

Abstract
Lin28A and Lin28B selectively block the expression of let-7 microRNAs and function as oncogenes in a variety of human cancers. Lin28A recruits a TUTase (Zcchc11/TUTase4) to let-7 precursors to block processing by Dicer in the cell cytoplasm. Here we find that unlike Lin28A, Lin28B represses let-7 processing through a TUTase-independent mechanism. Lin28B functions in the nucleus by sequestering primary let-7 transcripts and inhibiting their processing by the Microprocessor. The inhibitory effects of Zcchc11 depletion on the tumorigenic capacity and metastatic potential of human cancer cells and xenografts is restricted to Lin28A-expressing tumors. Furthermore, the majority of human colon and breast tumors analyzed exclusively express either Lin28A or Lin28B. Lin28A is expressed in HER2-overexpressing breast tumors while Lin28B expression characterizes triple-negative breast tumors. Overall our results illuminate the distinct mechanisms by which Lin28A and Lin28B function, and have implications for the development of new strategies for cancer therapy.
Keywords: Lin-28, Lin28A, Lin28B, let-7, microRNA (miRNA), TUTase, Zcchc11, TUTase4, TUT4, Cancer
INTRODUCTION
Control of gene expression by microRNAs (miRNAs) is important for normal development. Altered miRNA expression is linked with various diseases including cancer (Small and Olson, 2011). miRNA biogenesis begins with transcription of primary transcripts (pri-miRNAs) that contain a stem-loop structure. In the cell nucleus, pri-miRNAs are processed by Microprocessor, containing the ribonuclease Drosha and its essential cofactor DGCR8 (Denli et al., 2004; Gregory et al., 2004). Microprocessor cleaves the double-stranded RNA towards the base of the stem-loop to release a 60–80 nucleotide (nt) precursor (pre-miRNA) that is exported to the cell cytoplasm and cleaved by Dicer to generate a 22 nt duplex (Hutvagner et al., 2001; Krol et al., 2010). One RNA strand is bound by Argonaute and incorporated into the RNA-induced silencing complex (RISC) (Gregory et al., 2005; Liu et al., 2004). Base-pairing between the miRNA and target mRNA guides RISC to complementary transcripts leading to gene repression through mRNA degradation and/or translational repression (Krol et al., 2010).
Altered miRNA expression is directly associated with cancer initiation, progression, and metastasis and is observed in a wide variety of human malignancies (Di Leva and Croce, 2010). The let-7 miRNA family members act as tumor suppressors by inhibiting expression of oncogenes and key regulators of mitogenic pathways including RAS, MYC, and HMGA2 (Bussing et al., 2008). Let-7 is downregulated in numerous different cancers and low let-7 correlates with poor prognosis (Boyerinas et al., 2010; Shell et al., 2007; Takamizawa et al., 2004). Restoration of let-7 expression effectively inhibits cancer growth in mouse models of lung and breast cancers (Barh et al., 2010; Esquela-Kerscher et al., 2008; Slack, 2009; Trang et al., 2010; Yu et al., 2007a). In humans, there are twelve let-7 family members (let-7a-1, -2, -3; let-7b; let-7c; let-7d; let-7e; let-7f-1, -2; let-7g; let-7i; miR-98) located at eight different chromosomal loci. Of note, many tumors are characterized by the coordinate downregulation of multiple let-7 miRNAs (Shell et al., 2007).
