampus based on its expression pattern (Besser et al., 2009). Here, using pharmacological and genetic tools, we firmly establish GPR39 as the molecular determinant behind neuronal mZnR function. Importantly, we report that the physiological activation of mZnR/GPR39 in CA3 neurons leads to an increase in surface expression and activity of the K+/Cl− cotransporter 2 (KCC2), the principal Cl− outward transporter critical for the maintenance of hyperpolarizing GABAA reversal potentials (Rivera et al., 1999; Woo et al., 2002).
Materials and Methods
Slice preparation and stimulation.
Experimental procedures were performed in accordance with a protocol approved by the committee for the Ethical Care and Use of Animal in Experiments at the Faculty of Health Sciences at Ben-Gurion University. Hippocampal slices were obtained from postnatal day 12 (P12)–P15 mice of either sex, following protocols aimed to preserve mossy fiber structure (Amaral and Witter, 1989; Bischofberger et al., 2006; Amaral et al., 2007). In one series of control experiments we used slices obtained from P6 mice (Fig. 1A). Transverse slices (400 μm) were kept in artificial CSF (ACSF) at room temperature. To test the effect of extracellular Zn2+, slices were transferred to NaH2PO4-free ACSF to prevent precipitation of the metal (Besser et al., 2009). Slices were then either pretreated with physiologically relevant concentrations of Zn2+ (200 μm) for 2 min (Vogt et al., 2000; Qian and Noebels, 2005), or mossy fibers were subjected to electrical stimulation (a train of 10 pulses at 66 Hz; Master-8 stimulator unit, A.M.P.I.) (Qian and Noebels, 2005; Frederickson et al., 2006; Besser et al., 2009). Both stimuli were previously shown to induce similar ZnR-dependent Ca2+ responses in the postsynaptic cells in CA3 (Besser et al., 2009). Following exogenous Zn2+ treatment, the solution was replaced with Zn2+-free ACSF solution, and slices were quickly placed in the recording chamber. Imaging of KCC2 activity always started within 2 min following the end of the Zn2+-treatment. The mossy fiber stimulated slices were kept in the recording chamber and perfused with Zn2+-free ACSF solution for ∼2 min. Since KCC2 may undergo rapid recycling, this timing was carefully maintained for all paradigms used. Inhibitors of the Gq-coupled receptor, PLC and MEK1/2 (YM-254890, U73122, U0126), a signaling pathway activated by the ZnR, were applied 30 min before application of Zn2+.
Figure 1.

Extracellular Zn2+ upregulates NH4+-dependent influx mediated by KCC2 activity. A, To assess KCC2 transport activity, we monitored BCECF fluorescence in acute mouse hippocampal slices using the NH4+ transport paradigm. Application of NH4Cl (5 mm) induces initial alkalinization (due to passive diffusion of NH3 through the cell membrane). Subsequent NH4+ influx via KCC2, acting in reverse mode, induces acidification of the cells, and the initial rate of acidification represents KCC2 activity (see Materials and Methods). This paradigm was applied to slices obtained from 6- or 14-d-old mice (n = 4 slices). Inset shows the signal in the region marked by the box, where the linear regression curve was fitted to represent KCC2 activity. Note that for clarity of the inset only every second measurement is shown. Averaged rates of NH4+ influx were 0.0006 ± 0.0004 (R/R0/s) in P6 slices and −0.0004 ± 0.0003 (R/R0/s) in P14 slices (*p < 0.05). B, Intracellular Zn2+ concentration in slices loaded with FluoZin-3, following (at indicated times) electrical stimulation (Besser et al., 2009), addition of Zn2+ (200 μm) and depolarization using ACSF containing 50 mm KCl (replacing NaCl) in the presence of Zn2+ (200 μm). Only following depolarization, in the presence of Zn2+, a rise in fluorescence was observed (n = 3 slices). C, Averaged traces from BCECF-loaded slices (n = 8 slices), imaged with or without application of NH4Cl (5 mm) in the presence or absence of DIOA (100 μm). Slices were pretreated with extracellular Zn2+ (200 μm, 2 min) or maintained in ACSF (vehicle) and imaged within 2 min. In the box, the signal from the region marked in the graph is depicted, and the linear regression curve that was fitted to represent KCC2 activity is shown. Note that for clarity only every second measurement is shown. Inset shows bright-field (left) and BCECF fluorescence (right) images of the CA3 region that was monitored; SO stratum oriens, SL stratum lucidum. D, Averaged rate of acidification due to steady-state NH4+ influx ±SEM (n = 8 slices, *p < 0.05 compared with vehicle control). The rate of NH4+ influx is enhanced following pretreatment with extracellular Zn2+, and is blocked by DIOA (100 μm) or furosemide (100 μm) but not by the Na+/K+/Cl− cotransporter inhibitor bumetanide (1 μm). E, Shown are representative traces from slices exposed to NH4Cl that were calibrated to pHi, using the calibration curve (see Materials and Methods). Each trace is an average of 7–12 ROIs within the somatic CA3 region from a single slice. Slices were treated with Zn2+ (200 μm, 2 min) or maintained in ACSF (vehicle) as marked.
Fluorescence imaging.
Slices were loaded with either Fura-2 AM (25 μm, TefLabs) or BCECF AM [2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein AM] (25 μm, TefLabs) for 20 min or with MQAE [N-(ethoxycarbonylmethyl)-6-methoxy-quinolinium bromide] (5 mm, TefLabs) or FluoZin-3 AM (25 μm, Invitrogen) for 30 min, all AM dyes were applied in the presence of 0.02% pluronic acid. Slices were then washed for at least 20 min in ACSF (Beierlein et al., 2002; Besser et al., 2009). Fluorescent imaging measurements, focusing on CA3, were acquired every 3 s (Imaging Workbench 4, INDEC BioSystems; and polychrome monochromator, TILL Photonics) using a 10× objective (Olympus BX51) with 4 × 4 binning of the image (SensiCam, PCO). Under these conditions, minimal bleaching was observed and the fluorescence signal from a large number of CA3 neurons could be acquired (Besser et al., 2009). In each slice 9–15 traces from individual regions of interest (ROIs) were acquired in the pyramidal cell layer [stratum pyramidale (SP)], established by a bright-field image acquired before each measurement. All ROIs were randomly selected within the somatic SP region as observed in the bright-field image (Fig. 1C). All selected ROIs in a slice were included in subsequent analyses.
Synaptic Zn2+ release in the CA3 was detected using the extracellular Zn2+-sensitive fluorescent dye Newport Green DCF (2 μm; Invitrogen) (Frederickson et al., 2006), F being the intensity of the fluorescent signal obtained using 480 nm excitation and a 535 nm bandpass emission filter (Chroma Technology). Intracellular Zn2+ measurements were performed using FluoZin-3-loaded slices; F is the intensity of the fluorescent signal obtained using the same filters used for Newport Green. Intracellular Ca2+ signals were monitored in slices loaded with Fura-2. The loading procedure used in these experiments is similar to that used previously by our group, shown to effectively label neuronal cells within the CA3 region (Besser et al., 2009). Fluorescence signals are represented as a ratio, R, of the signal obtained using excitation of 340 nm/380 nm wavelengths and a 510 nm emission bandpass filter (Chroma Technology). Baseline fluorescent signals were obtained by averaging over the initial 10 s period (F0 for Newport Green and R0 for Fura-2) and subsequent fluorescence levels were normalized to this value (Hershfinkel et al., 2001), thus minimizing variations related to dye loading (Reynolds, 2001). Averaged traces of ROIs from at least three independent experiments are presented as R/R0 or F/F0 ± SEM, the number of slices averaged is marked n in the figure legend.
For measurements of KCC2 activity two independent paradigms were used. In each of these paradigms slices were loaded with a specific fluorescent indicator, then treated with extracellular Zn2+, or maintained in ACSF in control experiments, and imaged, within 2 min wash in ACSF, to determine rates of KCC2 activity. In one paradigm, slices were loaded with the H+ indicator BCECF. R is the ratio of the emitted signal obtained using excitation wavelength of 440 nm/480 nm and a bandpass emission filter at 535 nm (Chroma Technology) and R0 is the initial baseline as described above. Slices were perfused with ACSF to obtain a baseline and then NH4Cl (5 mm) was added, replacing KCl in the ACSF. Alkalinization of the cells following passive entry of NH3 into the cells was initially observed, as reported earlier (Titz et al., 2006); the time required to reach a steady concentration of NH3 within the cells varied, likely due to different permeation times within the slice. Extracellular NH4+ serves as a surrogate to K+ and reversal of KCC2 activity induces its transport into the cells leading to a pronounced decrease of cellular pH following the alkalinization phase (Titz et al., 2006). This acidification in the presence of NH4Cl, results from NH4+ influx at steady state and represents KCC2 activity. Rates of steady-state NH4+ influx were determined by monitoring the initial 75 s period of the decrease in intracellular pH (Shin et al., 2004; Hershfinkel et al., 2009), thus minimizing the exposure to NH4Cl (Bonnet and Wiemann, 1999) and the potential effects of pH regulatory mechanisms. BCECF fluorescence was calibrated using 10 μm nigericin in oxygenated (100% O2) solution containing the following (in mm): 118 KCl, 3 NaCl, 1 MgCl2, 1.5 CaCl2, 25 HEPES, and 10 glucose (Ritucci et al., 1996; Trapp et al., 1996; Ruusuvuori et al., 2004). Values of pHi were plotted as a function of the fluorescence signal and fitted to produce a calibration curve, which was used to calculate pHi in some slices exposed to the NH4Cl paradigm (Fig. 1F). In a second paradigm, slices were loaded with the Cl− indicator MQAE. With this technique, F is the intensity of the fluorescent signal obtained at 360 nm excitation and 510 nm emission bandpass filter (Chroma Technology) and F0 is the initial baseline as describe above. Following baseline acquisition, ACSF containing a final concentration of 10 mm KCl was applied, leading to reverse, Cl− influx through KCC2 and quenching of MQAE fluorescence (Galeffi et al., 2004; Pond et al., 2004). Care was taken to apply KCl after thorough washout of Zn2+, thereby minimizing permeation of this metal ion via voltage-activated ion channels (Sensi et al., 2000). A 30 s period of perfusion with the KCl solution was allowed for KCl equilibration within the slice, and rates of Cl− influx during the subsequent 50 s period were determined. Calibration of MQAE fluorescence using the K+/H+ ionophore nigericin (10 μm) and the Cl−/OH− antiporter tributyltin chloride (10 μm) yielded a Stern–Volmer constant (Verkman, 1990) of 23 ± 1 m−1 (or Kd of ∼44 mm Cl−), in agreement with previous works (Marandi et al., 2002). Inhibitors of KCC2 or NKCC1 [dihydroindenyl-oxy-alkanoic acid (DIOA), bumetanide, furosemide] were added as indicated to the perfusing ACSF solutions. Representative traces from single slices as well as averaged traces of all ROIs are presented as R/R0 or F/F0 ± SEM (BCECF or MQAE, respectively), the number of slices averaged is marked n in the figure legend. Averaged initial rates of fluorescence change (fitted as described above) due to KCC2 activity ± SEM are presented in bar graphs. Statistical significance was determined using t test or ANOVA with post hoc Tukey comparisons where appropriate.
