

Abstract
The apoA-I molecule adopts a two-domain tertiary structure and the properties of these domains modulate the ability to form HDL particles. Thus, human apoA-I differs from mouse apoA-I in that it can form smaller HDL particles; the C-terminal α-helix is important in this process and human apoA-I is unusual in containing aromatic amino acids in the non-polar face of this amphipathic α-helix. To understand the influence of these aromatic amino acids and the associated high hydrophobicity, apoA-I variants were engineered in which aliphatic amino acids were substituted with or without causing a decrease in overall hydrophobicity. The variants human apoA-I (F225L/F229A/Y236A) and apoA-I (F225L/F229L/A232L/Y236L) were compared to wild-type (WT) apoA-I for their abilities to (1) solubilize phospholipid vesicles and form HDL particles of different sizes, and (2) mediate cellular cholesterol efflux and create nascent HDL particles via ABCA1. The loss of aromatic residues and concomitant decrease in hydrophobicity in apoA-I (F225L/F229A/Y236A) has no effect on protein stability, but reduces by a factor of about three the catalytic efficiencies (Vmax/Km) of vesicle solubilization and cholesterol efflux; also, relatively large HDL particles are formed. With apoA-I (F225L/F229L/A232L/Y236L) where the hydrophobicity is restored by the presence of only leucine residues in the helix non-polar face, the catalytic efficiencies of vesicle solubilization and cholesterol efflux are similar to those of WT apoA-I; this variant forms smaller HDL particles. Overall, the results show that the hydrophobicity of the non-polar face of the C-terminal amphipathic α-helix plays a critical role in determining apoA-I functionality but aromatic amino acids are not required.
Keywords: ATP Binding Cassette Transporter A1 (ABCA1), amphipathic α-helix, apolipoprotein A-I, cellular cholesterol efflux, high density lipoprotein (HDL), lipid solubilization
1. Introduction
A major reason why high density lipoprotein (HDL) exerts anti-atherogenic effects is that this lipoprotein mediates the reverse transport of excess cholesterol from peripheral tissues to the liver for eventual excretion from the body [1–2]. Plasma HDL is heterogeneous in that it exists as a collection of functionally distinct subspecies which, for example, differ in their ability to promote efflux of cholesterol from cells [2]. Consequently, the protective action of HDL against cardiovascular disease is not simply a function of its concentration in the circulation but also of its “quality”, as reflected in the proportions of the various subspecies present. In light of these issues, there is a need to understand the structural basis for HDL heterogeneity and, in particular, how the structure of the principal HDL protein, apolipoprotein (apo) A-I, affects the distribution of nascent HDL particles created by the interaction of apoA-I with the ATP-binding cassette transporter A1 (ABCA1) [1, 3].
The question of how apoA-I structure affects ABCA1-mediated cellular cholesterol efflux is a natural extension of the many pioneering contributions of Dr. John Oram and colleagues to our understanding of cholesterol efflux and the ABCA1 system. The realization that ABCA1 plays a critical role in cellular cholesterol efflux and HDL biogenesis rests upon the twin discoveries (1) by Oram and colleagues in 1995 that Tangier disease is associated with impaired removal of cellular cholesterol and phospholipid by apoA-I [4] and (2) by several laboratories independently that mutation of the ABCA1 transporter is the defect in Tangier disease (reviewed in [5]). Oram’s group was among the first to report that ABCA1 controls cholesterol efflux to apoA-I [6] and that the transporter is active in macrophages and promotes apoA-I binding to these cells [7]. The binding sites were characterized by electron microscopy and shown to involve structures that protrude from the cell surface [8]. John Oram’s laboratory also showed that ABCA1 activity redistributes cholesterol to cell surface domains [9] and that interaction of apoA-I with ABCA1 activates signaling molecules (reviewed in [10]). Importantly, these interactions were also demonstrated to be impaired by myeloperoxidase-induced oxidation of apoA-I [3, 11], reflecting the sensitivity of the ABCA1 system to alterations in apoA-I structure.
ABCA1-mediated efflux of cellular cholesterol to apoA-I involves binding involves of apoA-I to exovesiculated plasma membrane domains created by the phospholipid translocase activity of ABCA1 [12]. The subsequent microsolubilization of the lipids in these membrane domains by the detergent-like apoA-I molecule [13] leads to the formation of discoidal nascent HDL particles that contain cholesterol [14]. Both the rate of cholesterol efflux and the size distribution of the nascent HDL particles are sensitive to apoA-I structure. For instance, multiple laboratories have shown that the C-terminal lipid-binding domain of human apoA-I [1, 15] is needed for optimal cholesterol efflux [13, 16–21]. The C-terminal α-helix is the most non-polar segment of the human apoA-I molecule [15] and this hydrophobicity is presumed to be responsible for the major role this helix plays in promoting cholesterol efflux. Consistent with this concept, mouse apoA-I possesses a relatively polar C-terminal domain [22–23] and is less efficient at supporting ABCA1-mediated cholesterol efflux from cells [24]. Furthermore, human apoA-I forms smaller nascent HDL particles than mouse apoA-I suggesting that the properties of the C-terminal domain are particularly important in determining HDL particle size. Other comparisons of human and mouse apoA-I have demonstrated that the human protein can form smaller HDL3 particles whereas the mouse protein cannot [25–27]. The C-terminal α-helix in human apoA-I is more hydrophobic than the equivalent helix in mouse apoA-I because of the presence of aromatic rather than aliphatic amino acid residues in the non-polar face of the helix in the former case [23]. Given that aromatic side chains interact differently than aliphatic side chains with phospholipid (PL) bilayers and promote apolipoprotein anchoring to PL-water interfaces [28–32], we postulated that the aromatic residues (especially the phenylalanine residues) in the C-terminal α-helix are the particular structural feature enabling human apoA-I to efflux cholesterol efficiently and form smaller nascent HDL particles. To test this hypothesis, we replaced the aromatic residues in the C-terminal helix of human apoA-I with aliphatic amino acids and determined the consequences for cholesterol efflux and HDL particle formation. The results indicate that both the rate of cholesterol efflux and the sizes of the HDL particles are sensitive to the hydrophobicity of the apoA-I C-terminal α-helix; higher hydrophobicity (whether achieved by the presence of aromatic or aliphatic amino acids) enhances the rate of lipid solubilization and formation of smaller HDL particles. These findings provide new insight into how apoA-I structure modulates HDL heterogeneity and thereby the functionality of this lipoprotein.
2. Materials and methods
2.1 ApoA-I Variants
As described before, human apoA-I cDNA cloned into the pET32a(+) vector from Novagen was used to express wild-type (WT) human apoA-I protein as a His-tagged thioredoxin fusion protein [33–34]. A cDNA insert encoding the human apoA-I variant F225L/F229A/Y236A was engineered from the above pET32a(+) plasmid using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, CA) and the following primers: 5′-cctgagcgctctcgaggaggcgactaagaagctcaacac-3′ and 5′-gtgttgagcttcttagtcgcctcctcgagagcgctcagg-3′ for Y236A, 5′-cgtgctggagagcctgaaggtcagcttcctg-3′ and 5′-caggaagctgaccttcaggctctccagcacg-3′ for F225L, and 5′-ggagagcctgaaggtcagcgcgctgagcgctctcgagg-3′ and 5′-cctcgagagcgctcagcgcgctgaccttcaggctctcc-3′ for F229A after the F225L mutation was introduced. Human apoA-I cDNA encoding the variant F225L/F229L/A232L/Y236L was cloned from the pET32a(+) expression vector containing human apoA-I cDNA using the following primer: 5′-cgtgctggagagcctgaaggtcagcctgctgagcctgctcgaggagctgactaagaagc-3′ with the QuikChange Multi Site-directed Mutagenesis kit (Stratagene, CA).
