

Abstract
Objective
ATN-224 (choline tetrathiomolybdate) is an oral Cu2+/Zn2+-superoxide dismutase 1 (SOD1) inhibitor with preclinical antitumor activity. We hypothesized that ATN-224 may induce antitumor effects as an antiangiogenic agent at low dose-levels while possessing direct antitumor activity at higher dose-levels. The objective of this study was to screen its clinical activity in patients with biochemically recurrent hormone-naïve prostate cancer.
Methods
Biochemically-recurrent prostate cancer patients with prostate specific antigen doubling times (PSADT) <12 months, no radiographic evidence of metastasis, and no hormonal therapy within 6 months (with serum testosterone levels >150 ng/dL) were eligible. ATN-224 was administered at two dose-levels, 300 mg (n=23) or 30 mg (n=24) daily, by way of randomization. PSA progression was defined as a ≥50% increase (and >5 ng/mL) in PSA from baseline or post-treatment nadir. Endpoints included the proportion of patients who were free of PSA progression at 24 weeks, changes in PSA slope/PSADT, and safety. The study was not powered to detect differences between the two treatment groups.
Results
At 24 weeks, 59% (95% CI 33–82%) of men in the low-dose arm and 45% (95% CI 17–77%) in the high-dose arm were PSA progression-free. Median PSA progression-free survival was 30 weeks (95% CI 21–40+) and 26 weeks (95% CI 24–39+) in the low-dose and high-dose groups, respectively. Pre- and on-treatment PSA kinetics analyses showed a significant mean PSA slope decrease (p=0.006) and a significant mean PSADT increase (p=0.032) in the low-dose arm only. Serum ceruloplasmin levels, a biomarker for ATN-224 activity, were lowered in the high-dose group, but did not correlate with PSA changes.
Conclusions
Low-dose ATN-224 (30 mg daily) may have biologic activity in men with biochemically-recurrent prostate cancer, as suggested by an improvement in PSA kinetics. However, the clinical significance of PSA kinetics changes in this patient population remains uncertain. The absence of a dose-response effect also reduces enthusiasm, and there are currently no plans to further develop this agent in prostate cancer.
BACKGROUND
ATN-224 (choline tetrathiomolybdate) is an orally-available inorganic small molecule that inhibits the copper/zinc-dependent enzyme, superoxide dismutase 1 (Cu/Zn-SOD1), in endothelial and tumor cells[1–3]. It is a second-generation choline salt of a copper-binding compound tetrathiomolybdate, which has been shown to have efficacy as an antiangiogenic and antitumor agent in several murine models of cancer[4–7], and has been tested as an antineoplastic agent in several clinical trials[8, 9].
SOD1 is an abundant cytosolic enzyme that dismutates superoxide (O2−) into molecular oxygen and hydrogen peroxide (H2O2), a promiscuous second messenger essential to mitogen signaling. In the presence of ATN-224, the generation of H2O2 is suppressed through the inhibition of SOD1. Mitogen-induced kinase cascades are down-regulated leading to the inhibition of cell proliferation and/or apoptosis[3]. In addition, ATN-224 directly blocks the phosphorylation of the epidermal growth factor (EGF) and platelet-derived growth factor (PDGF) receptors, and inhibits the translocation of the p65 subunit of nuclear factor κB (NF-κB) to the nucleus. These actions may potentially explain ATN224’s antiangiogenic and apoptotic effects in tumor cells[4].
In addition to inhibiting SOD1, ATN-224 may also mediate antitumor effects by lowering systemic copper levels, which has been demonstrated to down-regulate the expression of numerous factors associated with tumor angiogenesis and progression, including vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF)-2, NF-κB, interleukin (IL)-6, and IL-8[5, 10].
The antiangiogenic and antitumor activities of ATN-224 exhibit substantially different dose-responses with maximal antiangiogenic activity observed at doses that are approximately 10-fold lower than those required to inhibit tumor progression in tumor xenograft models. This is consistent with in vitro studies where ATN-224 inhibits the proliferation of endothelial cells in culture at concentrations that are 5- to 10-fold lower than those required to inhibit proliferation of the most highly ATN224-sensitive tumor cell lines[3].
We hypothesized that ATN-224 would have the potential to delay the progression of prostate cancer by mechanisms that include both antiangiogenic and antitumor effects. A prostate cancer population with presumed lower tumor burden was chosen to test this hypothesis, using PSA parameters to define the primary endpoint. Two distinct dose-levels were examined in this study. The high-dose group received 300 mg/day for 2 weeks followed by a titrated dose targeting ceruloplasmin (Cp) levels within the range of 5–15 ng/mL; the low-dose group received one-tenth of this dose (30 mg/day).