The developmentally regulated RNA-binding protein Lin28 was found to selectively repress expression of let-7 miRNAs (Heo et al., 2008; Newman et al., 2008; Rybak et al., 2008; Viswanathan et al., 2008). This posttranscriptional regulation of let-7 by Lin28 is required for normal development and contributes to the pluripotent state by preventing let-7 mediated differentiation of embryonic stem cells (ESCs). Lin28 overexpression or let-7 inhibition with antisense RNAs promotes reprogramming of human and mouse fibroblasts to induced pluripotent stem cells (iPSCs) (Martinez and Gregory, 2010; Melton et al., 2010; Yu et al., 2007b). Unlike in C. elegans where a single Lin28 gene is responsible for repression of let-7 expression and control of developmental timing, the mammalian genome encodes two Lin28 paralogs, Lin28 (hereafter Lin28A) and Lin28B (Guo et al., 2006; Lehrbach et al., 2009; Moss et al., 1997; Van Wynsberghe et al., 2011; Viswanathan and Daley, 2010). Lin28B also represses expression of multiple let-7 members, and genome-wide association studies (GWAS) have linked Lin28B with the determination of human height and control of the age of onset of puberty and menopause; phenotypes that are recapitulated in a mouse model (Zhu et al., 2010). Activation of Lin28A/Lin28B occurs in several different primary human tumors and these tumors display low levels of let-7 expression (Iliopoulos et al., 2009; Viswanathan et al., 2009). Indeed Lin28A/Lin28B function as oncogenes that promote cellular transformation when ectopically expressed (Iliopoulos et al., 2009; Viswanathan et al., 2009; West et al., 2009). Importantly, this effect is abrogated when let-7 is reintroduced into these cells (Iliopoulos et al., 2009; Viswanathan et al., 2009). Therefore, Lin28-mediated cellular transformation is directly dependent on let-7 levels. Conversely, depletion of Lin28A or Lin28B in human cancer cells results in decreased cell proliferation (Chang et al., 2009; Iliopoulos et al., 2009; Viswanathan et al., 2009). Lin28A/Lin28B may contribute to the development of aggressive, poorly differentiated tumors since their expression is associated with advanced disease in hepatocellular carcinoma (HCC), chronic myeloid leukemia (CML), Wilms’ tumor, ovarian carcinoma, colon adenocarcinoma, and germ cell tumors (Dangi-Garimella et al., 2009; Guo et al., 2006; Iliopoulos et al., 2009; Ji and Wang, 2010; King et al., 2011; Liang et al., 2010; Lu et al., 2009; Oh et al.; Peng et al., 2010; Viswanathan et al., 2009; Wang et al., 2010; West et al., 2009; Yang et al., 2010), and is associated with poor clinical outcome and patient survival in HCC, colon, and ovarian cancer (King et al., 2011; Lu et al., 2009; Viswanathan et al., 2009). In the case of LIN28B, rare amplification or translocation events might explain activation in some cases (Viswanathan et al., 2009). A more common mechanism might be transcriptional activation by upstream factors. For example, c-Myc binds to both Lin28A and Lin28B loci and activates expression of these genes (Chang et al., 2009). In a breast cancer model, transient expression of Src oncoprotein results in a transformed cell line that forms self-renewing mammospheres harboring tumor initiating cells (Iliopoulos et al., 2009). The transformation process involves NF-κB activation leading to direct transcriptional upregulation of Lin28B, consequent let-7 loss, and de-repression of the let-7 target gene IL-6. Since IL-6 activates NF-κB, this regulatory circuit represents a positive feedback loop, providing a molecular link between inflammation and cancer.
Selective regulation of let-7 expression involves Lin28A binding to the terminal loop of let-7 precursors, a molecular recognition that requires both the cold-shock domain (CSD) and CCHC-type zinc finger RNA-binding domains of the Lin28A protein (Piskounova et al., 2008). Lin28A recruits the activity of a terminal uridylyltransferase (TUTase), Zcchc11 (also known as TUTase4 or TUT4) that inhibits pre-let-7 processing by Dicer and leads to the rapid decay of oligouridylated pre-let-7 RNAs (Hagan et al., 2009; Heo et al., 2009). Although both Lin28A and Lin28B can both recruit Zcchc11/TUT4 to uridylate pre-let-7 in vitro, the molecular mechanism of the Lin28B-mediated blockade of let-7 expression has yet to be determined (Heo et al., 2008; Heo et al., 2009). Here we investigate the regulation of let-7 expression by Lin28B. Surprisingly, we find that despite their high degree of homology, Lin28A and Lin28B function through distinct mechanisms. Depletion of Zcchc11 effects let-7 expression only in Lin28A-expressing cancer cells whereas Lin28B functions through a TUTase-independent mechanism. We find that Lin28A and Lin28B are differentially localized in cells with Lin28A predominantly cytoplasmic whereas Lin28B accumulates in the nucleus due to its functional nuclear localization signals where it binds pri-let-7 miRNAs to block processing by the Microprocessor. In contrast, Lin28A functions in the cytoplasm by blocking at the Dicer step and recruiting the TUTase to uridylate pre-let-7. Our findings identify Zcchc11 as a possible therapeutic target in Lin28A-expressing cancers. Accordingly, we demonstrate that Zcchc11 depletion selectively inhibits the tumorigenic capacity and metastatic potential of Lin28A- but not Lin28B-expresing human cancer cells and xenografts. Our results illuminate the distinct mechanisms by which Lin28A and Lin28B function and have broad implications for the development of new strategies for cancer therapy.