The human apoA-I variants encoded by these cDNA inserts were expressed in E. coli strain BL21-DE3 and purified according to previously published procedures [33–34]. Cleavage of the thioredoxin fusion protein with thrombin leaves the target apoA-I with two extra amino acids, Gly-Ser, at the amino terminus. The apoA-I preparations were at least 95% pure as assessed by SDS-PAGE and protein concentrations were determined by a Lowry procedure [35] or absorbance at 280nm [21].
2.2 Spectroscopy
Far-UV CD spectra to determine the α-helix contents of the apoA-I variants were obtained as described before [33, 36] using a Jasco 810 spectropolarimeter; the α-helix content was calculated from the molar ellipticity at 222nm [37]. The parameters describing the reversible thermal denaturation were obtained by monitoring the molar ellipticity at 222nm while heating the apoA-I sample at 10°/min over the temperature range 20–90°C [38]. The fluorescence emission spectra from 300 to 400nm of the tryptophan residues in apoA-I samples treated with increasing concentrations of guanidine hydrochloride (GdnHCl) were used to calculate the free energy and midpoint of denaturation, as described before [33]. To monitor the exposure of hydrophobic surface, 8-anilino-1-napthalenesulfonic acid (ANS) fluorescence spectra were collected from 400 to 600nm at an excitation wavelength of 395nm in the absence and presence of the apoA-I variants and analyzed as described previously [33].
2.3 Lipid Interactions
The abilities of the apoA-I variants to solubilize dimyristoyl phosphatidylcholine (DMPC) multilamellar vesicles (MLV) were compared by monitoring the decrease in absorbance at 325nm [39]. The 10 min timecourses were fitted to a mono-exponential decay equation. The 10 min decrease in absorbance was measured as a function of apoA-I concentration to obtain Km and Vmax values. In order to evaluate the effects of apoA-I C-terminal α-helix nonpolar face hydrophobicity on reconstituted HDL particle size, appropriate amounts of DMPC and cholesterol (5 mol %) in chloroform were dried, dispersed in Tris-buffered saline (pH 7.4) with vortexing and subjected to three temperature cycles between 0 and 37°C to ensure complete hydration. The resultant MLV were incubated at a 2/1 w/w lipid/protein ratio with apoA-I for 20 h at 26°C. The size-distributions of the discoidal HDL particles created [40] were analyzed by native 4–12% PAGE [24].
2.4 Cholesterol efflux from cells
Baby hamster kidney (BHK) cells expressing high levels of human ABCA1 [9, 41] (kindly provided by Dr. John Oram) were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (FBS) and 50 μg/ml gentamicin. For cholesterol efflux assays, fully confluent cell cultures were diluted 1/7 – 1/10, seeded in 24-well plates in the above medium and allowed to attach overnight, kept in DMEM/2.5% FBS/50 μg/ml gentamicin supplemented with [3H]cholesterol to 1 μCi/ml over the following night to label cellular cholesterol pools with the tracer and incubated in DMEM/0.2% BSA +/− 10nM mifepristone for 18–19h over the third night to induce ABCA1 expression. Thereafter, cells were washed once with MEM-Hepes and allowed to release lipids in DMEM/50 μg/ml gentamicin +/−mifepristone +/−apoA-I for 4h. At the end of the cholesterol efflux period, cell medium was collected and filtered using 0.45 μm filter plates, and cellular lipids were extracted with hexane/isopropanol (3:2 v/v) and precipitated in a scintillation vial by evaporating the solvent. [3H]cholesterol cpm in a fraction of the medium and in the lipid precipitate were determined by liquid scintillation counting. Cellular cholesterol efflux as %/4h was calculated by multiplying [3H]cholesterol cpm in the medium by 100 and dividing the product by the sum of [3H]cholesterol cpm values in the medium and cells. Cellular cholesterol efflux to apoA-I was calculated by subtracting the average % of cellular cholesterol efflux in −mifepristone/+apoA-I cells from the individual % values of cellular cholesterol efflux in +mifepristone/+apoA-I cells. Km and Vmax values were calculated by fitting the cholesterol efflux values obtained at different concentrations of apoA-I to the Michaelis-Menten equation (Graphpad Prism 4.0). The size distributions of nascent HDL particles present in the extra-cellular medium at the end of the efflux period were determined by concentrating the medium and subjecting it to native 4–12% PAGE, as described before [14, 24].
3. Results
The hydrophobicity of the C-terminal α-helix of human apoA-I was manipulated by altering the amino acid composition of the non-polar face of the amphipathic helix and the mutations that were introduced are summarized in Figure 1. The sequence of residues 220–241 of WT human apoA-I is shown as a helical wheel in Fig. 1A and the corresponding hydrophobicity parameters are listed in Table 1. It is apparent that this sequence is significantly more nonpolar than the equivalent region of mouse apoA-I (cf. Fig. 1D and Table 1); the reason for this is the presence of three aromatic residues F225, F229 and Y236 in the human peptide (shaded residues in Fig. 1A) that are not present in the mouse counterpart. To explore the influence of these aromatic amino acids on the properties of apoA-I, the mutations F225L/F229A/Y236A were introduced to give the sequence depicted in Fig. 1B; this human apoA-I variant contains the same residues as mouse apoA-I does at the equivalent positions (cf. the helical wheels in Fig. 1B and D). These mutations reduce the total hydrophobicity per residue of the peptide to −1.71 kcal/mol which is similar to the value of −1.76 kcal/mol seen for the equivalent segment of the WT mouse apoA-I molecule. To distinguish the effects of the reduction in hydrophobicity from any specific effects of deleting aromatic amino acids, the human apoA-I variant F225L/F229L/A232L/Y236L was created to eliminate the reduction in hydrophobicity. These mutations introduce four additional leucine residues into the sequence so that the entire nonpolar face of the amphipathic helix comprises leucine residues (Fig. 1C). As a consequence, both the total hydrophobicity and the hydrophobicity of the helix nonpolar face are greater than the values of these parameters for WT human apoA-I (220–241) (Table 1).
Figure 1.
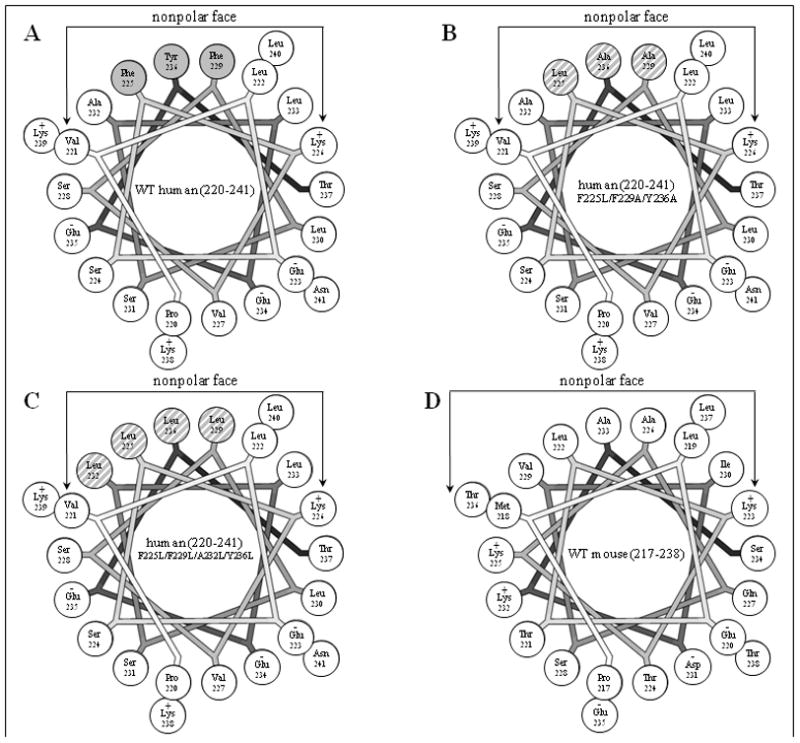
Helical wheel projections of the C-terminal amphipathic α-helical region of apoA-I variants. A. Residues 220–241 of WT human apoA-I; aromatic residues in the nonpolar face are shaded grey. B. Residues 220–241 of human apoA-I (F225L/F229A/Y236A); the mutated residues in the nonpolar face are grey-hatched. C. Residues 220–241 of human apoA-I (F225L/F229L/A232L/Y236L); the mutated residues in the nonpolar face are grey-hatched. D. Residues 217–238 of WT mouse apoA-I (this is the equivalent segment to residues 220–241 in human apoA-I because the mouse protein is three residues shorter [23]). The helical wheels are drawn with the Wheel program [54].