PATIENTS AND METHODS
Eligibility
Patients with histologically confirmed adenocarcinoma of prostate with evidence of biochemical relapse after local therapy were eligible. Patients had no detectable disease as assessed by physical examination and radiographic measures (bone scan and CT of the abdomen/pelvis) within 4 weeks of study entry, and had a PSA doubling time (PSADT) of <12 months. PSADT was calculated using the natural log of 2 (0.693) divided by the slope of the relationship of log PSA against time (in months). The last PSA level prior to enrollment had to be ≥2 ng/mL and rising over the prior value.
Patients had to be ≥18 years old, with life expectancy >6 months and Eastern Cooperative Oncology Group (ECOG) performance status ≤2, and with adequate hepatic, renal and marrow function, serum testosterone >150 ng/dL, no uncontrolled intercurrent illness, no known history of HIV disease or infectious hepatitis A, B or C, no active other malignancy, no history of malabsorption syndromes, and ability to swallow capsules.
Prior chemotherapy was allowed as long as the requirements for adequate organ and marrow function were met. Patients who had radiotherapy within 3 months or other investigational agents within 28 days prior to the first dose of ATN-224 were excluded. Patients who had hormone therapy within 6 months, were receiving steroids for active concurrent illness, any estrogen-like agents, or any hormonally active over-the-counter compounds or finasteride were excluded. Copper- or zinc-containing supplements were prohibited although diet was not regulated.
The institutional review board of each participating institution approved the study, and all participants gave written informed consent. The trial was conducted through the Department of Defense (DoD)/Prostate Cancer Foundation (PCF)-sponsored Prostate Cancer Clinical Trials Consortium (PCCTC). The members of the consortium worked with the manufacturer (Attenuon, LLC.) in the planning and design of this clinical study. Attenuon supported the research aspects of the study.
Study drug
ATN-224 is an oral investigational agent supplied by Attenuon, LLC. It was stored refrigerated (2–8°C) with stability for at least 24 months. It was administered together with an antacid to prevent ATN-224 from degrading in gastric acid and providing more consistent absorption. Patients took their assigned dose of ATN-224 within 30 minutes after breakfast and at least 1 hour after taking omeprazole 20 mg or another equivalent antacid. Other proton-pump inhibitors or histamine H2-receptor antagonists (i.e. ranitidine) were allowed if patients were unable to receive omeprazole or were already taking another antacid.
Study design
This was a randomized phase II multicenter study designed to assess the efficacy of two dose-levels of ATN-224 for the treatment of biochemically-recurrent prostate cancer. Patients were randomized (1:1) after confirmation of eligibility requirements. There were no stratification factors. Because patients in the high-dose group (but not the low-dose group) had doses titrated according to Cp levels, the study was not blinded.
A loading dose of 300 mg/day for 2 weeks followed by a titrated maintenance dose was the recommended starting dose from a phase I study[11]. For the high-dose group, the initial starting dose of ATN-224 was 300 mg orally once daily for two weeks. All patients, regardless of Cp levels, were dose-reduced to 150 mg daily for the next two weeks. Starting with the second 28-day cycle, the dose was titrated to a level that maintained Cp within the target range of 5–15 mg/dL. The need for dose adjustments on the basis of Cp was assessed every 2 weeks. Dose reductions for Cp were 40% and dose increases were 20% of the current dose. Patients could receive multiple dose increases, but dose increases could not occur more frequently than once every 2 weeks. There was no limit on the number of dose reductions or dose increases based solely on Cp level (in the absence of dose-limiting toxicities), although any patient whose Cp level fell below 5 mg/dL while receiving the lowest possible dose of 30 mg/day would be withdrawn from the study.
The initial starting dose of ATN-224 administered to the low-dose group was 30 mg orally once daily and Cp levels were monitored at the beginning of each cycle. However, no dose adjustments for serum Cp were made. If patients developed grade 3–4 toxicity, dosing of ATN-224 was temporarily suspended and then restarted at the same dose after the toxicity resolved to grade 1 or less.
Adverse events (AEs) were graded according to the Common Terminology Criteria for Adverse Events, version 3.0 (CTCAEv3). Patients with mild-to-moderate (grade 1–2) AEs related to ATN-224 were treated symptomatically with appropriate therapy and continued on therapy. Severe or life-threatening (grade 3–4) AEs related to ATN-224 that could be treated symptomatically to grade 2 or less received appropriate therapy without ATN-224 dose delay or adjustment. Dose adjustments downward were made for persistent grade 3–4 toxicities (>14 days) or if serum Cp fell below 5 mg/dL. For the high-dose cohort only, dose adjustments were made upward in the absence of dose-limiting toxicity if the target plasma Cp range (5 – 15 mg/dL) was not reached.