RESULTS
Lin28B regulates let-7 expression through a TUTase-independent mechanism
The paralogous RNA-binding proteins Lin28A and Lin28B have a high degree of sequence identity and conserved domain organization (Figure 1A) and both proteins selectively block let-7 expression (Newman et al., 2008; Viswanathan et al., 2008). We screened several human cancer cell lines and found that some express Lin28A, whereas others express Lin28B (Figure 1B). We did not observe co-expression of both Lin28A and Lin28B in any cell line suggesting that their expression may be mutually exclusive. We found ubiquitous Zcchc11 expression. Hela cells express Zcchc11 but neither Lin28A nor Lin28B. Since Lin28A-mediated repression of let-7 in mouse ESCs involves the TUTase Zcchc11 we next asked whether Lin28A and Lin28B function through the same mechanism to block let-7 processing. Previous reports have used recombinant Lin28A and Lin28B interchangeably in biochemical assays demonstrating that Lin28B is capable of enhancing Zcchc11 activity in vitro, however the physiological relevance of these observations remains unknown (Heo et al., 2009).
Figure 1. Lin28B regulates let-7 biogenesis through a TUTase-independent mechanism.
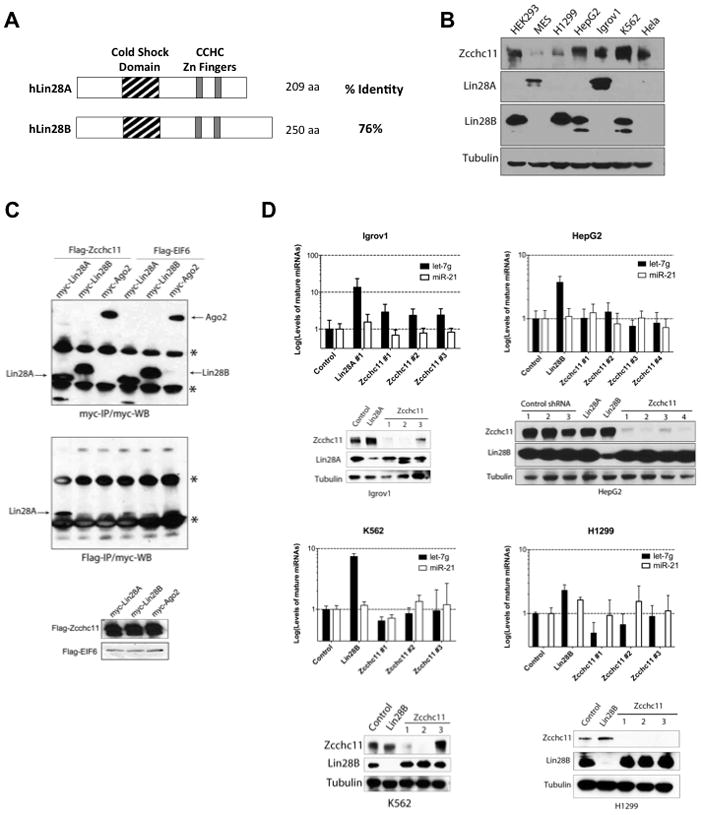
(A) Schematic representation of human Lin28A and Lin28B (B) Western Blot analysis of Zcchc11, Lin28A, and Lin28B in extracts prepared from human cancer cell lines. (C) Co-immunoprecipitation (co-IP): Hela cells were co-transfected with Human myc-Lin28A, myc-Lin28B, or myc-Ago2 with either Flag-Zcchc11 or Flag-EIF6. Flag-IP and Flag- and Myc-western blots were performed to detect expression and interaction respectively. See Also Figure S1. (D) Stable knockdown of Zcchc11 leads to upregulation of mature let-7g levels in Lin28A-expressing cells but not Lin28B-expressing cell lines. miRNA levels were measured by q.RT-PCR. Error bars represent SEM (n=3). Protein knockdown was monitored by Western blot.