Table 1.
ApoA-I C-terminal α-helix parameters
| α-helixa | Total hydrophobicity/residueb,c (kcal/mol) | Hydrophobic moment/residueb (kcal/mol) | Hydrophobicity of nonpolar face/residueb,c (kcal/mol) |
|---|---|---|---|
| WT human apoA-I (220–241) | −1.48 | 1.50 | 2.31 |
| human apoA-I (220–241) F225L/F229A/Y236A | −1.71 | 1.28 | 1.60 |
| human apoA-I (220–241) F225L/F229L/A232L/Y236L | −1.33 | 1.62 | 2.80 |
| WT mouse apoA-I (217–238) | −1.76 | 1.62 | 1.83 |
3.1 Structural characterization of apoA-I variants with altered C-terminal helix hydrophobicity
The mutations in the C-terminal helix of human apoA-I summarized in Fig. 1B and C do not significantly modify the α-helix content of the protein but they do alter the stability of the apoA-I molecule (Table 2). Thus, measurements of molar ellipticity at 222 nm across the temperature range 20–90°C (Fig. 2) show that the mutations F225L/F229A/Y236A and F225L/F229L/A232L/Y236L reduce the Tm of 59°C for WT apoA-I by 2–3°C (Table 2). Interestingly, the mutations F225L/F229A/Y236A and F225L/F229L/A232L/Y236L exert opposite effects on the cooperativity of thermal unfolding; the former mutations increase the Van’t Hoff enthalpy by 11 kcal/mol whereas the latter mutations decrease it to a similar extent (Table 2). The GdnHCl and urea denaturation data summarized in Fig. 3 and Table 3 confirm that whereas the F225L/F229A/Y236A mutation has no significant effect on apoA-I stability, the mutation F225L/F229L/A232L/Y236L significantly destabilizes the protein and reduces the cooperativity (m value) of unfolding. As reported before, the free energy of unfolding of apoA-I measured by urea denaturation is higher than the value derived by GdnHCl denaturation [22, 42]. Consistent with the F225L/F229L/A232L/Y236L mutations having a relatively large effect on apoA-I stability, more hydrophobic surface is exposed in this variant giving rise to somewhat enhanced ANS binding relative to the binding seen with WT apoA-I (Fig. 4 and Table 2). In comparison, this increase in ANS binding is less than that caused by mutations in the N-terminal domain that directly disrupt helix packing within the helix bundle [36, 43]. In contrast, the reduction in hydrophobicity associated with the mutation F225L/F229A/Y236A reduces ANS binding by a factor of 2. These observations agree with prior reports that the C-terminal α-helical region of apoA-I is involved in creating the hydrophobic ANS binding site [33, 36].
Table 2.
α-Helix content and parameters of thermal denaturation and ANS binding of human apoA-I C-terminal variants
| apoA-I variant | α-Helixa (%) | Tmb (°C) | ΔHvc (kcal/mol) | ANS fluorescenced |
|---|---|---|---|---|
| WT | 52 ± 1 | 59 | 33 | 1.0 |
| F225L/F229A/Y236A | 52 ± 2 | 57 | 44 | 0.5 |
| F225L/F229L/A232L/Y236L | 55 ± 4 | 56 | 23 | 1.1 |
Mean ± SD from at least three experiments.
The reproducibility in midpoint temperature Tm is ± 1.5°C.
Van’t Hoff enthalpy. Estimated error is ± 0.5kcal/mol.
Values are relative to WT. Estimated error is within ± 0.1.
Figure 2.
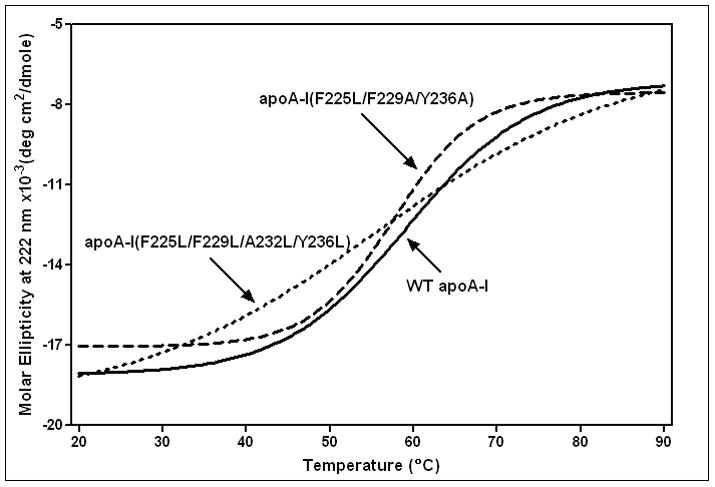
Thermal unfolding of WT human apoA-I and C-terminal α-helix variants monitored by the molar ellipticity at 222nm. The protein concentrations were 50μg/ml.
Figure 3.
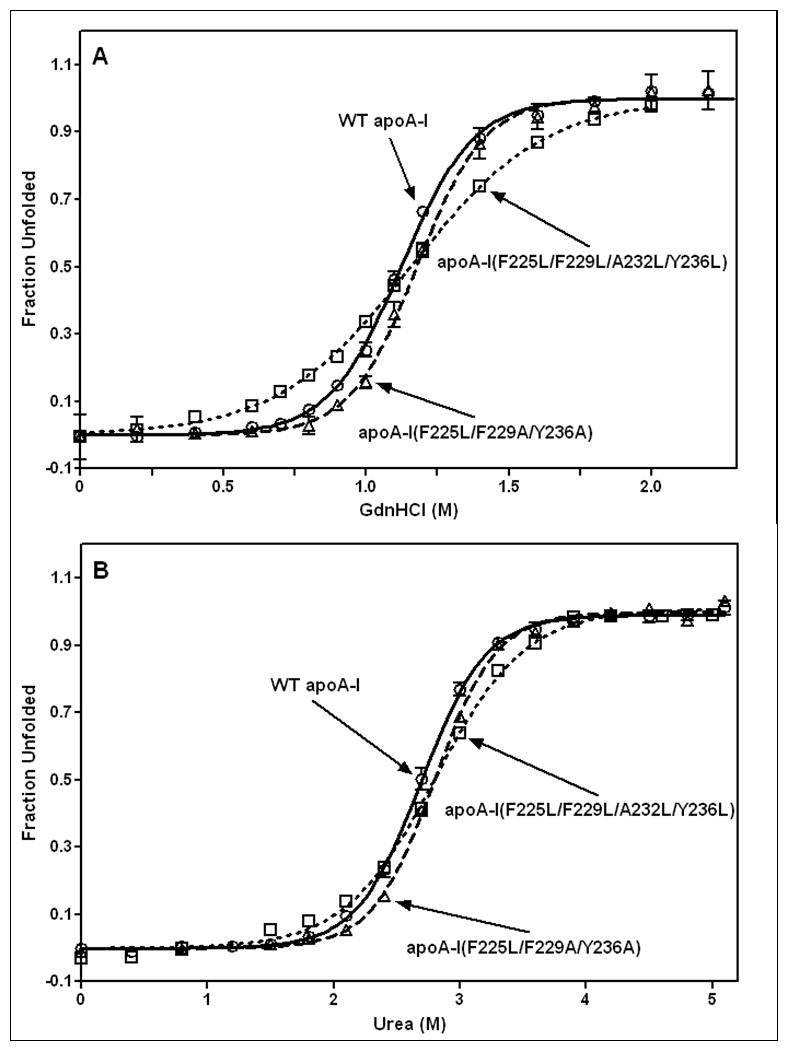
GdnHCl-induced (A) and urea-induced (B) denaturation of human apoA-I variants monitored by Trp fluorescence. WT apoA-I (o, solid line); apoA-I (F225L/F229A/Y236A) (Δ, dashed line); apoA-I (F225L/F229L/A232L/Y236L) (□, dotted line).