Baseline and follow-up evaluation
Baseline evaluation included history and physical examination, electrocardiogram (ECG), complete blood count, comprehensive metabolic profile, serum testosterone, and serum PSA. Digital rectal examination, bone scan, abdominal and pelvic CT scans were obtained within 4 weeks prior to study entry and then every 3 cycles. At least 3 PSA levels, drawn ≥28 days apart, were required in order to calculate the pre-treatment PSADT. All values within 6 months of the start of therapy were used for post-treatment PSADT determination. PSA levels were obtained at the beginning of each 28-day cycle and all values were used for calculation. PSA slope was calculated as the linear regression line of the natural log of PSA against time.
PSA progression was defined as an increase in serum PSA over baseline of ≥50% (and by >5 ng/mL), confirmed by a second PSA value obtained ≥28 days later. Disease progression was assigned to the earliest observed time when a clinical or radiographic evaluation showed progression. For patients who had not clinically/radiographically progressed by the last follow-up visit, they were censored at the date when the last tumor assessment revealed lack of progression.
Correlative studies
For patients in the high-dose group, serum for Cp was sent to a central laboratory for analysis. Cp levels were analyzed every other week until the Cp levels were within the target range (5–15 ng/mL) and the patient had been receiving a stable dose of ATN-224 for 4 weeks, at which point the Cp level was monitored every 4 weeks. Cp was monitored weekly if it was below 5 mg/dL. For patients in the low-dose group, Cp levels were monitored at the beginning of each cycle.
Statistical analysis
The primary objective of the study was to assess the proportion of patients in each treatment arm who did not experience PSA progression as defined above (i.e. remained PSA progression-free) at 24 weeks. The desired 95% confidence interval (CI) for this estimate was less than ±0.20. Because the 95% CI is largest when the proportion is 0.50, this was used to determine the minimum number of patients required. To this end, with 25 patients (in each group), the 95% CI would be less than ±0.20 using the Greenwood formula for standard error. Progression-free survival (PFS), an important secondary endpoint, was measured from the time of study drug initiation until the time of clinical/radiographic progression or death. The proportion of patients achieving a ≥25% reduction in PSA from baseline was also recorded. Pre- and on-study changes in PSADT and PSA slope were computed for each patient as detailed above. Descriptive statistics were used to report proportions, and time-to-event endpoints were estimated using the Kaplan-Meier method. Within-patient changes in PSADT/slope before and after study initiation were sought using paired t-tests. Comparisons between groups were performed as exploratory analyses only, because the study was not powered for inferential statistics.
Cp levels from the first four treatment cycles and last follow-up visit were examined to compare the low- and high-dose arms. A natural log transformation was applied to the Cp data before analysis. To account for the correlation among repeated measures collected over multiple treatment cycles for the same patient, generalized estimating equations (GEE)[12] were used for model estimation and hypothesis testing. A compound symmetric covariance structure was assumed for these regression models, implying that the aspect of covariance between repeated measures was due to patient contribution irrespective of time. The Cp levels from weeks one and three on cycles 1–4 and long term follow-up were modeled as a function of dose to test the global null hypothesis of equal mean Cp in the two study arms. Mean Cp levels across sites were similarly estimated and compared. Boxplots[13] used to display the log transformed data were created using R software[14]. All other statistical computations were performed using the SAS package[15]. All p-values are two-sided (with statistical significance set at <0.05), and all confidence intervals (CI) were at the 95% level.
RESULTS
Patient characteristics
Study participants were recruited at 6 centers from the PCCTC, namely Johns Hopkins Kimmel Cancer Center (Baltimore, MD), Oregon Health & Science University Knight Cancer Institute (Portland, OR), University of California San Francisco (San Francisco, CA), University of Wisconsin (Madison, WI), Memorial Sloan-Kettering Cancer Center (New York, NY) and M.D. Anderson Cancer Center (Houston, TX) between 12/2006 and 10/2008. Fifty-two patients were accrued but 1 patient did not receive any treatment and was not included in the efficacy or safety analyses. Four patients had pre-treatment PSA data that were not available, and these patients were also excluded from the efficacy analysis. Forty-seven patients (24, low-dose; 23, high-dose) had full information and are reported here. Median duration on study was 24.1 weeks in the low-dose arm, and 20.1 weeks in the high-dose arm. Patient characteristics are summarized in Table 1. As demonstrated, baseline PSA values appear balanced between the groups. However, Gleason sum was not well balanced, with the suggestion that patients in the high-dose arm had tumors of higher Gleason scores.
Table 1.