To begin to investigate whether both Lin28A and Lin28B function through a TUTase-dependent mechanism we performed co-immunoprecipitation (co-IP) experiments. Myc-tagged Lin28A, Lin28B, or Ago2 were co-expressed with either Flag-tagged Zcchc11 or Flag-EIF6 control (Figure 1C). Since the Lin28A-Zcchc11 interaction has been shown to be RNA-dependent we also co-expressed pri-let-7g (Heo et al., 2009). Consistent with earlier reports, myc-Lin28A was found to be associated with affinity-purified Flag-Zcchc11 (Heo et al., 2009). However, we were unable to detect a physical interaction between myc-Lin28B and Flag-Zcchc11. We performed additional co-IP experiments in which we titrated the amount of exogenously expressed Flag-Zcchc11. These experiments confirmed the specific physical interaction Zcchc11 and Lin28A, whereas myc-Lin28B was not detected in any of the Flag-Zcchc11 IPs (Supplementary Figure 1A). This was additionally confirmed by the co-IP of endogenous Lin28A in Igrov1 cells (Supplementary Figure 1B). Together, these results indicate that unlike Lin28A, we could not detect any physical interaction between Lin28B and Zcchc11.
Next, to address the functional requirement of Zcchc11 in the Lin28A- and Lin28B-mediated repression of let-7 expression we performed a series of knockdown experiments to deplete Zcchc11 in a panel of human cancer cell lines. We used shRNAs to deplete Lin28A or Zcchc11 expression in Igrov cells and measured the effect on let-7 expression by quantitative reverse transcription PCR (q.RT-PCR). As expected, depletion of Lin28A led to ~10-fold increase in let-7 levels. Knockdown of Zcchc11 with three independent shRNAs also led to elevated mature let-7 levels (Figure 1D). Therefore Zcchc11 is involved in the repression of let-7 expression in this Lin28A-expressing human cancer cell line as has been previously reported in mESCs and P19 embryonal carcinoma cells (Hagan et al., 2009; Heo et al., 2009). We performed analogous experiments in three different Lin28B-expressing cancer cell lines: HepG2, K562 and H1299 (Figure 1D) and found no significant effect on mature let-7 levels in any of the cell lines when Zcchc11 was depleted. In contrast, knockdown of Lin28B consistently led to the expected increase in mature let-7. Overall our results indicate that Zcchc11 negatively regulates let-7 expression in Lin28A- but not Lin28B-expressing cell lines suggesting that Lin28B employs a TUTase-independent mechanism to block let-7 processing.
Lin28B localizes to the nucleus
We sought potential explanations for the functional differences between Lin28A and Lin28B. We examined the subcellular localization of the endogenous Lin28A and Lin28B proteins by immunofluorescence assays (Figure 2A). Lin28A was mostly localized to the cytoplasm of Igrov1 cells whereas Lin28B localized to specific foci in the nucleus of H1299 cells where it co-localized with the nucleolar marker Fibrillarin. To further confirm the localization of Lin28B in the nucleoli, we performed immunofluorescence assays on H1299 cells in which Lin28B expression (or control) was stably knocked down by shRNA and showed that the observed nucleolar staining pattern is specific to Lin28B (Figure 2B). These data were further confirmed by biochemical fractionation and Western blot of both cell lines (Figure 2C). Consistent with published data we found Zcchc11 only in the cytoplasmic fraction in both the Lin28A- and Lin28B-expressing cell lines (Figure 2C). These data suggest that the divergence in the mechanisms by which Lin28A and Lin28B block let-7 biogenesis derives from their differential subcellular localization. The lack of physical and functional interactions between Zcchc11 and Lin28B is therefore likely due to their localization to distinct cellular compartments, even though recombinant Lin28B has the capacity to enhance Zcchc11 activity in vitro (Heo et al., 2009).
Figure 2. Lin28A and Lin28B are differentially localized within the cell.