Table 3.
Thermodynamic parameters for denaturation of apoA-I variants monitored by tryptophan fluorescence
| apoA-I variant | GdnHCl-denaturation
|
urea-denaturation
|
||||
|---|---|---|---|---|---|---|
| ΔG°D (kcal/mol) | m (kcal/mol)M−1 | D½ (M) | ΔG°D (kcal/mol) | m (kcal/mol)M−1 | D½ (M) | |
| WT | 5.0 ± 0.1 | 4.4 ± 0.1 | 1.14 ± 0.05 | 5.9 ± 0.2 | 2.2 ± 0.1 | 2.7 ± 0.2 |
| F225L/F229A/Y236A | 5.4 ± 0.3 | 4.6 ± 0.2 | 1.18 ± 0.07 | 6.1 ± 0.2 | 2.2 ± 0.1 | 2.8 ± 0.1 |
| F225L/F229L/A232L/Y236L | 2.9 ± 0.1 | 2.5 ± 0.1 | 1.16 ± 0.03 | 4.8 ± 0.2 | 1.7 ± 0.1 | 2.8 ± 0.1 |
Figure 4.
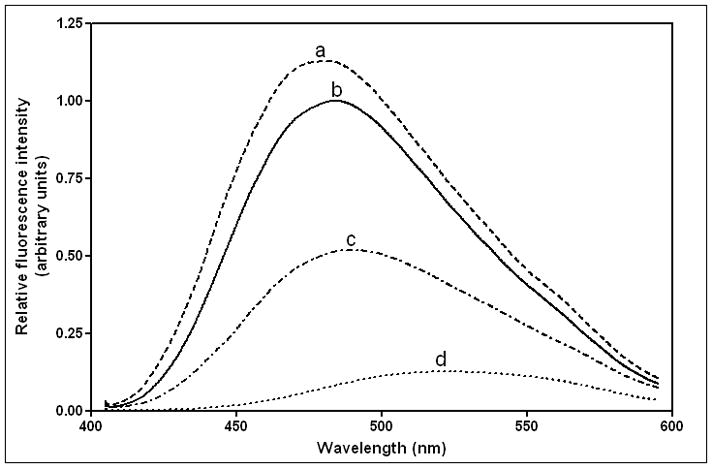
Fluorescence spectra of ANS (250 μM) in the presence of 50 μg/ml human apoA-I variants. (a) apoA-I (F225L/F229L/A232L/Y236L), (b) WT apoA-I, (c) apoA-I (F225L/F229A/Y236A), (d) free ANS.
3.2 Effects of altering C-terminal helix hydrophobicity on the lipid-solubilizing properties of apoA-I
It is well-established that apoA-I can effectively solubilize DMPC MLV to create discoidal HDL particles (for reviews, see [1, 44–45]). The concentration-dependence of MLV clearance by WT apoA-I shown in Fig. 5 is consistent with prior reports [22, 39, 42–43]; fitting these data to the Michaelis-Menten equation yields Vmax and Km values. The catalytic efficiency (Vmax/Km) for WT apoA-I for this process is 0.018 (fraction turbidity cleared in 10min) (μg apoA-I/ml)−1. This value is normalized to one for the purposes of comparison and, on this basis, the reduction in hydrophobicity introduced by the mutation F225L/F229A/Y236A reduces the relative catalytic efficiency to 0.3. Restoration of the C-terminal helix hydrophobicity by forming the variant apoA-I (F225L/F229L/A232L/Y236L) restores the ability to effectively solubilize DMPC MLV (Fig. 5) and the relative catalytic efficiency for this variant is 1.3.
Figure 5.
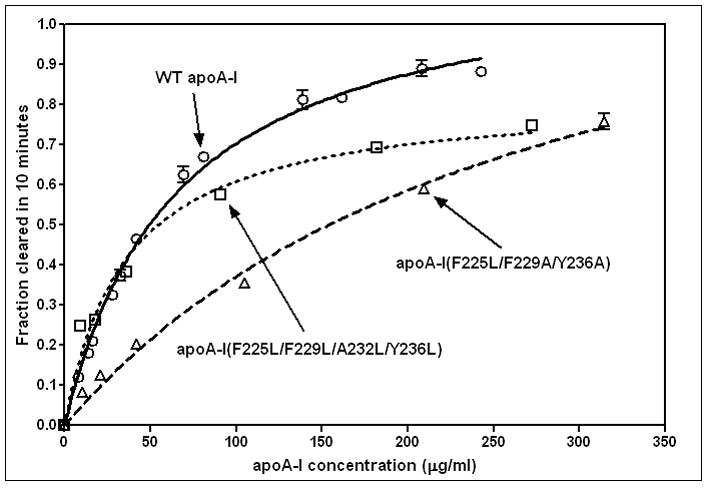
Effect of C-terminal α-helix nonpolar face hydrophobicity on the ability of apoA-I to solubilize DMPC MLV. The fractional decrease in absorbance of the MLV suspension (0.25mg DMPC/ml) at 325nm in 10min by different concentrations of apoA-I was measured as described in Materials and Methods. The data were fitted to the Michaelis-Menten equation to obtain the Vmax and Km values. WT apoA-I (o, solid line); apoA-I (F225L/F229A/Y236A) (Δ, dashed line); apoA-I (F225L/F229L/A232L/Y236L) (□, dotted line).
Previously, we have shown that the sizes of both nascent HDL and reconstituted HDL (formed by solubilization of PL MLV) are sensitive to alterations in apoA-I structure [22, 24]. For example, human and mouse apoA-I form HDL particles of different sizes, with human apoA-I favoring smaller particles. The results in Fig. 6 demonstrate how the hydrophobicity of the C-terminal α-helix contributes to this effect. When WT human apoA-I interacts with DMPC/5 mol% cholesterol MLV under the conditions chosen, the predominant HDL particle created has a hydrodynamic diameter of 13nm and there are minor species in the 9–11nm range (Fig. 6 Lane 1). Reducing the hydrophobicity of the C-terminal helix to create apoA-I (F225L/F229A/Y236A) leads to formation of 14nm and larger particles (Lane 2) while increasing the hydrophobicity by introducing leucine residues to give apoA-I (F225L/F229L/A232L/Y236L) reduces HDL size (the major species have diameters in the 12–13nm range and smaller 9nm particles are formed (Lane 3)).
Figure 6.