Patient characteristics at study entry
| Characteristic | Low dose | High dose |
|---|---|---|
|
| ||
| Patients (n) | 24 | 23 |
|
| ||
| Age (yrs) | ||
| Average | 63.8 | 64.1 |
| Range | 53 – 75 | 53 – 81 |
|
| ||
| Race (%) | ||
| White | 91 | 87 |
| Non-white | 9 | 13 |
|
| ||
| Gleason sum (%) | ||
| ≥ 7 | 14 | 45 |
| < 7 | 86 | 55 |
|
| ||
| Baseline PSA (ng/mL) | ||
| Mean +/− SD | 8.8 +/− 7.7 | 14.9 +/− 23.4 |
| Median | 6.5 | 6.7 |
| Range | 2.0 – 32.3 | 1.7 – 101.0 |
|
| ||
| Prior treatment | ||
| Prostatectomy only | 9 | 8 |
| Radiotherapy only | 7 | 6 |
| Prostatectomy and salvage radiotherapy | 8 | 9 |
Abbreviations: PSA = prostate-specific antigen
PSA parameters
Table 1A shows the results of the primary efficacy analysis, performed separately by treatment group and also combined. As demonstrated, not all patients remained on study for 24 weeks and many were unevaluable for the primary endpoint. Table 1B depicts PSA progression-free survival using Kaplan-Meier analysis, and also shows median PSA progression-free survival in each study arm. PSA kinetic changes are shown in Table 1C, listing pre-treatment PSA slope and PSADT values for each treatment group, as well as the on-treatment changes in these parameters after study drug initiation. As demonstrated, significant within-patient changes in PSA slope and PSADT were observed in men receiving low-dose ATN-224. A small minority of patients in both groups achieved PSA declines >25%, while one patient (in the high-dose arm) showed a >50% decrease in PSA.
Toxicity
Consistent with results from phase I clinical trials[11], ATN-224 was well tolerated (Table 3). The most frequent non-hematological adverse events were fatigue, gastrointestinal disorders, and skin reactions. Hematological toxicities included reversible grade 3–4 leucopenia. Three patients in the high-dose arm had to discontinue the study drug due to grade 3–4 toxicities (one due to leucopenia, one due to generalized rash, one due to elevated aminotransferases).
Table 3.
Summary of the most common adverse events possibly related to study drug
| Events | Any | Grade 3 | Grade 4 |
|---|---|---|---|
| Hematologic | |||
| Leucopenia | 11 (21%) | 4* (8%) | 2** (4%) |
| Lymphopenia | 3 (6%) | ||
| Anemia | 2 (4%) | ||
| Nonhematologic | |||
| Fatigue | 30 (58%) | ||
| Diarrhoea | 11 (21%) | ||
| Nausea | 10 (19%) | ||
| Dysgeusia | 8 (15%) | ||
| Flatulence | 7 (13%) | ||
| Dizziness | 5 (10%) | ||
| Rash | 5 (10%) | 2** (4%) | |
| Nasal congestion | 5 (10%) | ||
| Constipation | 4 (8%) | ||
| Dyspnoea | 4 (8%) | ||
| Elevated Alanine aminotransferase | 4 (8%) | 1** (2%) | |
Note:
1 event from low-dose and 3 events from high-dose group.
All events were from high-dose group.
Correlative studies
Figure 1 shows the log transformed Cp levels by cycle and week for the two study arms. The ordered pattern of Cp over the four cycles of the study and long-term follow-up was similar in the two dose-levels. Overall, the high-dose arm had lower on-study mean log Cp levels (mean=2.4 mg/dL, standard error=0.07) compared to the low-dose arm (mean=3.0 ng/dL, standard error=0.07), and this difference was statistically significant (χ2=18.96, p<0.0001). The GEE estimated difference in the mean log Cp levels (low-dose minus high-dose) was 0.59 ng/dL (95% CI: 0.39–0.78). Very similar results were obtained when restricting the analysis to the observed Cp levels at week one of the first four cycles of treatment (χ2=15.5, p<0.0001) with a GEE estimated difference in mean log Cp of 0.52 ng/dL (95% CI: 0.31–0.73). Although there were some minor differences in mean Cp across the six participating sites, the global test for this factor was not significant (χ2=8.44, p=0.13). Data from a single site with higher Cp values on average are identified in green and a site with lower Cp values on average is marked in red. Adjusting for study site did not alter the results of the Cp treatment arm comparisons.
Figure 1.

Correlation of Cp levels and PSA levels. A: Changes of Cp according to the two ATN-224 dose-levels, in study participants. Boxplots of Cp by study arm, low-dose (30 mg) and high-dose (300 mg), across four treatment cycles and long-term follow-up are shown. The horizontal line inside each box represents the median Cp level. Green points represent patients from a higher Cp value site and red points are patients from a lower Cp value site. B: GEE analysis of correlation between Cp and PSA. Different symbols are for different patients. The overall p-value for the correlation of log(Cp) with log(PSA) for all patients with values on the same date is 0.16.