Surface expression and immunoblotting.
Changes in KCC2 membrane expression were monitored as previously described (Rivera et al., 2002; Thomas-Crusells et al., 2003) using hippocampal slices. Acute slices were biotinylated (100 μm sulfo-NHS-Biotin, Pierce) in ACSF (45 min, at room temperature) and then unbound sulfo-NHS-biotin was scavenged using 1 μm lysine in ACSF (2 times). Some slices were also loaded with Fura-2 and the ZnR-dependent Ca2+ responses were monitored (see Fig. 5C). For surface expression analysis, biotinylated slices were incubated with Zn2+ (200 μm, 2 min) at room temperature in the presence or absence of the Gαq inhibitor YM-254890. Slices maintained in Zn2+-free ACSF for the same time interval were used as control, detecting the basal, time-dependent removal of biotinylated KCC2 from the surface membrane (Rivera et al., 2004; Lee et al., 2007; Zhao et al., 2008). Slices were washed in ACSF and ∼6 min following Zn2+ or control treatment, slices were lysed (1% Triton X-100, 0.1% SDS, 1 mm EDTA, 50 mm, NaCl, 20 mm Tris-HCl, pH 7.5, and protease inhibitors, Sigma) and Neutravidin Gel (Pierce) was added (overnight at 4°C), as previously described (Rivera et al., 2004). Samples were resolved on 7.5% SDS-PAGE followed by immunoblot analysis of KCC2 (C2366, Sigma) and transferrin receptor (13–6800, Invitrogen), a nonrelated surface protein which was used for normalization. Immunoblots were digitally imaged using ChemImager5 (Alpha-Innotech, Labtrade), and quantified using EZQuant-Gel software.
Figure 5.

Upregulation of KCC2 activity is mediated by mZnR signaling pathway and enhanced KCC2 surface expression. A, KCC2 activity was monitored in slices from WT mice loaded with BCECF, following stimulation of the mossy fibers in control slices (vehicle) or in slices pretreated with a Gαq inhibitor (YM-254890, 1 μm), a PLC inhibitor (U73122, 1 μm) or an ERK1/2 inhibitor (U0126, 1 μm); n = 7 slices. B, Averaged rate of NH4+ influx as monitored in A. Dashed line indicates the rate of transport in nonstimulated control slices taken from Figure 4A (n = 7 slices, **p < 0.01 compared with vehicle control without stimulation). C, Top, Slices were biotinylated, control slices were maintained in ACSF, and loaded with the intracellular Ca2+-sensitive dye, Fura-2. Then Zn2+ (200 μm) was applied and the Ca2+ response is shown in control and biotinylated slices. Bottom, Surface expression level of KCC2, monitored using surface biotinylation followed by immunoblotting of KCC2 or transferrin receptor (TrfR), which is a nonrelated membrane transporter used as control. Surface expression in vehicle-control slices or in slices treated with Zn2+ in the presence or absence of YM-254890 (1 μm). Densitometry analysis of KCC2 surface expression is shown to the right, normalized to TrfR expression (n = 4 slices, *p < 0.05 compared with vehicle control).
Electrophysiological recordings.
Whole-cell or cell-attached recordings from CA3 neurons were either made blindly (Hamill et al., 1981; Blanton et al., 1989) or under infrared-differential interference contrast microscopic control (Stuart et al., 1993). For blind recording, the slices were maintained in a small (300 μl) interface-type recording chamber (Haas et al., 1979); for visually controlled recording, slices were held submerged in a chamber on the fixed stage of an Axioskop FS microscope (Carl Zeiss). All recordings were made at 32 ± 1°C. Patch pipettes were manufactured from thick-walled borosilicate glass capillaries (1.5 mm o.d., Hilgenberg, Germany) and had resistances of 1.5–3.5 MΩ. For cell-attached recordings, pipettes containing 5 μm GABA in ACSF were coated to within ∼100 μm of the tip with Sylgard (Dow Corning). The pipette solution for whole-cell current-clamp experiments contained the following (in mm): 130 K-gluconate, 6 KCl, 2 MgCl2, 10 HEPES (potassium salt), pH 7.25. Leak subtraction and channel openings detection was made using pCLAMP 9.0 software (Molecular Devices).
Single GABAA channel openings were obtained in cell-attached patches using an Axopatch 200B amplifier (Molecular Devices), and whole-cell current-clamp voltage recordings from the soma were obtained using an Axoclamp-2B amplifier in bridge mode. Command voltage protocols were generated and single-channel data were acquired on-line with a Digidata 1320A A/D interface. Data were low-pass filtered at 2–5 kHz (−3 dB, 4-pole Bessel filter) and digitized at 10–20 kHz. Capacitive and leak currents were reduced before data acquisition using the built-in circuits of the amplifier. For whole-cell current-clamp recordings, data were low-pass filtered at 10 kHz (−3 dB, single-pole Bessel filter) and digitized at 50 kHz. Data were fitted using Origin 6.0 (OriginLab, Northampton, MA). Values are given as mean ± SD. For statistical analysis, a paired Student's t test was performed.
Genotyping of mice.
PCR was used to screen GPR39 genotypes from mouse biopsy samples (Moechars et al., 2006). Primers 5′-ACCCTCATCTTGGTGTACCT-3′ and 5′-ATGTAGCG CTCAAAGCTGAG-3′ amplified a 311 bp band from the wild-type allele, whereas primers 5′-GGAAvCTCTCACTCGACCTGGG-3′ and 5′-GCAGCGCAT CGCCTTCTATC-3′ amplified a 262 bp band from the knock-out allele.
Cell culture and GPR39 silencing.
Human neuroblastoma SH-SY5Y cells were cultured in DMEM as previously described (Kan et al., 2007). For gene silencing experiments, cells were cotransfected with silencing constructs, 3 μg of siGPR39 or siT1R3 using LipofectAMINE 2000 as directed by the manufacturer (Invitrogen), and imaged 48 h following transfection. The target sequence of the human GPR39 for siRNA was CCATGGAGTTCTACAGCATtt and that of human T1R3 was CUUAGGAUGAAGGGGGACUtt. ZnR activity, using Fura-2 (Besser et al., 2009), or KCC2 activity, using the BCECF and MQAE paradigms, were measured in Ringer's solution, using the same procedures described above.
Results
Extracellular Zn2+ upregulates KCC2 activity
We explored whether mZnR affected the activity of KCC2, a transporter strongly regulated by alterations in [Ca2+]i (Fiumelli et al., 2005). To monitor KCC2 activity we measured influx rates of NH4+, a surrogate ion for K+, using the pH-sensitive dye BCECF (Hershfinkel et al., 2009). In agreement with the previously described developmental expression of KCC2 (Rivera et al., 1999), NH4+ transport, measured as the rate of BCECF fluorescence quenching, was observed in slices from P14 mice, but not in slices from postnatal day 6 mice (Fig. 1A). To study the effect of mZnR, slices were treated with 200 μm Zn2+ for 2 min, a paradigm that activates mZnR signaling (Besser et al., 2009) without increasing [Zn2+]i (Fig. 1B). We then measured NH4+ transport rate, mediated by KCC2, in P12–P15 slices following ZnR activation or in controls that were maintained in nominally Zn2+-free ACSF (vehicle). We observed that extracellular Zn2+ treatment induced a pronounced increase the rate of NH4+ transport in CA3 neurons compared with vehicle control (Fig. 1C). Importantly, NH4+ transport following Zn2+ application was inhibited by the KCC2 inhibitors DIOA (100 μm) and furosemide (100 μm), but not by the Na+/K+/Cl− cotransporter inhibitor bumetanide (1 μm; Fig. 1D) suggesting that this transport is predominantly mediated by KCC2 activity. In a small subset of slices, we calibrated the decrease in BCECF fluorescence to changes intracellular pH using nigericin (Fig. 1E), as previously described (Trapp et al., 1996; Ruusuvuori et al., 2004). Following calibration, the rate of pH change in the Zn2+-treated slices was 0.002 ± 0.0003 pHi/s, while in control slices rates were only 0.0006 ± 0.0002 pHi/s (n = 3 for each group). Thus, in these experiments, the effects of Zn2+ on KCC2 were essentially identical to those described above.