Native polyacrylamide 4–12% gradient gels stained with Coomassie Brilliant Blue comparing the sizes of discoidal complexes formed by incubation of DMPC/5 mol % cholesterol MLV with human apoA-I variants. The migration positions of standard proteins of known hydrodynamic diameters are indicated on the left: Lane 1, WT apoA-I; Lane 2, apoA-I (F225L/F229A/Y236A); Lane 3, apoA-I (F225L/F229L/A232L/Y236L). Optical scans of lanes 1–3 are shown at the bottom of the figure.
3.3 ABCA1-mediated cellular cholesterol efflux to apoA-I variants with altered C-terminal helix hydrophobicity
Since the ability of apoA-I to accept cholesterol from ABCA1-expressing cells and create nascent HDL particles depends on its lipid-solubilizing capability (for reviews, see [1, 3, 10]), we examined how sensitive this function of apoA-I is to the alterations in C-terminal helix sequence and hydrophobicity summarized in Fig. 1 and Table 1. Fig. 7 shows the effects of the mutations on the kinetics of cholesterol efflux from BHK cells (Vmax and Km values [24]) parallel their effects on the kinetics of DMPC MLV solubilization (Fig. 5). Thus, the mutation F225L/F229A/Y236A reduces the ability to mediate cholesterol efflux (relative catalytic efficiency = 0.4) compared to the value of 1.0 for WT apoA-I. Restoration of the hydrophobicity of the C-terminal helix by introducing the mutations F225L/F229L/A232L/Y236L restores the ability to support ABCA1-mediated cholesterol efflux (relative catalytic efficiency = 0.9).
Figure 7.
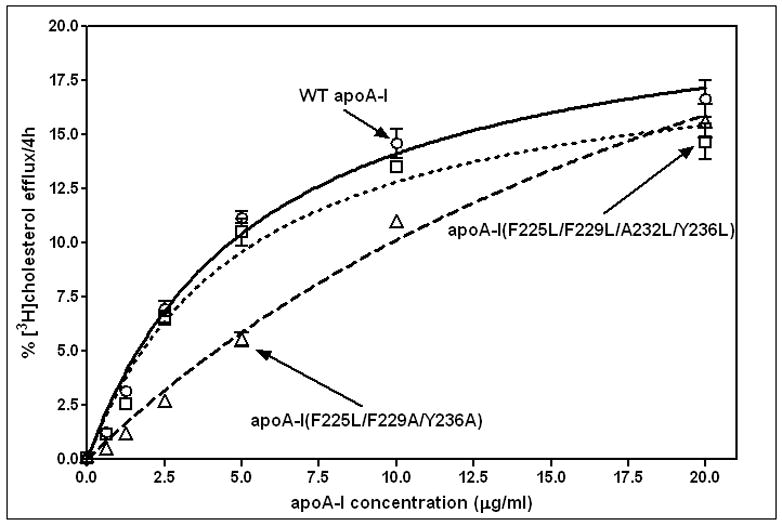
Effects of apoA-I C-terminal α-helix nonpolar face hydrophobicity on the concentration-dependence of cholesterol efflux via ABCA1 from BHK cells. The efflux of [3H]cholesterol in 4h to different concentrations of apoA-I variants was measured as described in Materials and Methods. The Vmax and Km values were calculated by fitting the data to the Michaelis-Menten equation. WT apoA-I (o, solid line); apoA-I (F225L/F229A/Y236A) (Δ, dashed line); apoA-I (F225L/F229L/A232L/Y236L) (□, dotted line).
The effects of altering C-terminal helix hydrophobicity on the size distribution of nascent HDL particles created when the apoA-I variants are incubated with ABCA1-expressing cells are summarized in Fig. 8. As was observed in the DMPC MLV solubilization experiments (Fig. 6), introduction of the mutations F225L/F229A/Y236A into human apoA-I alters the nascent HDL particle size distribution; in this case, formation of the major population migrating as a band at ~ 12nm diameter is prevented (cf. Fig. 8, Lanes 1 and 2). In contrast, introduction of the mutations F225L/F229L/A232L/Y236L restores formation of this size of nascent HDL particle (Fig. 8, Lane 3).
Figure 8.

Blots stained with anti-apoA-I of native polyacrylamide 4–12% gradient gels of medium collected after incubation of BHK cells with apoA-I variants. The cholesterol efflux experiments were performed as described in Fig. 7 and the concentrated medium containing nascent HDL particles was subjected to electrophoresis in the gradient gel. The migration positions of standard proteins of known hydrodynamic diameters are indicated on the left: Lane 1, WT human apoA-I; Lane 2, human apoA-I (F225L/F229A/Y236A); Lane 3, human apoA-I (F225L/F229L/A232L/Y236L).
4. Discussion
The anti-atherogenic properties of HDL are a function of both the quantity and quality of this lipoprotein in circulation [46]. The quality of HDL, for example with respect to its ability to promote cellular cholesterol efflux, is affected by the relative amounts of HDL subspecies present [2]. Thus, it is important to understand the factors that modulate HDL heterogeneity. It is known that nascent HDL particles are heterogeneous with respect to size and composition [14, 47–50] and here we extend prior work on this topic by examining the influence of apoA-I C-terminal α-helix hydrophobicity on nascent HDL particle formation. For reasons that are not understood fully, variations in the properties of the apoA-I N- and C-terminal tertiary structure domains [22, 33] are associated with formation of HDL particles of different sizes [22]. As mentioned in the Introduction, the C-terminal domain of human apoA-I is more hydrophobic than that of mouse apoA-I but it is not known how this structural feature contributes to the different properties of the two apoA-I molecules, especially with respect to ability to mediate cellular cholesterol efflux and HDL particle formation. It is noteworthy that human apoA-I contains aromatic residues in the C-terminal α-helix whereas mouse apoA-I does not (cf. Fig. 1A, D). The presence of three aromatic residues in positions 225, 229 and 236 (Fig. 1) is a feature of primate apoA-I, with aliphatic amino acids being present at these sites in most other mammalian apoA-I [23]. Importantly, both the presence of aromatic residues [29, 31] and their positioning in the nonpolar face of amphipathic α-helices [30, 32] can modulate apolipoprotein-PL membrane interactions. Here, we investigate how the presence of aromatic residues in the C-terminal α-helix affects the HDL-forming properties of the apoA-I molecule.
4.1 Influence of C-terminal helix hydrophobicity on apoA-I structure
The C-terminal α-helix in apoA-I is critical for lipid binding because it mediates the initial interaction [15, 33]. The segment spanning residues 220–241 is the most non-polar part of the human apoA-I molecule [15] so it is unsurprising that hydrophobic surface exposure (Fig. 4) and lipid-solubilization ability (Fig. 5) are sensitive to mutations in the non-polar face of the C-terminal amphipathic α-helix. Importantly, the data in Fig. 2 and 3 indicate that mutations in the C-terminal α-helix not only exert local effects but also affect the properties of the separately folded N-terminal helix bundle domain. Since the tryptophan residues in human apoA-I are all located in the helix bundle domain (at positions 8, 50, 72 and 108), the differences in free energy of stabilization derived from tryptophan fluorescence measurements (Table 3) reflect alterations in helix bundle conformation [42]. It is apparent that the C-terminal helix interacts with the N-terminal helix bundle and that this domain-domain interaction affects the ability of the bundle to unfold. Substitution of the aromatic residues at positions 225, 229 and 236 and the concomitant decrease in hydrophobicity in the apoA-I variant (F225L/F229A/Y236A) has little effect on helix bundle stability (Table 3); it follows that aromatic residues in the C-terminal α-helix of WT human apoA-I do not play an essential role in the domain-domain interaction. In contrast, increasing the hydrophobicity by locating leucine residues at all positions in the helix nonpolar face (Fig. 1C) (variant apoA-I (F225L/F229L/A232L/Y236L)) interferes with helix bundle packing and destabilizes the bundle (Table 3). The fact that the free energy of unfolding measured by GdnHCl denaturation (where electrostatic interactions between amino acids are shielded by the ionic denaturant [51]) is lower for the apoA-I variant than for WT apoA-I is consistent with nonpolar interactions in the helix bundle being perturbed by the increased hydrophobicity of the nonpolar face of the C-terminal amphipathic α-helix. This change in the C-terminal α-helix also reduces the free energy of unfolding somewhat when urea is the denaturant (Table 3) indicating that electrostatic interactions (which are not shielded when urea is the denaturant [51]) in the helix bundle are also reduced by modification of the hydrophobicity of the C-terminal helix. The alterations in N-terminal helix bundle packing and stability presumably arise because the positioning of the two domains with respect to one another in the apoA-I molecule is modified when the hydrophobicity of the C-terminal helix nonpolar face is changed.