A scatter plot of log Cp level against log PSA level is shown in Figure 1B. These data are plotted with different symbols by patient, and indicate that these two parameters are correlated. The GEE regression model: log(Cp) = 2.42 + 0.097 * log(PSA) indicates that the log Cp levels tend to increase as the log PSA increases (β1 = 0.097; 95% CI: −0.023 to 0.22). Quantitatively, for a 10% increase in PSA, one could expect a 0.93% increase in Cp. Adjusting for the correlation between observations within the same patient, the p-value for this correlation of log(Cp) with log(PSA) for everyone with values on the same date is 0.16. Cp appears to be quite variable along most of the range of PSA values, except at the very high end of the range. In addition, when the PSA variable is dichotomized (e.g. above and below the median value of 2.3 ng/mL), the relationship is no longer significant.
DISCUSSION
Biochemically-relapsed, non-metastatic, hormone-naïve prostate cancer represents a unique disease state [16, 17]. As many as 70,000 men per year in the United States fall into this category where a rising PSA is the only manifestation of illness [17, 18]. In men with PSA progression after local therapy, management options include surveillance, initiation of androgen deprivation therapy (ADT), dietary intervention, or clinical trial participation. A subset of patients may benefit from salvage radiation therapy. However, no definite survival benefit has been demonstrated with the early institution of ADT in this population. Given the adverse effects of ADT, many men choose to avoid or delay medical castration for a rising PSA alone. Developing alternative options for patients with “PSA-only” prostate cancer is therefore highly desirable.
Evaluating drugs to treat prostate cancer poses unique challenges. The natural history of the disease often spans decades. The Prostate-Specific Antigen Working Group (PSAWG) has described eligibility criteria and trial design considerations for patients in the state of a rising PSA after local therapy [19]. While changes in PSA have not been endorsed as a surrogate marker of clinical benefit or survival, modulation of PSA levels is considered to be an indicator of biological drug effects that could help screen for agents that might alter the natural history of the disease. The current study adhered to the concepts described by the PSAWG and was designed to determine the proportion of patients who remained PSA progression-free for 24 weeks. This may help us predict clinical responses in patients with biochemically-relapsed prostate cancer.
We designed this randomized phase II study in order to screen for clinical activity of ATN-224, and to guide the conduct of future studies. Overall, 54% of evaluable patients were PSA progression-free at 24 weeks suggesting that ATN-224 may have biological activity in this patient population. When examining each dose-level separately, it appeared that the low-dose arm had greater effects on PSA parameters than the high-dose arm. However, differences between the two arms cannot be interpreted, as the study lacked power for comparative efficacy analyses. In addition, it is not known if modulation of PSA parameters is related to true clinical benefits. Interpretation of the results is further complicated by the baseline patient characteristic analysis suggesting that there were less high-Gleason patients in the low-dose group.
Data from a retrospective cohort of patients with clinically localized prostate cancer treated with either radical prostatectomy or radiation therapy suggested that a short post-treatment PSADT (<3 months) is a useful surrogate endpoint for all-cause mortality and prostate cancer-specific mortality after biochemical recurrence[20]. In men who had a rising PSA after radical prostatectomy, a PSADT shorter than the median value of 10 months was the most significant predictor for metastatic progression[21]. Similarly, a short post-treatment PSADT after radiation therapy predicted progression to metastatic disease[22, 23]. The PSA velocity was also related significantly to survival in men with rising PSA after external beam radiotherapy[24].
In this study, we found that compared to pre-treatment values, post-treatment PSADT was statistically longer after low-dose ATN 224 treatment but not after high-dose therapy. PSA slope was also significantly decreased post-treatment in the low-dose arm. One explanation for this could be the different baseline populations as mentioned above. However, it is also impossible to prove that the same effects could not have been seen with a placebo treatment. To this end, in a placebo-controlled trial evaluating the effect of celecoxib on PSADT in a similar patient population, 20% of 40 men in the placebo group had post-treatment PSADT ≥200% of baseline PSADT[25]. A separate study of rosiglitazone versus placebo in a similar population showed that 40% of men in the placebo group had a post-treatment PSADT >150% of baseline[26]. In our study, 8 patients (33%) in low-dose group and 3 men (13%) in the high-dose group had PSADT >200% of baseline after 6 months of treatment. Certainly there are differences in baseline PSADT characteristics in our study compared to these prior trials (e.g. PSADTs were between 6 and 24 months[25] or <24 months[26] in the other studies, and <12 months in the current study), and therefore direct comparisons of our study with the prior trials is not possible. Therefore, in the absence of a placebo-control, it is difficult to interpret the significance of the PSA changes reported here.
Surrogate endpoints for future studies evaluating investigational agents in this patient population need to be validated. PSADT and PSA kinetics changes may be acceptable for phase II studies, but would have questionable significance in larger phase III studies in the absence of clinical endpoints (e.g. metastasis-free survival or overall survival). Metastasis-free survival as a surrogate for the survival endpoint is currently being discussed in the clinical research community and may be reasonable for studies requiring prolonged follow-up. Interestingly, a preliminary evaluation of several PSA measures in the “PSA-only” population seems to suggest that changes in PSA kinetics induced by treatments with a variety of experimental agents correlate significantly with metastatic progression[27], although these data require prospective confirmation.