We next studied the effect of mZnR activation on KCC2 by directly monitoring the rate of [Cl−] transport (Hershfinkel et al., 2009), using the fluorescent dye MQAE following application of 10 mm KCl, which reverses KCC2 transport and results in Cl− influx. Similar to what we observed using the BCECF-based method, extracellular Zn2+ application (200 μm, 2 min) significantly enhanced the Cl− influx rates in CA3 neurons compared with the influx rate monitored in controls (Fig. 2A–C). The KCC2 inhibitor DIOA (100 μm) completely blocked Cl− influx in the Zn2+-treated slices, suggesting that both the basal and Zn2+-enhanced Cl− transport was mediated by KCC2 (Fig. 2B,C). These results, combined with those described earlier, strongly suggest that extracellular Zn2+, under conditions associated with ZnR activation (Hershfinkel et al., 2001; Besser et al., 2009), can produce pronounced enhancement of KCC2 activity. It is noteworthy that in a recent study we demonstrated that a rise in intracellular Zn2+ can strongly attenuate KCC2-mediated activity, both in a recombinant expression system and in dissociated cortical neurons (Hershfinkel et al., 2009). We therefore confirmed that this phenomenon could also be discerned in brain slices. To raise intracellular Zn2+ in hippocampal slices (P12–P15), before imaging KCC2 activity, we applied Zn2+ (100 μm) together with the Zn2+ ionophore pyrithione (ZnPyr; 5 μm). Slices were washed in Zn2+-free ACSF and then imaged for KCC2 activity using the MQAE paradigm as in Figure 2, A and B. In contrast to the actions of extracellular Zn2+, and in agreement with our prior work (Hershfinkel et al., 2009), the ZnPyr treatment induced a significant attenuation of the rate of Cl− transport in CA3 neurons (Fig. 2D).
Figure 2.

Extracellular Zn2+ upregulates Cl−-dependent influx mediated by KCC2 activity. A, Representative traces from slices loaded with the Cl−-sensitive dye MQAE (Hershfinkel et al., 2009), each trace is an average of 7–12 ROIs within the somatic CA3 region of one slice. Slices were treated with Zn2+ (200 μm, 2 min) or maintained in ACSF (vehicle). Within 2 min of the Zn2+ treatment slices were imaged and 10 mm KCl was added as indicated. B, Averaged traces from slices treated with or without 10 mm KCl. Shown are traces from control slices (vehicle, n = 7 slices), slices pretreated with extracellular Zn2+ (200 μm, 2 min, n = 7 slices), slices treated with Zn2+ in the presence of the KCC2 inhibitor DIOA (100 μm, n = 10 slices) or control slices, without KCl (n = 9 slices). Inset shows the CA3 region that was imaged (left, bright-field; right, fluorescent images); SO stratum oriens, SL stratum lucidum. In the box, the signal from the region marked in the graph is depicted, and the linear regression curve that was fitted to represent KCC2 activity is shown. Note that for clarity every second measurement is shown. C, Averaged rates ± SEM (n = 7 slices for vehicle or Zn2+-treated, and 10 slices for Zn2+ + DIOA) of Cl− influx as monitored in B (*p < 0.05, **p < 0.01 compared with vehicle control). D, The effect of intracellular Zn2+ rise on KCC2 activity was determined in MQAE-loaded slices, as in A. Shown are averaged traces from control slices (vehicle) or slices pretreated with Zn2+ (100 μm) in the presence of its ionophore pyrithione (5 μm), marked as ZnPyr (n = 5 slices). Averaged rates of Cl− influx were 0.033 ± 0.004 (−%F/F0/s) in vehicle control slices and 0.017 ± 0.002 (−%F/F0/s) in the ZnPyR-treated slices (*p < 0.05).
Extracellular Zn2+ induces alterations in EGABA
By modulating the Cl− gradient, KCC2 strongly influences the reversal potential of GABAA receptor-mediated ionic currents (EGABA) (Rivera et al., 2004; Lee et al., 2005; Blaesse et al., 2009). We tested whether the increased KCC2 activity induced by extracellular Zn2+ altered EGABA in cell-attached patch recordings from the somata of CA3 neurons (Chudotvorova et al., 2005; Lee et al., 2005; Tyzio et al., 2006). With 5 μm GABA in the pipette solution, single or multiple GABAA channel openings were observed in all patches tested (n = 4), while no such openings were seen in the absence of GABA (n = 4), or with GABA in the presence of picrotoxin (100 μm, n = 3). EGABA values were initially extrapolated by monitoring channel opening while applying slow (40 mV/s) depolarizing voltage ramps (Fig. 3A). In control conditions, currents through the GABAA channels reversed very near the resting membrane potential Vr (EGABA= −2.3 ± 0.5 mV relative to Vr), in agreement with previous observations (Tyzio et al., 2006). However, following treatment with 200 μm Zn2+ (2 min) EGABA in the same patches shifted to more negative values (−17 ± 2 mV relative to Vr, n = 4) for the duration of the recordings (≥5 min following Zn2+ application). The negative Zn2+-induced shift of EGABA was also calculated by measuring individual channel amplitudes in each cell-attached patch before and after Zn2+ exposure (Fig. 3B). The same results were obtained when EGABA values were averaged from binned amplitudes across all patches (Fig. 3C, n = 4). In all, Zn2+ exposure induced a shift in EGABA of ∼−15 mV without changing the channel slope conductance, as determined from linear fits to the I–V curves of the single open channels. Importantly, Zn2+ did not affect the resting membrane potential measured under whole-cell recording conditions in a separate group of neurons (Vr = −79.0 ± 5.3 mV, control; −78.6 ± 5.0 mV, Zn2+; n = 4). Using the Stern-Volmer equation (see Materials and Methods), we estimated that Zn2+ exposure led to a 10 ± 1 mm decrease in [Cl−]i. Alterations in the Cl− gradient of this magnitude would be expected to induce ∼a −20 mV change in EGABA, in very close agreement to our experimental findings.
Figure 3.
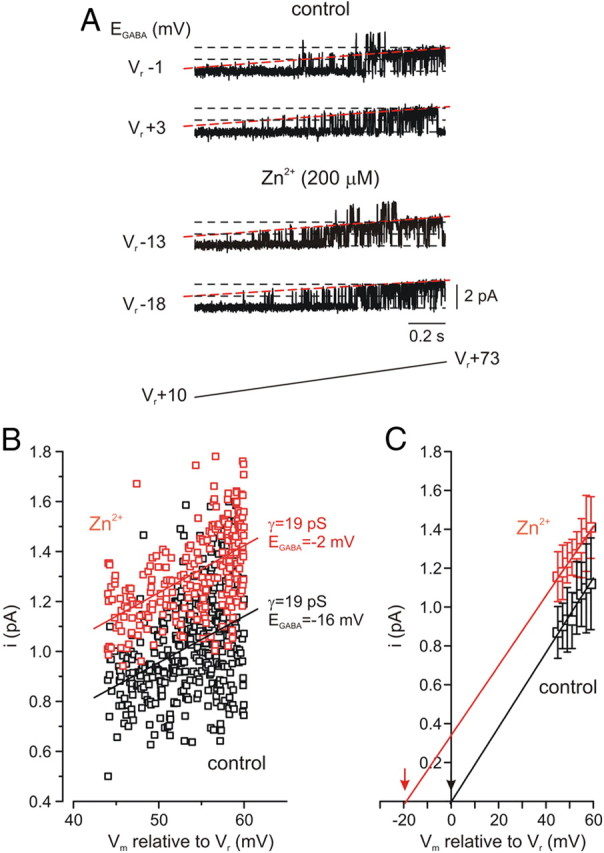
Extracellular Zn2+ induces a hyperpolarizing shift in GABAA reversal potential. A, In cell-attached recordings with 5 μm GABA in a patch pipette, GABAA channel reversal potential was measured by delivering slow depolarizing voltage ramps from 10 to 73 mV relative to the membrane resting potential (Vr +10 to Vr +73 mV) every 15 s (n = 4 cells). Shown are two consecutive ramp current sweeps from a patch which contained GABAA channels, under control conditions (top) and the same patch 5 min after addition of Zn2+ (200 μm, 2 min) to the bath (bottom), with the respective extrapolated EGABA values for each ramp to the left. Sweeps are leak-subtracted and digitized at 20 kHz through a low-pass filter of 2 kHz (−3 dB). Dashed lines (red) are drawn through the closed and single-channel open state. B, Effect of Zn2+ application on I–V relationships of the current through a single GABAA channel-containing patch. Amplitudes of channel opening plotted against voltage in control (vehicle treated, black) and following Zn2+ application (red), from a representative cell. The straight lines are linear fits of the data. Notice that the channel slope conductance remains unchanged (19 pS), the extrapolated EGABA, however, shifted from the membrane resting potential from −2 to −16 mV following Zn2+ application. C, Mean unitary current amplitudes binned with a step of 2 mV and averaged over all patches. Note that the x-axis intercepts of the two fitted lines (indicated by arrows) give the reversal potential values of Vr −2.3 ± 0.5 mV in control and Vr −17 ± 2 mV following Zn2+ application.