4.2 Influence of apoA-I C-terminal helix hydrophobicity on lipid solubilization and HDL particle formation
As mentioned earlier, the presence of aromatic residues can enhance the lipid binding properties of amphipathic α-helices and the consequences of removing these residues from the C-terminal helix of WT apoA-I appear consistent with this concept. Thus, the ability of the variant apoA-I (F225L/F229A/Y236A) to solubilize DMPC MLV and mediate cellular cholesterol efflux is diminished (Fig. 5, 7). Furthermore, this variant cannot form HDL particles as small as those created by WT human apoA-I when reconstituted from DMPC MLV (Fig. 6). Also, the size distribution of nascent HDL particles formed when this variant is incubated with ABCA1-expressing cells is different to that formed by WT apoA-I (Fig. 8). However, the requirement for aromatic residues is not absolute because restoration of hydrophobicity by substituting aliphatic leucine residues in the nonpolar face of the helix (variant apoA-I (F225L/F229L/A232L/Y236L)) is sufficient to essentially restore the HDL-forming properties to those of WT human apoA-I (Fig. 5–8). Apparently, if the nonpolar face is sufficiently hydrophobic, the helix can insert effectively into defects in a PL bilayer and destabilize the membrane leading to fragmentation and formation of discoidal HDL particles [1, 13, 39–40]. The enhanced insertion and higher surface concentration of apoA-I that occurs when the C-terminal helix is made more hydrophobic leads to formation of smaller HDL particles (Fig. 6). This effect presumably explains why human apoA-I forms HDL2 and HDL3 in plasma whereas mouse apoA-I forms a population of larger HDL particles [22, 25].
In summary and conclusion, the hydrophobicity of the C-terminal helix in human apoA-I is high and this enables this protein to solubilize PL vesicles efficiently and form smaller HDL particles than those formed by mouse apoA-I. Small reductions in C-terminal helix hydrophobicity are sufficient to slow the rate of solubilization and prevent formation of smaller HDL particles in the HDL3 size range. The relatively high hydrophobicity of the segment spanning residues 220–241 of human apoA-I is due to the presence in the non-polar face of the amphipathic helix of aromatic amino acids which are not present in the corresponding segment of apoA-I from mammals other than primates. In principle, relative to aliphatic amino acids, aromatic amino acids can modulate helix-PL interactions differently due to two effects, (1) their intrinsically high hydrophobicity and (2) the presence of pi electrons in the aromatic side-chains. The present results show that replacement of aromatic residues without maintaining the high hydrophobicity characteristic of the C-terminal helix of human apoA-I leads to a loss of lipid-solubilizing ability, but replacement while maintaining the high hydrophobicity does not affect functionality. It follows that it is the high intrinsic hydrophobicity rather than aromaticity per se of the phenylalanine residues in the C-terminal α-helix of human apoA-I that confers different lipoprotein-forming properties on human apoA-I. Overall, the results support the concept that increasing the C-terminal α-helix hydrophobicity is likely to enhance the ability of apoA-I to promote reverse cholesterol transport (cf. [52]).
Highlights.
Apolipoprotein (apo) A-I structure and function.
ApoA-I interacts with ABCA1 to mediate efflux of cellular cholesterol.
ApoA-I solubilizes lipids to form HDL particles.
Increased apoA-I α-helix hydrophobicity enhances formation of smaller HDL particles.
Acknowledgments
This work was supported by NIH Grant HL22633, American Heart Association Postdoctoral Fellowship 10POST3630047 (NNL), and Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (22590046).
Abbreviations
- ABCA1
ATP binding cassette transporter A1
- ANS
8-anilino-1-napthalenesulfonic acid
- apo
apolipoprotein
- BHK
baby hamster kidney
- DMPC
dimyristoyl phosphatidylcholine
- FBS
fetal bovine serum
- GdnHCl
guanidine hydrochloride
- MEM
minimum essential medium
- MLV
multilamellar vesicle
- PL
phospholipid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lund-Katz S, Phillips MC. High density lipoprotein structure-function and role in reverse cholesterol transport. Subcell Biochem. 2010;51:183–227. doi: 10.1007/978-90-481-8622-8_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rothblat GH, Phillips MC. High-density lipoprotein heterogeneity and function in reverse cholesterol transport. Curr Opin Lipidol. 2010;21:229–238. doi: 10.1097/mol.0b013e328338472d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oram JF, Heinecke JW. ATP-binding cassette transporter A1: A cell cholesterol exporter that protects against cardiovascular disease. Physiol Rev. 2005;85:1343–1372. doi: 10.1152/physrev.00005.2005. [DOI] [PubMed] [Google Scholar]
- 4.Francis GA, Knopp RH, Oram JF. Defective removal of cellular cholesterol and phospholipids by apolipoprotein A-I in tangier disease. J Clin Invest. 1995;96:78–87. doi: 10.1172/JCI118082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oram JF. Tangier Disease and ABCA1. Biochim Biophys Acta. 2000;1529:321–330. doi: 10.1016/s1388-1981(00)00157-8. [DOI] [PubMed] [Google Scholar]
- 6.Lawn RM, Wade DP, Garvin MR, Wang X, Schwartz K, Porter JG, Seilhamer JJ, Vaughan AM, Oram JF. The Tangier Disease Gene Product ABC1 Controls the Cellular Apolipoprotein-Mediated Lipid Removal Pathway. J Clin Invest. 1999;104:R25–R31. doi: 10.1172/JCI8119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oram JF, Lawn RM, Garvin MR, Wade DP. ABCA1 is the cAMP-inducible Apolipoprotein Receptor that Mediates Cholesterol Secretion from Macrophages. J Biol Chem. 2000;275:34508–34511. doi: 10.1074/jbc.M006738200. [DOI] [PubMed] [Google Scholar]
- 8.Lin G, Oram JF. Apolipoprotein Binding to Protruding Membrane Domains During Removal of Excess Cellular Cholesterol. Atherosclerosis. 2000;149:359–370. doi: 10.1016/s0021-9150(99)00503-1. [DOI] [PubMed] [Google Scholar]
- 9.Vaughan AM, Oram JF. ABCA1 redistributes membrane cholesterol independent of apolipoprotein interactions. J Lipid Res. 2003;44:1373–1380. doi: 10.1194/jlr.M300078-JLR200. [DOI] [PubMed] [Google Scholar]