SOD1 as a target for cancer therapeutics should be revisited as well. Studies have shown that over-expression of SOD in vitro increases cell differentiation, decreases cell growth and proliferation, and can reverse a malignant phenotype[28, 29]. Other in vivo studies showed that SOD could be highly expressed in aggressive human solid tumors[30–32]. Furthermore, high SOD expression has occasionally been associated with a poor prognosis and with resistance to cytotoxic drugs and radiation[33, 34]. It is suggested that most of the apparent conflicts between the above in vitro and in vivo observations can be reconciled by considering the net redox status of tumor cells in different environments[35]. It is possible that the baseline net cell redox balance will determine the sensitivity of cancer cells to a SOD1 inhibitor such as ATN-224. Regrettably, evaluation of SOD1 expression was not conducted as part of this trial.
ATN-224 may also mediate antitumor effects by lowering systemic copper levels, as reflected by serum Cp. Although ATN-224 decrease Cp level as expected, there was only a weak correlation between Cp levels and PSA alterations. SOD1 activity in erythrocytes has also been proposed as a biomarker for copper status in humans[36, 37]. It may give us some information to explain why the low-dose ATN-224 group achieved a higher degree of PSA progression-free survival.
This study has several limitations. First, there was no placebo control in this trial. It has been reported that in similar patient populations, placebo-treated patients can have stable disease for many weeks. In addition, it is not possible to assess whether post-treatment PSA changes would have also occurred to the same degree on a placebo arm, due to factors such as more frequent PSA measurements captured on-treatment with a consequent regression of PSADT values towards the mean. Second, the baseline patient factors in this study were not all well balanced between groups, with more high-risk patients observed in the high-dose group. This makes interpretation of the data difficult, especially because Gleason sum has been shown to be a predictor of progression and survival. It is possible that ATN-224 may be more efficacious in patients with low-risk disease. Third, the sample size here was not large enough to permit an adequately-powered comparative analysis between the two study arms, and thus this study is unable to provide data to suggest which dose may be more efficacious in terms of PSA parameters.
In conclusion, ATN-224 therapy was associated with significant post-treatment changes in PSA parameters compared to pre-treatment values in men with PSA-recurrent non-metastatic prostate cancer when used at a dose of 30 mg/day but not at the higher dose. Further analyses (perhaps using pooled data from several comparable studies) are required to determine whether changes in PSA kinetics (PSA doubling time, slope, velocity) correlate with clinically-relevant endpoints in this patient population. In addition, placebo-controlled trials may aid the interpretation of future trials evaluating non-hormonal investigational drugs in this patient population.
Table 2.
Modulation of PSA parameters. A: PSA progression-free rate at 24 weeks, shown separately by treatment group and also combined. B: Kaplan-Meier analysis of PSA progression-free survival, analyzed by treatment group. C: Pre-treatment and on-study PSA kinetics (PSADT and PSA slope) parameters.
| Table 2A | ||
|---|---|---|
| PSA progression-free rate at 24 weeks | ||
| Dose level (No. of evaluable patients) | No. achieving primary endpoint (%) | 95% CI |
| Low-dose (17) | 10 (59%) | 33 – 82% |
| High-dose (11) | 5 (45%) | 17 – 77% |
| All (28) | 15 (54%) | 36 – 71% |
| Table 2B |
|---|
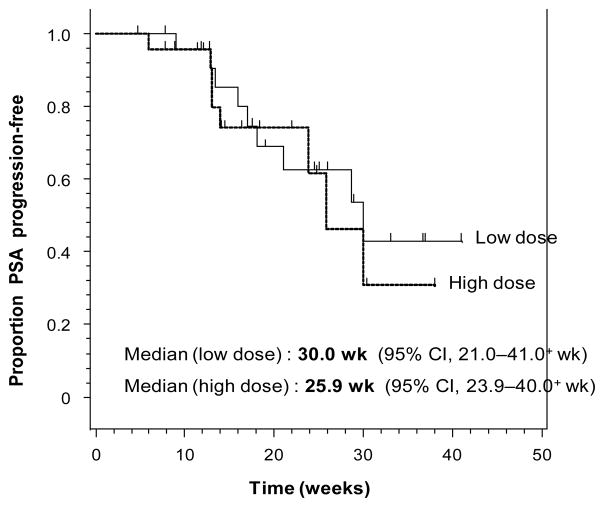
|
| Table 2C | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cohorts | N | Mean PSA slope | Mean PSADT (months) | >25% PSA decline from baseline (%) | ||||
| Baseline value | On-study change* (p value) | 95% CI for change | Baseline value | On-study change* (p value) | 95% CI for change | |||
| Low-dose | 24 | 0.221 | −0.083 (0.006) | (−0.141, −0.026) | 4.8 | +4.1 (0.032) | (+0.4, +7.8) | 3/24 (13%) |
| High-dose | 23 | 0.198 | −0.017 (0.663) | (−0.097, +0.063) | 5.2 | −1.7 (0.705) | (−10.7, +7.4) | 4/23 (17%) |
compared to baseline value
Acknowledgments
We are grateful to the PCCTC Coordinating Center, Ting Wang, and all the research nurses and study coordinators.