Synaptically released Zn2+ upregulates KCC2 activity
We then asked whether the synaptically released Zn2+ could also regulate KCC2 activity. Electrical stimulation of mossy fibers (a train of 10 pulses at 66 Hz), previously shown to trigger Zn2+ release (Qian and Noebels, 2005) and mZnR activation (Besser et al., 2009), significantly enhanced NH4+ influx rates in BCECF-loaded hippocampal slices compared with the influx rates in nonstimulated, control, slices (Fig. 4A,B). While the mossy fiber stimulation triggers local release of synaptic-Zn2+ we observe a significant, albeit probably underestimated, metabotropic Ca2+ response in the postsynaptic region where the ROIs are selected (Besser et al., 2009) and in accordance, a significant change in KCC2 activity is monitored. In fact, mossy fiber stimulation enhanced KCC2 transport in CA3 neurons to a degree similar to that obtained following exogenous Zn2+ exposure (Fig. 1). To determine the role of the synaptic Zn2+ we studied KCC2 activity in slices from ZnT3 KO mice. These mice lack synaptic Zn2+, but otherwise exhibit little alterations in glutamatergic synaptic transmission in this hippocampal region (Lopantsev et al., 2003). Control slices from ZnT3 KO and wild-type mice showed similar basal KCC2 activity rates (Fig. 4A,B), suggesting that the lack of ZnT3 does not affect intracellular Zn2+ levels, which could affect KCC2 activity (Hershfinkel et al., 2009). In slices obtained from ZnT3 KO mice, mossy fiber stimulation had no effect on NH4+ influx rates. Our previous work suggests that ZnR activity can be triggered in slices from ZnT3 KO mice with exogenous Zn2+ (Besser et al., 2009). We therefore asked whether KCC2 activity could be upregulated in slices from ZnT3 KO mice upon addition of Zn2+. Slices from ZnT3 KO mice were treated with Zn2+ (200 μm, 2 min) and KCC2 activity was measured using BCECF. Application of Zn2+ did induce an increase in NH4+ transport rate in this preparation (Fig. 4A,B), indicating that the signaling cascade linking ZnR activation to increased KCC2 activity remains intact in the absence of vesicular Zn2+. Finally, additional experiments were performed using MQAE. Stimulation of slices obtained from ZnT3 KO animals induced a lower Cl− influx rate compared with that of stimulated or Zn2+-treated slices from WT mice (Fig. 4C,D). Together, these results demonstrate that KCC2 activity in CA3 neurons is increased by stimulus-dependent release of synaptic Zn2+ from mossy fibers.
Figure 4.

KCC2 activity is upregulated by synaptic Zn2+ released from mossy fibers. A, KCC2 activity measured in BCECF-loaded slices, as in Figure 1. Slices from ZnT3 KO mice lacking synaptic Zn2+ (open symbols) or WT mice (filled symbols) were monitored under control nonstimulated conditions or 2 min following electrical stimulation of the mossy fibers (a train of 10 pulses at 66 Hz) that induces release of synaptic Zn2+ (n = 6), or following application of Zn2+ (200 μm, 2 min, n = 6 slices). B, Averaged rates of NH4+ influx ± SEM as monitored in A (n = 6 slices, **p < 0.01 compared with WT control). C, KCC2-dependent Cl− transport monitored in MQAE-loaded slices, as in Figure 2, using 10 mm KCl to reverse KCC2 activity. Slices from ZnT3 KO mice (open symbols) or WT mice (filled symbols) were monitored following stimulation of the mossy fibers (a train of 10 pulses at 66 Hz) or following application of Zn2+ (200 μm, 2 min) (n = 5 slices). D, Averaged rates ± SEM of Cl− influx as monitored in C (n = 5 slices, **p < 0.01 compared with the stimulated ZnT3 KO). The rate of ion influx, using the NH4+ or Cl− transport, is enhanced following mossy fiber stimulation in slices from WT mice compared with ZnT3 KO mice, indicating that KCC2 activity is upregulated by the release of synaptic Zn2+.
Zn2+ upregulates KCC2 activity via ZnR signaling and increase in KCC2 surface expression
Synaptically released Zn2+ acts via a Gq-protein-coupled receptor mZnR to trigger intracellular Ca2+ release followed by activation of ERK1/2 (Besser et al., 2009). We tested whether activation of this pathway is linked to the upregulation of KCC2 activity following synaptic Zn2+ release. Application of either the Gαq inhibitor YM-254890 (1 μm) or the PLC inhibitor U73122 (1 μm), both of which block mZnR-mediated intracellular Ca2+ rises (Besser et al., 2009), prevented mossy fiber stimulation-dependent increases in KCC2 activity (Fig. 5A,B). This suggests that mZnR-dependent rise in intracellular Ca2+ is essential for upregulation of KCC2 activity. mZnR-dependent Ca2+ rises also trigger intracellular signaling leading to activation of mitogen-activated protein kinase (MAPK) pathways (Hershfinkel et al., 2007). Phosphorylation has been also implicated in regulation of KCC2 activity (Rinehart et al., 2009). Inhibition of ERK1/2 MAPK activation by U0126 (1 μm) completely reversed the Zn2+-dependent upregulation of KCC2 activity (Fig. 5A,B). These results indicate that increased KCC2 activity, triggered by synaptic Zn2+, is mediated by a MAPK pathway previously shown to be activated by a mZnR in neurons (Besser et al., 2009).
Rapid endocytosis followed by degradation, or recycling of KCC2 protein to the plasma membrane is an important regulatory mechanism for the activity of the cotransporter (Rivera et al., 2002, 2004; Wake et al., 2007). We therefore asked whether mZnR activation affects these processes by assaying KCC2 residual surface expression level using a biotinylation assay (see Materials and Methods). Following biotin labeling of surface proteins, mZnR activity was triggered (200 μm Zn2+, 2 min) and slices were maintained in ACSF for ∼6 min until lysed and mixed with Neutravidin. It is important to note here that the Zn2+-dependent Ca2+ responses, monitored using Fura-2, were essentially intact following treatment of slices with the biotinylation reagent (Fig. 5C, inset), indicating that this procedure did not appear to compromise ZnR signaling. Since biotinylated surface KCC2 (BT-KCC2) can undergo rapid endocytosis, even under baseline conditions (Rivera et al., 2004; Zhao et al., 2008), control slices were maintained in ACSF for the same time period as Zn2+-treated slices. Comparison of control and Zn2+-treated slices showed that residual levels of BT-KCC2 were significantly enhanced in Zn2+-treated slices (Fig. 5C). This suggests that Zn2+ reduces the rate of endocytosis and degradation of KCC2 and/or promotes the rapid recycling of this protein to the cell membrane. Either way, there is a net increase in surface expression of KCC2 following Zn2+ stimulation, a process that can account for the observed increases in cotransporter activity observed in this study. Importantly, the effects of Zn2+ on the surface expression of KCC2 were prevented by YM-254890 (1 μm; Fig. 5C), which blocks the ZnR-dependent intracellular Ca2+ rise (Besser et al., 2009). This strongly suggests that Zn2+-dependent overall increase in KCC2 surface expression is induced by the activation of a Gq-coupled mZnR.
GPR39 mediates ZnR signaling in CA3 neurons
Our previous work suggested that the Gq-coupled receptor GPR39 may mediate mZnR activity, and demonstrated that this protein is strongly expressed in CA3 neurons (Besser et al., 2009). Here, we used hippocampal slices from GPR39 KO mice (Moechars et al., 2006) (Fig. 6A) to evaluate whether this receptor is, in fact, the mZnR. First, we determined whether synaptic Zn2+ release was similar in GPR39 KO and WT mice using the nonpermeant form of the Zn2+-sensitive dye Newport Green (Frederickson et al., 2006). Following mossy fiber stimulation (a train of 10 pulses at 66 Hz), changes of Newport Green fluorescence (2 μm, Fig. 6B,C) in the CA3 were nearly identical in slices from WT and GPR39 KO mice, but, as expected, were absent in slices from ZnT3 KO mice (Cole et al., 1999; Carter et al., 2011). As such, the lack of GPR39 does not influence synaptic Zn2+ release.
Figure 6.

GPR39 mediates mZnR-dependent Ca2+ response in CA3 hippocampal neurons. A, Analysis of GPR39 transcripts was determined using PCR. The WT allele gives a 311 bp and the targeted GPR39 KO allele gives a 262 bp PCR product. This identifies GPR39−/− (KO) or GPR39+/+ (WT) mice. B, Synaptic Zn2+ release was determined using the nonpermeant form of Newport Green (Frederickson et al., 2006). Extracellular Zn2+-dependent changes in Newport Green (2 μm) fluorescence were monitored in the CA3 region following stimulation of the mossy fibers in slices from WT mice, GPR39 KO mice or ZnT3 KO mice, lacking synaptic Zn2+. Inset is a schematic model of the experimental setup. C, Averaged stimulation-dependent rise of Newport Green fluorescence, normalized to the initial fluorescence, monitored in slices from WT, GPR39 KO mice (n = 6 slices) or ZnT3 KO mice (n = 4 slices, **p < 0.01 compared with the GPR39 WT). D, Slices from WT or GPR39 KO mice were loaded with Fura-2, an intracellular Ca2+-sensitive dye. Intracellular Ca2+ rise was monitored following stimulation of the mossy fibers (a train of 10 pulses at 66 Hz, at the indicated time) in the presence of ACSF solution (vehicle) or ACSF containing the extracellular Zn2+ chelator CaEDTA (100 μm) (n = 7 slices). E, Averaged change of Ca2+ rises as monitored in D (n = 7 slices, **p < 0.01 compared with stimulated WT in ACSF). Note that the residual Ca2+ response in the slices from GPR39 KO mice is not attenuated further by CaEDTA and is similar to the response observed in the presence of CaEDTA in slices from WT mice.