- 10.Tang C, Oram JF. The cell cholesterol exporter ABCA1 as a protector from cardiovascular disease and diabetes. Biochim Biophys Acta. 2009;1791:563–572. doi: 10.1016/j.bbalip.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 11.Shao B, Tang C, Heinecke JW, Oram JF. Oxidation of apolipoprotein A-I by myeloperoxidase impairs the initial interactions with ABCA1 required for signaling and cholesterol export. J Lipid Res. 2010;51:1849–1858. doi: 10.1194/jlr.M004085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vedhachalam C, Duong PT, Nickel M, Nguyen D, Dhanasekaran P, Saito H, Rothblat GH, Lund-Katz S, Phillips MC. Mechanism of ATP-binding cassette transporter A1-mediated cellular lipid efflux to apolipoprotein A-I and formation of high density lipoprotein particles. J Biol Chem. 2007;282:25123–25130. doi: 10.1074/jbc.M704590200. [DOI] [PubMed] [Google Scholar]
- 13.Gillotte KL, Zaiou M, Lund-Katz S, Anantharamaiah GM, Holvoet P, Dhoest A, Palgunachari MN, Segrest JP, Weisgraber KH, Rothblat GH, Phillips MC. Apolipoprotein-mediated plasma membrane microsolubilization. J Biol Chem. 1999;274:2021–2028. doi: 10.1074/jbc.274.4.2021. [DOI] [PubMed] [Google Scholar]
- 14.Duong PT, Collins HL, Nickel M, Lund-Katz S, Rothblat GH, Phillips MC. Characterization of nascent HDL particles and microparticles formed by ABCA1-mediated efflux of cellular lipids to apoA-I. J Lipid Res. 2006;47:832–843. doi: 10.1194/jlr.M500531-JLR200. [DOI] [PubMed] [Google Scholar]
- 15.Saito H, Lund-Katz S, Phillips MC. Contributions of domain structure and lipid interaction to the functionality of exchangeable human apolipoproteins. Prog Lipid Res. 2004;43:350–380. doi: 10.1016/j.plipres.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 16.Sviridov D, Pyle LE, Fidge N. Efflux of cellular cholesterol and phospholipid to apolipoprotein A-I mutants. J Biol Chem. 1996;271:33277–33282. doi: 10.1074/jbc.271.52.33277. [DOI] [PubMed] [Google Scholar]
- 17.Burgess JW, Frank PG, Franklin V, Liang P, McManus DC, Desforges M, Rassart E, Marcel YL. Deletion of the C-Terminal Domain of Apolipoprotein A-I Impairs Cell Surface Binding and Lipid Efflux in Macrophage. Biochemistry. 1999;38:14524–14533. doi: 10.1021/bi990930z. [DOI] [PubMed] [Google Scholar]
- 18.Panagotopulos SE, Witting SR, Horace EM, Hui DY, Maiorano JN, Davidson WS. The role of apolipoprotein A-I helix 10 in apolipoprotein-mediated cholesterol efflux via the ATP-binding cassette transporter ABCA1. J Biol Chem. 2002;277:39477–39484. doi: 10.1074/jbc.M207005200. [DOI] [PubMed] [Google Scholar]
- 19.Favari E, Bernini F, Tarugi P, Franceschini G, Calabresi L. The C-terminal domain of apolipoprotein A-I is involved in ABCA1-driven phospholipid and cholesterol efflux. Biochem Biophys Res Comm. 2002;299:801–805. doi: 10.1016/s0006-291x(02)02745-6. [DOI] [PubMed] [Google Scholar]
- 20.Chroni A, Liu T, Gorshkova I, Kan HY, Uehara Y, von Eckardstein A, Zannis VI. The central helices of apoA-I can promote ATP-binding cassette transporter AI (ABCA1)-mediated lipid efflux. J Biol Chem. 2003;278:6719–6730. doi: 10.1074/jbc.M205232200. [DOI] [PubMed] [Google Scholar]
- 21.Vedhachalam C, Liu L, Nickel M, Dhanasekaran P, Anantharamaiah GM, Lund-Katz S, Rothblat G, Phillips MC. Influence of apo A-I structure on the ABCA1-mediated efflux of cellular lipids. J Biol Chem. 2004;279:49931–49939. doi: 10.1074/jbc.M406924200. [DOI] [PubMed] [Google Scholar]
- 22.Tanaka M, Koyama M, Dhanasekaran P, Nguyen D, Nickel M, Lund-Katz S, Saito H, Phillips MC. Influence of tertiary structure domain properties on the functionality of apolipoprotein A-I. Biochemistry. 2008;47:2172–2180. doi: 10.1021/bi702332b. [DOI] [PubMed] [Google Scholar]
- 23.Brouillette CG, Anantharamaiah GM, Engler JA, Borhani DW. Structural models of human apolipoprotein A-I: a critical analysis and review. Biochim Biophys Acta. 2001;1531:4–46. doi: 10.1016/s1388-1981(01)00081-6. [DOI] [PubMed] [Google Scholar]
- 24.Vedhachalam C, Chetty PS, Nickel M, Dhanasekaran P, Lund-Katz S, Rothblat GH, Phillips MC. Influence of apolipoprotein (Apo) A-I structure on nascent high density lipoprotein (HDL) particle size distribution. J Biol Chem. 2010;285:31965–31973. doi: 10.1074/jbc.M110.126292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rubin EM, Ishida BY, Clift SM, Krauss RM. Expression of human apolipoprotein A-I in transgenic mice results in reduced plasma levels of murine apolipoprotein A-I and the appearance of two new high density lipoprotein size subclasses. Proc Natl Acad Sci USA. 1991;88:434–438. doi: 10.1073/pnas.88.2.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reschly EJ, Sorci-Thomas M, Davidson WS, Meredith SC, Reardon C, Getz GS. Apolipoprotein A-I alpha-helices 7 and 8 modulate high density lipoprotein subclass distribution. J Biol Chem. 2002;277:9645–9654. doi: 10.1074/jbc.M107883200. [DOI] [PubMed] [Google Scholar]
- 27.Carnemolla R, Ren X, Biswas TK, Meredith SC, Reardon CA, Wang J, Getz GS. The specific amino acid sequence between helices 7 and 8 influences the binding specificity of human apolipoprotein A-I for high density lipoprotein (HDL) subclasses: A potential for HDL preferential generation. J Biol Chem. 2008;283:15779–15788. doi: 10.1074/jbc.M710244200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang G, Pierens GK, Treleaven WD, Sparrow JT, Cushley RJ. Conformations of human apolipoprotein E (263–286) and E (267–289) in aqueous solutions of sodium dodecyl sulfate by CD and 1H NMR. Biochemistry. 1996;35:10358–10366. doi: 10.1021/bi960934t. [DOI] [PubMed] [Google Scholar]
- 29.Datta G, Chaddha M, Hama S, Navab M, Fogelman AM, Garber DW, Mishra VK, Epand RM, Epand RF, Lund-Katz S, Phillips MC, Segrest JP, Anantharamaiah GM. Effects of increasing hydrophobicity on the physical-chemical and biological properties of a class A amphipathic helical peptide. J Lipid Res. 2001;42:1096–1104. [PubMed] [Google Scholar]
- 30.Datta G, Epand RF, Epand RM, Chaddha M, Kirksey MA, Garber DW, Lund-Katz S, Phillips MC, Hama S, Navab M, Fogelman AM, Palgunachari MN, Segrest JP, Anantharamaiah GM. Aromatic residue position on the nonpolar face of class A amphipathic helical peptides determines biological activity. J Biol Chem. 2004;279:26509–26517. doi: 10.1074/jbc.M314276200. [DOI] [PubMed] [Google Scholar]