Funding Support: DOD W81XWH-09-1-0149, NCI 5P30CA006973
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Donate F, et al. Identification of biomarkers for the antiangiogenic and antitumour activity of the superoxide dismutase 1 (SOD1) inhibitor tetrathiomolybdate (ATN-224) Br J Cancer. 2008;98(4):776–83. doi: 10.1038/sj.bjc.6604226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lowndes SA, et al. Copper chelator ATN-224 inhibits endothelial function by multiple mechanisms. Microvasc Res. 2009;77(3):314–26. doi: 10.1016/j.mvr.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Juarez JC, et al. Copper binding by tetrathiomolybdate attenuates angiogenesis and tumor cell proliferation through the inhibition of superoxide dismutase 1. Clin Cancer Res. 2006;12(16):4974–82. doi: 10.1158/1078-0432.CCR-06-0171. [DOI] [PubMed] [Google Scholar]
- 4.Pan Q, Bao LW, Merajver SD. Tetrathiomolybdate inhibits angiogenesis and metastasis through suppression of the NFkappaB signaling cascade. Mol Cancer Res. 2003;1(10):701–6. [PubMed] [Google Scholar]
- 5.Pan Q, et al. Copper deficiency induced by tetrathiomolybdate suppresses tumor growth and angiogenesis. Cancer Res. 2002;62(17):4854–9. [PubMed] [Google Scholar]
- 6.Lowndes SA, Harris AL. Copper chelation as an antiangiogenic therapy. Oncol Res. 2004;14(11–12):529–39. doi: 10.3727/0965040042707952. [DOI] [PubMed] [Google Scholar]
- 7.Hassouneh B, et al. Tetrathiomolybdate promotes tumor necrosis and prevents distant metastases by suppressing angiogenesis in head and neck cancer. Mol Cancer Ther. 2007;6(3):1039–45. doi: 10.1158/1535-7163.MCT-06-0524. [DOI] [PubMed] [Google Scholar]
- 8.Brewer GJ, et al. Treatment of metastatic cancer with tetrathiomolybdate, an anticopper, antiangiogenic agent: Phase I study. Clin Cancer Res. 2000;6(1):1–10. [PubMed] [Google Scholar]
- 9.Redman BG, et al. Phase II trial of tetrathiomolybdate in patients with advanced kidney cancer. Clin Cancer Res. 2003;9(5):1666–72. [PubMed] [Google Scholar]
- 10.Juarez JC, et al. Superoxide dismutase 1 (SOD1) is essential for H2O2-mediated oxidation and inactivation of phosphatases in growth factor signaling. Proc Natl Acad Sci U S A. 2008;105(20):7147–52. doi: 10.1073/pnas.0709451105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lowndes SA, et al. Phase I study of copper-binding agent ATN-224 in patients with advanced solid tumors. Clin Cancer Res. 2008;14(22):7526–34. doi: 10.1158/1078-0432.CCR-08-0315. [DOI] [PubMed] [Google Scholar]
- 12.Liang KYZSL. Longitudinal data analysis using generalized linear models. Biometrika. 1986;73:13–22. [Google Scholar]
- 13.Tukey JW. Exploratory Data Analysis. Reading, MA: Addison Wesley Publishing Co., Inc; 1977. [Google Scholar]
- 14.R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria: 2004. [Google Scholar]
- 15.SAS Institute Inc. SAS User=s Guide: Statistics, Version. 5. Cary, NC: SAS Institute Inc; 1985. [Google Scholar]
- 16.Scher HI, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008;26(7):1148–59. doi: 10.1200/JCO.2007.12.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moul JW, Banez LL, Freedland SJ. Rising PSA in nonmetastatic prostate cancer. Oncology (Williston Park) 2007;21(12):1436–45. discussion 1449, 1452, 1454. [PubMed] [Google Scholar]
- 18.Moul JW. Prostate specific antigen only progression of prostate cancer. J Urol. 2000;163(6):1632–42. [PubMed] [Google Scholar]
- 19.Scher HI, et al. Eligibility and outcomes reporting guidelines for clinical trials for patients in the state of a rising prostate-specific antigen: recommendations from the Prostate-Specific Antigen Working Group. J Clin Oncol. 2004;22(3):537–56. doi: 10.1200/JCO.2004.07.099. [DOI] [PubMed] [Google Scholar]
- 20.D’Amico AV, et al. Surrogate end point for prostate cancer-specific mortality after radical prostatectomy or radiation therapy. J Natl Cancer Inst. 2003;95(18):1376–83. doi: 10.1093/jnci/djg043. [DOI] [PubMed] [Google Scholar]