We then evaluated mossy-fiber stimulation-dependent activation of the metabotropic pathway, leading to release of Ca2+, using slices loaded with Fura-2. While mossy fiber stimulation triggered pronounced intracellular Ca2+ responses in CA3 neurons in slices from WT mice (Besser et al., 2009), these responses were attenuated by ∼50% in GPR39-defficient slices (Fig. 6D,E). The residual response is most likely induced by group I metabotropic glutamate receptor activation (Bianchi et al., 1999; Kapur et al., 2001). Moreover, any residual Ca2+ responses observed in GPR39 KO slices were unaffected by extracellular Zn2+ chelation with CaEDTA (150 μm; Fig. 6D,E). This chelator, however, reduced mossy fiber stimulus-evoked Ca2+ responses in WT slices to levels nearly identical to those observed in the GPR39 KO slices. These results are very similar to those previously observed in ZnT3 KO mice (Besser et al., 2009), and reinforce the notion that synaptically released Zn2+ via activation of a mZnR is responsible for a substantial component of metabotropic Ca2+ responses in CA3 neurons. Importantly, we demonstrate here that GPR39 is critical for Zn2+-dependent Ca2+ responses as no responses were observed in slices obtained from animals lacking this receptor, indicating, for the first time, that endogenous GPR39 mediates mZnR signaling in the hippocampus. In fact, this strongly implies that the mZnR and GPR39 are one and the same molecule.
Finally, we compared the rates of NH4+ transport under control conditions and following either Zn2+ exposure or mossy fiber stimulation in slices from GPR39 KO mice and WT littermates. Electrical stimulation of the mossy fibers (a train of 10 pulses at 66 Hz) or application of exogenous Zn2+ (200 μm, 2 min) resulted in robust increase in KCC2 activity in slices obtained from WT controls (Fig. 7A,B). Strikingly however, neither stimulus could alter KCC2 activity in slices from GPR39 KO mice (Fig. 7A–C). In agreement, a similar stimulatory effect of extracellular Zn2+ on KCC2 activity was observed in the SHSY-5Y neuronal cell line, which exhibits GPR39-mediated mZnR activity (Fig. 7D, inset) (Besser et al., 2009). Finally, siRNA-mediated silencing of GPR39 effectively abolished Zn2+-dependent Ca2+ release (Fig. 7D, inset) and the upregulation of KCC2 activity in the SHSY-5Y cells (Fig. 7D). These data also argues for the identity of GPR39 as the zinc receptor.
Figure 7.

GPR39 mediates Zn2+-dependent upregulation of KCC2 activity. A, KCC2 activity was studied using the BCECF paradigm. NH4+ influx rate was monitored in slices from WT (filled symbols) or GPR39 KO (open symbols) mice loaded with BCECF. BCECF fluorescence changes were monitored in slices following mossy fiber stimulation (a train of 10 pulses at 66 Hz) or in control nonstimulated slices (n = 6 slices). B, Slices from WT (filled symbols) or GPR39 KO (open symbols) mice (as in A) were pretreated with extracellular Zn2+ (200 μm, 2 min) or controls (n = 6 slices) and NH4+ influx rate was monitored. C, Averaged rates of NH4+ influx as monitored in A-B (n = 6 slices, *p < 0.05 compared with WT control). In slices from GPR39 KO mice, application of exogenous Zn2+ or synaptic Zn2+ release by mossy fiber stimulation did not affect KCC2 activity, which was similar to the level of the transporter activity in control slices obtained from WT mice. D, SHSY-5Y cells were transfected with siRNA constructs for silencing GPR39 (Besser et al., 2009) or a nonrelated G-protein-coupled receptor (siT1R3, control) and subjected to the MQAE paradigm to monitor KCC2 activity. Cells were treated with or without Zn2+ (200 μm, 2 min) and KCC2 activity was monitored in the presence or absence of DIOA (n = 5, *p < 0.05 compared with vehicle siT1R3, control). Zn2+-dependent Ca2+ rise was monitored using Fura-2 (see inset) and was attenuated in the siGPR39 transfected cells.
Discussion
GPR39, a previously orphan G-protein-coupled receptor, was first proposed to mediate obestatin signaling and regulate food intake (Zhang et al., 2005). However, the absence of GPR39 expression in the hypothalamus (Jackson et al., 2006) and the identification of Zn2+, rather than obestatin, as its putative endogenous ligand (Yasuda et al., 2007), left this receptor without known physiological roles. The localization of GPR39 to CA3 neurons (Besser et al., 2009), as well as the results presented here using a genetic model (Moechars et al., 2006), indicate that GPR39 is responsible for endogenous mZnR-dependent metabotropic responses in the hippocampus. We propose that a fundamental role for mZnR/GPR39 in CA3 is to initiate signaling affecting the excitability of postsynaptic neurons following activity-dependent release of vesicular Zn2+ from mossy fiber terminals. Interestingly, synaptic Zn2+-dependent activation of ERK1/2 was recently implicated in hippocampal-dependent contextual discrimination (Sindreu et al., 2011). Our previous results suggest that mZnR-dependent signaling induced activation of ERK1/2 in the postsynaptic cells in the hippocampus (Besser et al., 2009), hence the identification of GPR39 as the receptor mediating Zn2+-dependent ERK1/2 activation may, in future work, implicate GPR39 as a novel signaling component for memory formation.
The present study and our pervious results (Hershfinkel et al., 2009) demonstrate that synaptic and intracellular Zn2+ have opposite effects on KCC2 activity. Synaptically released, extracellular Zn2+, acting via a mZnR, enhances KCC2 activity and surface expression, thereby inducing a hyperpolarizing shift in GABAA reversal potential. Activation of ERK1/2 by extracellular Zn2+ is essential for the upregulation of KCC2 activity, suggesting that phosphorylation of KCC2 may be required to enhance its activity. Previous studies have shown that direct phosphorylation of KCC2 regulates its activity (Strange et al., 2000; Rinehart et al., 2009; Kahle et al., 2010) and may also lead to enhanced KCC2 surface expression (Lee et al., 2007, 2010; Wake et al., 2007; Watanabe et al., 2009). In contrast to the effects of extracellular Zn2+, increases in intracellular Zn2+, triggered either by a Zn2+ ionophore or by injurious stimuli such as oxygen-glucose deprivation, inhibit KCC2 activity (Hershfinkel et al., 2009), possibly by directly interacting with the cotransporter. This leads to a depolarizing shift in the GABAA reversal potential, which may contribute to the injurious process (Buzsàki et al., 1989; Dietz et al., 2008; Papp et al., 2008). Seemingly consistent with such a mechanism, Zip-deficient mice, which lack Zn2+ influx mediating transporters, were shown to have reduced sensitivity to kainate-induced neuronal death (Qian et al., 2011). Because cytoplasmic free Zn2+ ions will interact equally well with existing or newly inserted plasma membrane KCC2, the rise in intracellular Zn2+ would be expected to induce an overall attenuation of KCC2 activity, overriding the upregulation of this transporter by extracellular Zn2+, which is, in fact what we observe.
Synaptic Zn2+ accumulation is developmentally regulated, this ion is observed in synaptic vesicles only after the first postnatal week (Frederickson et al., 1981; Nitzan et al., 2002), somewhat resembling developmental KCC2 expression (Lu et al., 1999; Rivera et al., 1999; Lee et al., 2005). While in the mouse hippocampus some vesicular Zn2+ is observed at postnatal day 6, the levels of this metal are substantially increased during the 2–3 postnatal weeks (Nitzan et al., 2002; Liguz-Lecznar et al., 2005). Thus, the results obtained in this study using the mossy fiber stimulation of mice on postnatal days 12–15 may be enhanced in adult mice. Interestingly, under pathophysiological conditions, changes in both synaptic Zn2+ levels (Doering et al., 2007; Carter et al., 2011) and KCC2 expression (Huberfeld et al., 2007; Khirug et al., 2010) are also observed. BDNF, interictal-like activity and stress induce a positive shift in EGABA mediated by reduction of KCC2 surface expression (Rivera et al., 2002, 2004; Wake et al., 2007). Moreover, certain forms of intracellular Ca2+ signaling can also induce a depolarizing shift in EGABA that is dependent on KCC2 activity or surface expression, including coincident presynaptic and postsynaptic activity, repetitive postsynaptic firing (Woodin et al., 2003; Fiumelli et al., 2005), and release of Ca2+ via the ryanodine receptor (Fiumelli et al., 2005). In contrast, activation of a different intracellular Ca2+ signaling pathway, triggered by group I mGluRs, induces a pronounced hyperpolarizing shift in GABAA reversal potential, which is sensitive to KCC2 inhibitors (Banke and Gegelashvili, 2008). As shown here, a neuronal mZnR, which triggers Ca2+ release via the same pathway as group I mGluRs, similarly enhances KCC2 activity and renders the inhibitory drive for GABA more effective. We show that the metabotropic Ca2+ response and its downstream ERK1/2 activation are essential for mZnR-dependent upregulation of KCC2 activity. Furthermore, our results using the Gαq inhibitor indicate that metabotropic Ca2+ responses triggered by mZnR are required for enhancing KCC2 surface expression. While mZnR activation may regulate KCC2 endocytosis, degradation, or its recycling back to the membrane our, data clearly indicate that this results in upregulation of KCC2 activity. Importantly, we show that a brief change in extracellular Zn2+ activates metabotropic signaling that is sufficient to rapidly upregulate KCC2 activity in the hippocampus. Such robust effects on CA3 pyramidal cells signaling was previously shown to result from similar stimulation protocols in the mossy fibers, also dependent on the IP3 pathway (Scott et al., 2008). Thus, synaptically released Zn2+, acting via mZnR signaling, may be a significant component of the dynamic regulation of KCC2 in neurons.