- 31.James PF, Dogovski C, Dobson RC, Bailey MF, Goldie KN, Karas JA, Scanlon DB, O’Hair RA, Perugini MA. Aromatic residues in the C-terminal helix of human apoC-I mediate phospholipid interactions and particle morphology. J Lipid Res. 2009;50:1384–1394. doi: 10.1194/jlr.M800529-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Handattu SP, Garber DW, Horn DC, Hughes DW, Berno B, Bain AD, Mishra VK, Palgunachari MN, Datta G, Anantharamaiah GM, Epand RM. ApoA-I mimetic peptides with differing ability to inhibit atherosclerosis also exhibit differences in their interactions with membrane bilayers. J Biol Chem. 2007;282:1980–1988. doi: 10.1074/jbc.M606231200. [DOI] [PubMed] [Google Scholar]
- 33.Saito H, Dhanasekaran P, Nguyen D, Holvoet P, Lund-Katz S, Phillips MC. Domain structure and lipid interaction in human apolipoproteins A-I and E: A general model. J Biol Chem. 2003;278:23227–23232. doi: 10.1074/jbc.M303365200. [DOI] [PubMed] [Google Scholar]
- 34.Morrow JA, Arnold KS, Weisgraber KH. Functional characterization of apolipoprotein E isoforms overexpressed in escherichia coli. Protein Expr Purif. 1999;16:224–230. doi: 10.1006/prep.1999.1069. [DOI] [PubMed] [Google Scholar]
- 35.Markwell MAK, Haas SM, Bieber LL, Tolbert NE. A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples. Anal Biochem. 1978;87:206–210. doi: 10.1016/0003-2697(78)90586-9. [DOI] [PubMed] [Google Scholar]
- 36.Tanaka M, Dhanasekaran P, Nguyen D, Ohta S, Lund-Katz S, Phillips MC, Saito H. Contributions of the N- and C-terminal helical segments to the lipid-free structure and lipid interaction of apolipoprotein A-I. Biochemistry. 2006;45:10351–10358. doi: 10.1021/bi060726t. [DOI] [PubMed] [Google Scholar]
- 37.Alexander ET, Tanaka M, Kono M, Saito H, Rader DJ, Phillips MC. Structural and functional consequences of the Milano mutation (R173C) in human apolipoprotein A-I. J Lipid Res. 2009;50:1409–1419. doi: 10.1194/jlr.M800578-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Acharya P, Segall ML, Zaiou M, Morrow JA, Weisgraber K, Phillips MC, Lund-Katz S, Snow JW. Comparison of the stabilities and unfolding pathways of human apolipoprotein E isoforms by differential scanning calorimetry and circular dichroism. Biochim Biophys Acta. 2002;1584:9–19. doi: 10.1016/s1388-1981(02)00263-9. [DOI] [PubMed] [Google Scholar]
- 39.Segall ML, Dhanasekaran P, Baldwin F, Anantharamaiah GM, Weisgraber K, Phillips MC, Lund-Katz S. Influence of apoE domain structure and polymorphism on the kinetics of phospholipid vesicle solubilization. J Lipid Res. 2002;43:1688–1700. doi: 10.1194/jlr.m200157-jlr200. [DOI] [PubMed] [Google Scholar]
- 40.Massey JB, Pownall HJ. Cholesterol is a determinant of the structures of discoidal high density lipoproteins formed by the solubilization of phospholipid membranes by apolipoprotein A-I. Biochim Biophys Acta. 2008;1781:245–253. doi: 10.1016/j.bbalip.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oram JF, Vaughan AM, Stocker R. ATP-binding cassette transporter A1 mediates cellular secretion of alpha-tocopherol. J Biol Chem. 2001;276:39898–39902. doi: 10.1074/jbc.M106984200. [DOI] [PubMed] [Google Scholar]
- 42.Koyama M, Tanaka M, Dhanasekaran P, Lund-Katz S, Phillips MC, Saito H. Interaction between the N- and C-Terminal Domains Modulates the Stability and Lipid Binding of Apolipoprotein A-I. Biochemistry. 2009;48:2529–2537. doi: 10.1021/bi802317v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanaka M, Dhanasekaran P, Nguyen D, Nickel M, Takechi Y, Lund-Katz S, Phillips MC, Saito H. Influence of N-terminal helix bundle stability on the lipid-binding properties of human apolipoprotein A-I. Biochim Biophys Acta. 2011;1811:25–30. doi: 10.1016/j.bbalip.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pownall HJ, Massey JB, Sparrow JT, Gotto AM. Lipid-protein interactions and lipoprotein reassembly. In: Gotto AM, editor. Plasma Lipoproteins. Elsevier Science Publishers B.V.; Amsterdam: 1987. pp. 95–127. [Google Scholar]
- 45.Jonas A. Lipid-binding properties of apolipoproteins. In: Rosseneu M, editor. Structure and function of apolipoproteins. CRC Press; Boca Raton: 1992. pp. 217–250. [Google Scholar]
- 46.Duffy D, Rader DJ. Update on strategies to increase HDL quantity and function. Nat Rev Cardiol. 2009;6:455–463. doi: 10.1038/nrcardio.2009.94. [DOI] [PubMed] [Google Scholar]
- 47.Liu L, Bortnick AE, Nickel M, Dhanasekaran P, Subbaiah PV, Lund-Katz S, Rothblat GH, Phillips MC. Effects of apolipoprotein A-I on ATP-binding cassette transporter A1-mediated efflux of macrophage phospholipid and cholesterol. J Biol Chem. 2003;278:42976–42984. doi: 10.1074/jbc.M308420200. [DOI] [PubMed] [Google Scholar]
- 48.Denis M, Haidar B, Marcil M, Bouvier M, Krimbou L, Genest J., Jr Molecular and cellular physiology of apolipoprotein A-I lipidation by the ATP-binding cassette transporter A1 (ABCA1) J Biol Chem. 2004;279:7384–7394. doi: 10.1074/jbc.M306963200. [DOI] [PubMed] [Google Scholar]
- 49.Krimbou L, Hassan HH, Blain S, Rashid S, Denis M, Marcil M, Genest J. Biogenesis and speciation of nascent apoA-I-containing particles in various cell lines. J Lipid Res. 2005;46:1668–1677. doi: 10.1194/jlr.M500038-JLR200. [DOI] [PubMed] [Google Scholar]
- 50.Mulya A, Lee JY, Gebre AK, Thomas MJ, Colvin PL, Parks JS. Minimal lipidation of pre-beta HDL by ABCA1 results in reduced ability to interact with ABCA1. Arterioscler Thromb Vasc Biol. 2007;27:1828–1836. doi: 10.1161/ATVBAHA.107.142455. [DOI] [PubMed] [Google Scholar]
- 51.Monera OD, Kay CM, Hodges RS. Protein denaturation with guanidine hydrochloride or urea provides a different estimate of stability depending on the contributions of electrostatic interactions. Protein Sci. 1994;3:1984–1991. doi: 10.1002/pro.5560031110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alexander ET, Vedhachalam C, Sankaranarayanan S, de la Llera-Moya M, Rothblat GH, Rader DJ, Phillips MC. Influence of Apolipoprotein A-I Domain Structure on Macrophage Reverse Cholesterol Transport in Mice. Arterioscler Thromb Vasc Biol. 2011;31:320–327. doi: 10.1161/ATVBAHA.110.216226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palgunachari MN, Mishra VK, Lund-Katz S, Phillips MC, Adeyeye SO, Alluri S, Anantharamaiah GM, Segrest JP. Only the two end helixes of eight tandem amphipathic helical domains of human apo A-I have significant lipid affinity. Arterioscler Thromb Vasc Biol. 1996;16:328–338. doi: 10.1161/01.atv.16.2.328. [DOI] [PubMed] [Google Scholar]
- 54.Jones MK, Anantharamaiah GM, Segrest JP. Computer programs to identify and classify amphipathic alpha helical domains. J Lipid Res. 1992;33:287–296. [PubMed] [Google Scholar]