- 21.Pound CR, et al. Natural history of progression after PSA elevation following radical prostatectomy. Jama. 1999;281(17):1591–7. doi: 10.1001/jama.281.17.1591. [DOI] [PubMed] [Google Scholar]
- 22.Patel A, et al. Recurrence patterns after radical retropubic prostatectomy: clinical usefulness of prostate specific antigen doubling times and log slope prostate specific antigen. J Urol. 1997;158(4):1441–5. doi: 10.1016/s0022-5347(01)64238-1. [DOI] [PubMed] [Google Scholar]
- 23.Zagars GK, Pollack A. Kinetics of serum prostate-specific antigen after external beam radiation for clinically localized prostate cancer. Radiother Oncol. 1997;44(3):213–21. doi: 10.1016/s0167-8140(97)00123-0. [DOI] [PubMed] [Google Scholar]
- 24.D’Amico AV, et al. Determinants of prostate cancer-specific survival after radiation therapy for patients with clinically localized prostate cancer. J Clin Oncol. 2002;20(23):4567–73. doi: 10.1200/JCO.2002.03.061. [DOI] [PubMed] [Google Scholar]
- 25.Smith MR, et al. Celecoxib versus placebo for men with prostate cancer and a rising serum prostate-specific antigen after radical prostatectomy and/or radiation therapy. J Clin Oncol. 2006;24(18):2723–8. doi: 10.1200/JCO.2005.03.7804. [DOI] [PubMed] [Google Scholar]
- 26.Smith MR, et al. Rosiglitazone versus placebo for men with prostate carcinoma and a rising serum prostate-specific antigen level after radical prostatectomy and/or radiation therapy. Cancer. 2004;101(7):1569–74. doi: 10.1002/cncr.20493. [DOI] [PubMed] [Google Scholar]
- 27.Antonarakis E, Lin J, Keizman D, Carducci MA, Eisenberger MA. The effect of changes in PSA kinetics on metastasis-free survival (MFS) in patients with PSA-recurrent prostate cancer (PC) treated with nonhormonal agents: Combined analysis of three randomized trials. J Clin Oncol. 2010;28:15s. (suppl; abstr 4549) [Google Scholar]
- 28.Zhang Y, et al. Overexpression of copper zinc superoxide dismutase suppresses human glioma cell growth. Cancer Res. 2002;62(4):1205–12. [PubMed] [Google Scholar]
- 29.Yoshizaki N, et al. Suppressive effect of recombinant human Cu, Zn-superoxide dismutase on lung metastasis of murine tumor cells. Int J Cancer. 1994;57(2):287–92. doi: 10.1002/ijc.2910570226. [DOI] [PubMed] [Google Scholar]
- 30.Satomi A, et al. Significance of superoxide dismutase (SOD) in human colorectal cancer tissue: correlation with malignant intensity. J Gastroenterol. 1995;30(2):177–82. doi: 10.1007/BF02348662. [DOI] [PubMed] [Google Scholar]
- 31.Svensk AM, et al. Differential expression of superoxide dismutases in lung cancer. Am J Clin Pathol. 2004;122(3):395–404. doi: 10.1309/A45Q-HB0Q-RRX6-CT9A. [DOI] [PubMed] [Google Scholar]
- 32.Crnogorac-Jurcevic T, et al. Expression profiling of microdissected pancreatic adenocarcinomas. Oncogene. 2002;21(29):4587–94. doi: 10.1038/sj.onc.1205570. [DOI] [PubMed] [Google Scholar]
- 33.Janssen AM, et al. Superoxide dismutases in relation to the overall survival of colorectal cancer patients. Br J Cancer. 1998;78(8):1051–7. doi: 10.1038/bjc.1998.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Janssen AM, et al. Superoxide dismutases in gastric and esophageal cancer and the prognostic impact in gastric cancer. Clin Cancer Res. 2000;6(8):3183–92. [PubMed] [Google Scholar]
- 35.Kinnula VL, Crapo JD. Superoxide dismutases in malignant cells and human tumors. Free Radic Biol Med. 2004;36(6):718–44. doi: 10.1016/j.freeradbiomed.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 36.Uauy R, et al. Red cell superoxide dismutase activity as an index of human copper nutrition. J Nutr. 1985;115(12):1650–5. doi: 10.1093/jn/115.12.1650. [DOI] [PubMed] [Google Scholar]
- 37.Milne DB. Copper intake and assessment of copper status. Am J Clin Nutr. 1998;67(5 Suppl):1041S–1045S. doi: 10.1093/ajcn/67.5.1041S. [DOI] [PubMed] [Google Scholar]