As KCC2 transport activity is a major determinant of the neuronal GABAA-inhibitory drive, this transporter is closely associated with seizure activity (Woo et al., 2002; Huberfeld et al., 2007). mZnR-mediated upregulation of KCC2 activity may thus partly account for the reported anticonvulsive actions of Zn2+ (Elsas et al., 2009). Indeed, the fact that synaptic Zn2+ can profoundly influence inhibitory drive is concordant with the enhanced susceptibility to kainate-triggered seizures present in ZnT3 KO mice (Cole et al., 2000). The release of vesicular Zn2+ could serve to limit kainate-triggered epileptic activity by rendering GABAA receptor inhibitory drive more effective through mZnR signaling via upregulation of KCC2 activity. The enhanced kainate-triggered seizure activity observed in ZnT3 KO animals could then be explained by the lack of mZnR-mediated regulation of KCC2. If synaptic Zn2+ acts via regulation of the Cl− gradient, the presence or absence of vesicular Zn2+ may not be critical for regulating epileptic activity when GABAA receptor function is pharmacologically restricted. Interestingly, ZnT3 KO or WT mice exhibit similar susceptibility to seizures when these are generated by GABAA receptor blockers (Cole et al., 2000). The link between synaptic release of Zn2+ and modulation of GABA function described by our work may seem inconsistent with the known, subunit-dependent direct inhibitory actions of Zn2+ on GABAA receptors (Ben-Ari and Cherubini, 1991; Smart et al., 1991, 2004). Yet, Zn2+ released from excitatory synapses may have more pronounced effects on the overall function of mature inhibitory connections by altering KCC2 activity, as evidenced by the reported anticonvulsive properties of the metal.
Zinc is selectively sequestered into a subset of glutamate-containing synaptic vesicles by ZnT3 (Palmiter et al., 1996; Wenzel et al., 1997; Cole et al., 1999; Sindreu et al., 2003) and is released in an activity and Ca2+-dependent manner from hippocampal mossy fiber terminals (Qian and Noebels, 2005, 2006). Our previous study demonstrated that Zn2+ activates mZnR function in CA3 neurons and that GPR39 is localized to these cells (Besser et al., 2009). Here, we show that mZnR activity is mediated by GPR39, and is absent following GPR39 knockdown. This mZnR activation subsequently affects the neuronal Cl− gradient. The fact that both the endogenous transmitter, released from mossy fibers, and its exogenous application produce similar physiological effects, both of which are blocked by drugs that inhibit mZnR function, satisfies the “identity of action” and the “pharmacological identity” neurotransmitter identification criteria (Werman, 1966). Moreover, Zn2+ reuptake via specific transporters (Belloni-Olivi et al., 2009), or the previously reported rapid desensitization of mZnR induced by Zn2+ itself (Besser et al., 2009), may provide the mechanisms required for the inactivation of this pathway. Thus, the mZnR-dependent upregulation of KCC2 activity provides compelling physiological evidence establishing Zn2+ as a neurotransmitter in the mammalian brain. The results presented here elucidate a physiological function for GPR39, a neuronal receptor for synaptically released Zn2+. By increasing membrane expression and activity of KCC2, thereby reshaping the Cl− gradient, mZnR renders GABAA receptor-mediated inhibitory drive more effective.
Footnotes
This work was supported by the US-Israel Binational Science Foundation (Grant BSF2007121 to M.H. and E.A.), by the Israel Science Foundation (Grant 585/05 to M.H.), and by the US National Institutes of Health (Grant NS043277 to E.A.). We thank R. Palmiter from the University of Washington for the ZnT3 KO mice and advice. The GPR39 KO mice were kindly provided by D. Moechars from Johnson & Johnson Pharmaceutical Research and Development, a Division of Janssen Pharmaceutica. The Gαq inhibitor YM-254890 was a kind gift from Astellas Pharma Inc. We thank Drs. K. Kandler, P. Rosenberg, and E. Levitan for helpful discussions and suggestions.
References
- Aizenman E, Stout AK, Hartnett KA, Dineley KE, McLaughlin B, Reynolds IJ. Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release. J Neurochem. 2000;75:1878–1888. doi: 10.1046/j.1471-4159.2000.0751878.x. [DOI] [PubMed] [Google Scholar]
- Amaral DG, Witter MP. The three-dimensional organization of the hippocampal formation: a review of anatomical data. Neuroscience. 1989;31:571–591. doi: 10.1016/0306-4522(89)90424-7. [DOI] [PubMed] [Google Scholar]
- Amaral DG, Scharfman HE, Lavenex P. The dentate gyrus: fundamental neuroanatomical organization (dentate gyrus for dummies) Prog Brain Res. 2007;163:3–22. doi: 10.1016/S0079-6123(07)63001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banke TG, Gegelashvili G. Tonic activation of group I mGluRs modulates inhibitory synaptic strength by regulating KCC2 activity. J Physiol. 2008;586:4925–4934. doi: 10.1113/jphysiol.2008.157024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beierlein M, Fall CP, Rinzel J, Yuste R. Thalamocortical bursts trigger recurrent activity in neocortical networks: layer 4 as a frequency-dependent gate. J Neurosci. 2002;22:9885–9894. doi: 10.1523/JNEUROSCI.22-22-09885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belloni-Olivi L, Marshall C, Laal B, Andrews GK, Bressler J. Localization of zip1 and zip4 mRNA in the adult rat brain. J Neurosci Res. 2009;87:3221–3230. doi: 10.1002/jnr.22144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Cherubini E. Zinc and GABA in developing brain. Nature. 1991;353:220–225. doi: 10.1038/353220a0. [DOI] [PubMed] [Google Scholar]
- Besser L, Chorin E, Sekler I, Silverman WF, Atkin S, Russell JT, Hershfinkel M. Synaptically released zinc triggers metabotropic signaling via a zinc-sensing receptor in the hippocampus. J Neurosci. 2009;29:2890–2901. doi: 10.1523/JNEUROSCI.5093-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi R, Young SR, Wong RK. Group I mGluR activation causes voltage-dependent and -independent Ca2+ rises in hippocampal pyramidal cells. J Neurophysiol. 1999;81:2903–2913. doi: 10.1152/jn.1999.81.6.2903. [DOI] [PubMed] [Google Scholar]
- Bischofberger J, Engel D, Li L, Geiger JR, Jonas P. Patch-clamp recording from mossy fiber terminals in hippocampal slices. Nat Protoc. 2006;1:2075–2081. doi: 10.1038/nprot.2006.312. [DOI] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Blanton MG, Lo Turco JJ, Kriegstein AR. Whole cell recording from neurons in slices of reptilian and mammalian cerebral cortex. J Neurosci Methods. 1989;30:203–210. doi: 10.1016/0165-0270(89)90131-3. [DOI] [PubMed] [Google Scholar]
- Blasco-Ibáñez JM, Poza-Aznar J, Crespo C, Marqués-Marí AI, Gracia-Llanes FJ, Martínez-Guijarro FJ. Chelation of synaptic zinc induces overexcitation in the hilar mossy cells of the rat hippocampus. Neurosci Lett. 2004;355:101–104. doi: 10.1016/j.neulet.2003.10.053. [DOI] [PubMed] [Google Scholar]
- Bonnet U, Wiemann M. Ammonium prepulse: effects on intracellular pH and bioelectric activity of CA3-neurones in guinea pig hippocampal slices. Brain Res. 1999;840:16–22. doi: 10.1016/s0006-8993(99)01687-x. [DOI] [PubMed] [Google Scholar]
- Buzsàki G, Freund TF, Bayardo F, Somogyi P. Ischemia-induced changes in the electrical activity of the hippocampus. Exp Brain Res. 1989;78:268–278. doi: 10.1007/BF00228898. [DOI] [PubMed] [Google Scholar]
- Carter RE, Aiba I, Dietz RM, Sheline CT, Shuttleworth CW. Spreading depression and related events are significant sources of neuronal Zn2+ release and accumulation. J Cereb Blood Flow Metab. 2011;31:1073–1084. doi: 10.1038/jcbfm.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudotvorova I, Ivanov A, Rama S, Hübner CA, Pellegrino C, Ben-Ari Y, Medina I. Early expression of KCC2 in rat hippocampal cultures augments expression of functional GABA synapses. J Physiol. 2005;566:671–679. doi: 10.1113/jphysiol.2005.089821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole TB, Wenzel HJ, Kafer KE, Schwartzkroin PA, Palmiter RD. Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc Natl Acad Sci U S A. 1999;96:1716–1721. doi: 10.1073/pnas.96.4.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole TB, Robbins CA, Wenzel HJ, Schwartzkroin PA, Palmiter RD. Seizures and neuronal damage in mice lacking vesicular zinc. Epilepsy Res. 2000;39:153–169. doi: 10.1016/s0920-1211(99)00121-7. [DOI] [PubMed] [Google Scholar]
- Dietz RM, Weiss JH, Shuttleworth CW. Zn2+ influx is critical for some forms of spreading depression in brain slices. J Neurosci. 2008;28:8014–8024. doi: 10.1523/JNEUROSCI.0765-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering P, Danscher G, Larsen A, Bruhn M, Søndergaard C, Stoltenberg M. Changes in the vesicular zinc pattern following traumatic brain injury. Neuroscience. 2007;150:93–103. doi: 10.1016/j.neuroscience.2007.09.066. [DOI] [PubMed] [Google Scholar]
- Elsas SM, Hazany S, Gregory WL, Mody I. Hippocampal zinc infusion delays the development of afterdischarges and seizures in a kindling model of epilepsy. Epilepsia. 2009;50:870–879. doi: 10.1111/j.1528-1167.2008.01913.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiumelli H, Cancedda L, Poo MM. Modulation of GABAergic transmission by activity via postsynaptic Ca2+-dependent regulation of KCC2 function. Neuron. 2005;48:773–786. doi: 10.1016/j.neuron.2005.10.025. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Howell GA, Frederickson MH. Zinc dithizonate staining in the cat hippocampus: relationship to the mossy-fiber neuropil and postnatal development. Exp Neurol. 1981;73:812–823. doi: 10.1016/0014-4886(81)90214-4. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Giblin LJ, 3rd, Rengarajan B, Masalha R, Frederickson CJ, Zeng Y, Lopez EV, Koh JY, Chorin U, Besser L, Hershfinkel M, Li Y, Thompson RB, Krezel A. Synaptic release of zinc from brain slices: factors governing release, imaging, and accurate calculation of concentration. J Neurosci Methods. 2006;154:19–29. doi: 10.1016/j.jneumeth.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Fukahori M, Itoh M. Effects of dietary zinc status on seizure susceptibility and hippocampal zinc content in the El (epilepsy) mouse. Brain Res. 1990;529:16–22. doi: 10.1016/0006-8993(90)90806-m. [DOI] [PubMed] [Google Scholar]
- Galeffi F, Sah R, Pond BB, George A, Schwartz-Bloom RD. Changes in intracellular chloride after oxygen-glucose deprivation of the adult hippocampal slice: effect of diazepam. J Neurosci. 2004;24:4478–4488. doi: 10.1523/JNEUROSCI.0755-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesh R, Janakiraman L. Serum zinc levels in children with simple febrile seizure. Clin Pediatr (Phila) 2008;47:164–166. doi: 10.1177/0009922807306165. [DOI] [PubMed] [Google Scholar]
- Goldberg HJ, Sheehy EM. Fifth day fits: an acute zinc deficiency syndrome? Arch Dis Child. 1982;57:633–635. doi: 10.1136/adc.57.8.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas HL, Schaerer B, Vosmansky M. A simple perfusion chamber for the study of nervous tissue slices in vitro. J Neurosci Methods. 1979;1:323–325. doi: 10.1016/0165-0270(79)90021-9. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hershfinkel M, Moran A, Grossman N, Sekler I. A zinc-sensing receptor triggers the release of intracellular Ca2+ and regulates ion transport. Proc Natl Acad Sci U S A. 2001;98:11749–11754. doi: 10.1073/pnas.201193398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershfinkel M, Silverman WF, Sekler I. The zinc sensing receptor, a link between zinc and cell signaling. Mol Med. 2007;13:331–336. doi: 10.2119/2006-00038.Hershfinkel. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershfinkel M, Kandler K, Knoch ME, Dagan-Rabin M, Aras MA, Abramovitch-Dahan C, Sekler I, Aizenman E. Intracellular zinc inhibits KCC2 transporter activity. Nat Neurosci. 2009;12:725–727. doi: 10.1038/nn.2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosie AM, Dunne EL, Harvey RJ, Smart TG. Zinc-mediated inhibition of GABA(A) receptors: discrete binding sites underlie subtype specificity. Nat Neurosci. 2003;6:362–369. doi: 10.1038/nn1030. [DOI] [PubMed] [Google Scholar]
- Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, Rivera C. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci. 2007;27:9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson VR, Nothacker HP, Civelli O. GPR39 receptor expression in the mouse brain. Neuroreport. 2006;17:813–816. doi: 10.1097/01.wnr.0000215779.76602.93. [DOI] [PubMed] [Google Scholar]
- Kahle KT, Rinehart J, Lifton RP. Phosphoregulation of the Na-K-2Cl and K-Cl cotransporters by the WNK kinases. Biochim Biophys Acta. 2010;1802:1150–1158. doi: 10.1016/j.bbadis.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan I, Ben-Zur T, Barhum Y, Levy YS, Burstein A, Charlow T, Bulvik S, Melamed E, Offen D. Dopaminergic differentiation of human mesenchymal stem cells—utilization of bioassay for tyrosine hydroxylase expression. Neurosci Lett. 2007;419:28–33. doi: 10.1016/j.neulet.2007.03.070. [DOI] [PubMed] [Google Scholar]
- Kapur A, Yeckel M, Johnston D. Hippocampal mossy fiber activity evokes Ca2+ release in CA3 pyramidal neurons via a metabotropic glutamate receptor pathway. Neuroscience. 2001;107:59–69. doi: 10.1016/s0306-4522(01)00293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khirug S, Ahmad F, Puskarjov M, Afzalov R, Kaila K, Blaesse P. A single seizure episode leads to rapid functional activation of KCC2 in the neonatal rat hippocampus. J Neurosci. 2010;30:12028–12035. doi: 10.1523/JNEUROSCI.3154-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laube B, Kuhse J, Rundström N, Kirsch J, Schmieden V, Betz H. Modulation by zinc ions of native rat and recombinant human inhibitory glycine receptors. J Physiol. 1995;483:613–619. doi: 10.1113/jphysiol.1995.sp020610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Chen CX, Liu YJ, Aizenman E, Kandler K. KCC2 expression in immature rat cortical neurons is sufficient to switch the polarity of GABA responses. Eur J Neurosci. 2005;21:2593–2599. doi: 10.1111/j.1460-9568.2005.04084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH, Walker JA, Williams JR, Goodier RJ, Payne JA, Moss SJ. Direct protein kinase C-dependent phosphorylation regulates the cell surface Stability and activity of the potassium chloride cotransporter KCC2. J Biol Chem. 2007;282:29777–29784. doi: 10.1074/jbc.M705053200. [DOI] [PubMed] [Google Scholar]
- Lee HH, Jurd R, Moss SJ. Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol Cell Neurosci. 2010;45:173–179. doi: 10.1016/j.mcn.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liguz-Lecznar M, Nowicka D, Czupryn A, Skangiel-Kramska J. Dissociation of synaptic zinc level and zinc transporter 3 expression during postnatal development and after sensory deprivation in the barrel cortex of mice. Brain Res Bull. 2005;66:106–113. doi: 10.1016/j.brainresbull.2005.03.019. [DOI] [PubMed] [Google Scholar]
- Lopantsev V, Wenzel HJ, Cole TB, Palmiter RD, Schwartzkroin PA. Lack of vesicular zinc in mossy fibers does not affect synaptic excitability of CA3 pyramidal cells in zinc transporter 3 knockout mice. Neuroscience. 2003;116:237–248. doi: 10.1016/s0306-4522(02)00570-5. [DOI] [PubMed] [Google Scholar]
- Lu J, Karadsheh M, Delpire E. Developmental regulation of the neuronal-specific isoform of K-Cl cotransporter KCC2 in postnatal rat brains. J Neurobiol. 1999;39:558–568. [PubMed] [Google Scholar]
- Madry C, Betz H, Geiger JR, Laube B. Supralinear potentiation of NR1/NR3A excitatory glycine receptors by Zn2+ and NR1 antagonist. Proc Natl Acad Sci U S A. 2008;105:12563–12568. doi: 10.1073/pnas.0805624105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marandi N, Konnerth A, Garaschuk O. Two-photon chloride imaging in neurons of brain slices. Pflugers Arch. 2002;445:357–365. doi: 10.1007/s00424-002-0933-7. [DOI] [PubMed] [Google Scholar]
- Moechars D, Depoortere I, Moreaux B, de Smet B, Goris I, Hoskens L, Daneels G, Kass S, Ver Donck L, Peeters T, Coulie B. Altered gastrointestinal and metabolic function in the GPR39-obestatin receptor-knockout mouse. Gastroenterology. 2006;131:1131–1141. doi: 10.1053/j.gastro.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Nitzan YB, Sekler I, Hershfinkel M, Moran A, Silverman WF. Postnatal regulation of ZnT-1 expression in the mouse brain. Brain Res Dev Brain Res. 2002;137:149–157. doi: 10.1016/s0165-3806(02)00437-6. [DOI] [PubMed] [Google Scholar]
- Palmiter RD, Cole TB, Quaife CJ, Findley SD. ZnT-3, a putative transporter of zinc into synaptic vesicles. Proc Natl Acad Sci U S A. 1996;93:14934–14939. doi: 10.1073/pnas.93.25.14934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Vergnano AM, Barbour B, Casado M. Zinc at glutamatergic synapses. Neuroscience. 2009;158:126–136. doi: 10.1016/j.neuroscience.2008.01.061. [DOI] [PubMed] [Google Scholar]
- Papp E, Rivera C, Kaila K, Freund TF. Relationship between neuronal vulnerability and potassium-chloride cotransporter 2 immunoreactivity in hippocampus following transient forebrain ischemia. Neuroscience. 2008;154:677–689. doi: 10.1016/j.neuroscience.2008.03.072. [DOI] [PubMed] [Google Scholar]
- Pond BB, Galeffi F, Ahrens R, Schwartz-Bloom RD. Chloride transport inhibitors influence recovery from oxygen-glucose deprivation-induced cellular injury in adult hippocampus. Neuropharmacology. 2004;47:253–262. doi: 10.1016/j.neuropharm.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Qian J, Noebels JL. Visualization of transmitter release with zinc fluorescence detection at the mouse hippocampal mossy fibre synapse. J Physiol. 2005;566:747–758. doi: 10.1113/jphysiol.2005.089276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Noebels JL. Exocytosis of vesicular zinc reveals persistent depression of neurotransmitter release during metabotropic glutamate receptor long-term depression at the hippocampal CA3-CA1 synapse. J Neurosci. 2006;26:6089–6095. doi: 10.1523/JNEUROSCI.0475-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Xu K, Yoo J, Chen TT, Andrews G, Noebels JL. Knockout of Zn transporters Zip-1 and Zip-3 attenuates seizure-induced CA1 neurodegeneration. J Neurosci. 2011;31:97–104. doi: 10.1523/JNEUROSCI.5162-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman PT, Hartnett KA, Aras MA, Levitan ES, Aizenman E. Regulation of apoptotic potassium currents by coordinated zinc-dependent signalling. J Physiol. 2009;587:4393–4404. doi: 10.1113/jphysiol.2009.176321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds IJ. Measurement of cation movement in primary cultures using fluorescent dyes. Curr Protoc Neurosci Chapter. 2001;7 doi: 10.1002/0471142301.ns0711s04. Unit7.11. [DOI] [PubMed] [Google Scholar]
- Rinehart J, Maksimova YD, Tanis JE, Stone KL, Hodson CA, Zhang J, Risinger M, Pan W, Wu D, Colangelo CM, Forbush B, Joiner CH, Gulcicek EE, Gallagher PG, Lifton RP. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell. 2009;138:525–536. doi: 10.1016/j.cell.2009.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritucci NA, Erlichman JS, Dean JB, Putnam RW. A fluorescence technique to measure intracellular pH of single neurons in brainstem slices. J Neurosci Methods. 1996;68:149–163. doi: 10.1016/0165-0270(96)00051-9. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, Kokaia Z, Airaksinen MS, Voipio J, Kaila K, Saarma M. BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J Cell Biol. 2002;159:747–752. doi: 10.1083/jcb.200209011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipilä S, Payne JA, Minichiello L, Saarma M, Kaila K. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. 2004;24:4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruusuvuori E, Li H, Huttu K, Palva JM, Smirnov S, Rivera C, Kaila K, Voipio J. Carbonic anhydrase isoform VII acts as a molecular switch in the development of synchronous gamma-frequency firing of hippocampal CA1 pyramidal cells. J Neurosci. 2004;24:2699–2707. doi: 10.1523/JNEUROSCI.5176-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott R, Lalic T, Kullmann DM, Capogna M, Rusakov DA. Target-cell specificity of kainate autoreceptor and Ca2+-store-dependent short-term plasticity at hippocampal mossy fiber synapses. J Neurosci. 2008;28:13139–13149. doi: 10.1523/JNEUROSCI.2932-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sensi SL, Yin HZ, Weiss JH. AMPA/kainate receptor-triggered Zn2+ entry into cortical neurons induces mitochondrial Zn2+ uptake and persistent mitochondrial dysfunction. Eur J Neurosci. 2000;12:3813–3818. doi: 10.1046/j.1460-9568.2000.00277.x. [DOI] [PubMed] [Google Scholar]
- Sensi SL, Paoletti P, Bush AI, Sekler I. Zinc in the physiology and pathology of the CNS. Nat Rev Neurosci. 2009;10:780–791. doi: 10.1038/nrn2734. [DOI] [PubMed] [Google Scholar]
- Shin JH, Namkung W, Choi JY, Yoon JH, Lee MG. Purinergic stimulation induces Ca2+-dependent activation of Na+-K+-2Cl− cotransporter in human nasal epithelia. J Biol Chem. 2004;279:18567–18574. doi: 10.1074/jbc.M400639200. [DOI] [PubMed] [Google Scholar]
- Sindreu CB, Varoqui H, Erickson JD, Pérez-Clausell J. Boutons containing vesicular zinc define a subpopulation of synapses with low AMPAR content in rat hippocampus. Cereb Cortex. 2003;13:823–829. doi: 10.1093/cercor/13.8.823. [DOI] [PubMed] [Google Scholar]
- Sindreu C, Palmiter RD, Storm DR. Zinc transporter ZnT-3 regulates presynaptic Erk1/2 signaling and hippocampus-dependent memory. Proc Natl Acad Sci U S A. 2011;108:3366–3370. doi: 10.1073/pnas.1019166108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart TG, Moss SJ, Xie X, Huganir RL. GABAA receptors are differentially sensitive to zinc: dependence on subunit composition. Br J Pharmacol. 1991;103:1837–1839. doi: 10.1111/j.1476-5381.1991.tb12337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart TG, Hosie AM, Miller PS. Zn2+ ions: modulators of excitatory and inhibitory synaptic activity. Neuroscientist. 2004;10:432–442. doi: 10.1177/1073858404263463. [DOI] [PubMed] [Google Scholar]
- Strange K, Singer TD, Morrison R, Delpire E. Dependence of KCC2 K-Cl cotransporter activity on a conserved carboxy terminus tyrosine residue. Am J Physiol Cell Physiol. 2000;279:C860–C867. doi: 10.1152/ajpcell.2000.279.3.C860. [DOI] [PubMed] [Google Scholar]
- Stuart GJ, Dodt HU, Sakmann B. Patch-clamp recordings from the soma and dendrites of neurons in brain slices using infrared video microscopy. Pflugers Arch. 1993;423:511–518. doi: 10.1007/BF00374949. [DOI] [PubMed] [Google Scholar]
- Takeda A, Tamano H, Nagayoshi A, Yamada K, Oku N. Increase in hippocampal cell death after treatment with kainate in zinc deficiency. Neurochem Int. 2005;47:539–544. doi: 10.1016/j.neuint.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Thomas-Crusells J, Vieira A, Saarma M, Rivera C. A novel method for monitoring surface membrane trafficking on hippocampal acute slice preparation. J Neurosci Methods. 2003;125:159–166. doi: 10.1016/s0165-0270(03)00050-5. [DOI] [PubMed] [Google Scholar]
- Titz S, Hormuzdi S, Lewen A, Monyer H, Misgeld U. Intracellular acidification in neurons induced by ammonium depends on KCC2 function. Eur J Neurosci. 2006;23:454–464. doi: 10.1111/j.1460-9568.2005.04583.x. [DOI] [PubMed] [Google Scholar]
- Trapp S, Lückermann M, Brooks PA, Ballanyi K. Acidosis of rat dorsal vagal neurons in situ during spontaneous and evoked activity. J Physiol. 1996;496:695–710. doi: 10.1113/jphysiol.1996.sp021720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyzio R, Cossart R, Khalilov I, Minlebaev M, Hübner CA, Represa A, Ben-Ari Y, Khazipov R. Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science. 2006;314:1788–1792. doi: 10.1126/science.1133212. [DOI] [PubMed] [Google Scholar]
- Verkman AS. Development and biological applications of chloride-sensitive fluorescent indicators. Am J Physiol. 1990;259:C375–388. doi: 10.1152/ajpcell.1990.259.3.C375. [DOI] [PubMed] [Google Scholar]
- Vogt K, Mellor J, Tong G, Nicoll R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron. 2000;26:187–196. doi: 10.1016/s0896-6273(00)81149-6. [DOI] [PubMed] [Google Scholar]
- Wake H, Watanabe M, Moorhouse AJ, Kanematsu T, Horibe S, Matsukawa N, Asai K, Ojika K, Hirata M, Nabekura J. Early changes in KCC2 phosphorylation in response to neuronal stress result in functional downregulation. J Neurosci. 2007;27:1642–1650. doi: 10.1523/JNEUROSCI.3104-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Wake H, Moorhouse AJ, Nabekura J. Clustering of neuronal K+-Cl− cotransporters in lipid rafts by tyrosine phosphorylation. J Biol Chem. 2009;284:27980–27988. doi: 10.1074/jbc.M109.043620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel HJ, Cole TB, Born DE, Schwartzkroin PA, Palmiter RD. Ultrastructural localization of zinc transporter-3 (ZnT-3) to synaptic vesicle membranes within mossy fiber boutons in the hippocampus of mouse and monkey. Proc Natl Acad Sci U S A. 1997;94:12676–12681. doi: 10.1073/pnas.94.23.12676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werman R. Criteria for identification of a central nervous system transmitter. Comp Biochem Physiol. 1966;18:745–766. doi: 10.1016/0010-406x(66)90209-x. [DOI] [PubMed] [Google Scholar]
- Woo NS, Lu J, England R, McClellan R, Dufour S, Mount DB, Deutch AY, Lovinger DM, Delpire E. Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus. 2002;12:258–268. doi: 10.1002/hipo.10014. [DOI] [PubMed] [Google Scholar]
- Woodin MA, Ganguly K, Poo MM. Coincident pre- and postsynaptic activity modifies GABAergic synapses by postsynaptic changes in Cl- transporter activity. Neuron. 2003;39:807–820. doi: 10.1016/s0896-6273(03)00507-5. [DOI] [PubMed] [Google Scholar]
- Yasuda S, Miyazaki T, Munechika K, Yamashita M, Ikeda Y, Kamizono A. Isolation of Zn2+ as an endogenous agonist of GPR39 from fetal bovine serum. J Recept Signal Transduct Res. 2007;27:235–246. doi: 10.1080/10799890701506147. [DOI] [PubMed] [Google Scholar]
- Zhang JV, Ren PG, Avsian-Kretchmer O, Luo CW, Rauch R, Klein C, Hsueh AJ. Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin's effects on food intake. Science. 2005;310:996–999. doi: 10.1126/science.1117255. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang H, Li J, Jimenez DA, Levitan ES, Aizenman E, Rosenberg PA. Peroxynitrite-induced neuronal apoptosis is mediated by intracellular zinc release and 12-lipoxygenase activation. J Neurosci. 2004;24:10616–10627. doi: 10.1523/JNEUROSCI.2469-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang H, Li J, Dong L, Xu P, Chen W, Neve RL, Volpe JJ, Rosenberg PA. Intracellular zinc release and ERK phosphorylation are required upstream of 12-lipoxygenase activation in peroxynitrite toxicity to mature rat oligodendrocytes. J Biol Chem. 2006;281:9460–9470. doi: 10.1074/jbc.M510650200. [DOI] [PubMed] [Google Scholar]
- Zhao B, Wong AY, Murshid A, Bowie D, Presley JF, Bedford FK. Identification of a novel di-leucine motif mediating K(+)/Cl(−) cotransporter KCC2 constitutive endocytosis. Cell Signal. 2008;20:1769–1779. doi: 10.1016/j.cellsig.2008.06.011. [DOI] [PubMed] [Google Scholar]