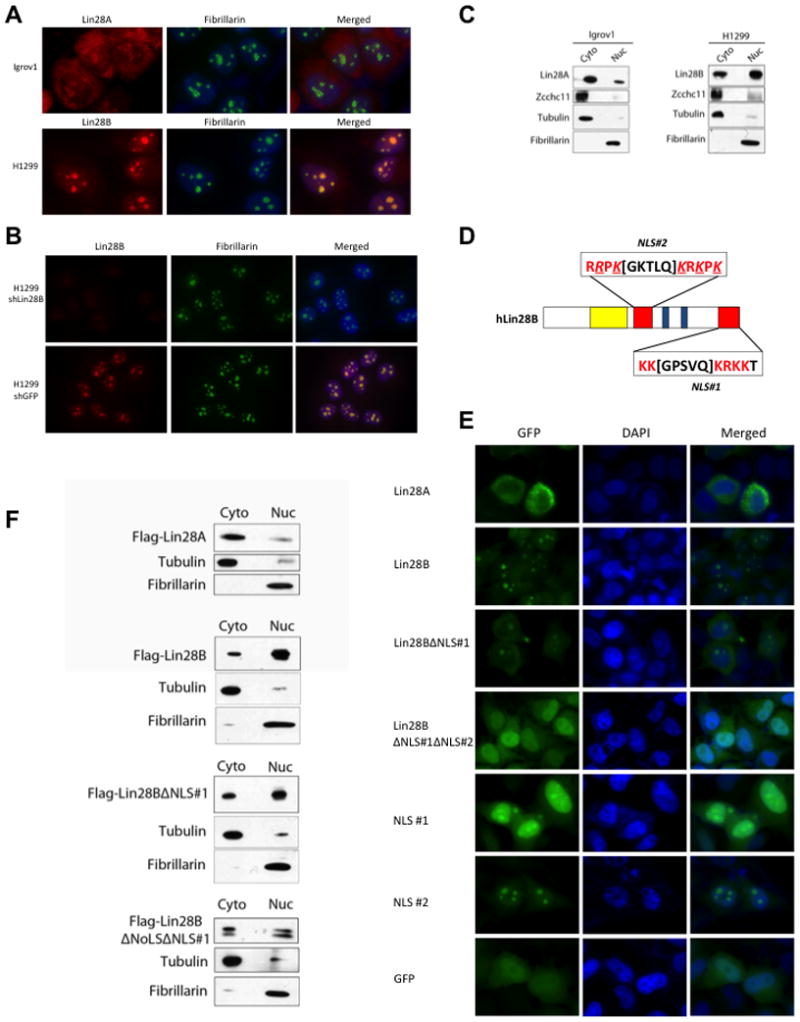
(A) Immunofluorescence detection of endogenous Lin28A in Igrov1 and Lin28B in H1299 cell lines. Fibrillarin, a known nucleolar protein, was used as a positive control. (B) Immunofluorescence analysis of control- and Lin28B-knockdown H1299 cell lines. (C) Biochemical fractionation of Igrov1 and H1299 cell lines. Endogenous Lin28A, Lin28B, and Zcchc11 in each fraction were detected by western blot. Fibrillarin was used as a nuclear marker; Tubulin was used as a cytoplasmic marker. (D) Schematic of nuclear localization signals (NLS) in the Lin28B protein. An Arginine as well as several Lysines that were replaced by Glycines are underlined and italicized (E) Localization of GFP-Lin28 fusion proteins in Hela cells. (F) Fractionation of Flag-Lin28 proteins, exogenously expressed in Hela cells. Proteins were detected by Flag western blot.
Lin28B contains functional nuclear localization signals
Lin28B protein has an extended C-terminus compared to Lin28A which upon closer inspection contains a putative bipartite nuclear localization signal (NLS), KK[GPSVQ]KRKK. Another potential NLS, RRPK[GKTLQ]KRKPK, was identified in the linker region that connects the two functional RNA-binding domains (Figure 2D). To test the function of these putative NLS we generated constructs for the expression of a series of GFP fusion proteins. We transiently transfected Hela cells with these constructs and analyzed the subcellular localization of the GFP-Fusion proteins by microscopy (Figure 2E). Consistent with the localization of endogenous Lin28A in Igrov1 cells, we found Lin28A-GFP localized mainly to the cytoplasm. Lin28B-GFP predominantly localized to specific foci in the nucleus, again recapitulating the nucleolar localization of endogenous Lin28B observed in H1299 cells. When we exogenously expressed the Lin28BΔNLS#1 truncation we observed increased signal in the cytoplasm, however some nucleolar localization still remained consistent with the presence of a second NLS. Indeed, the double mutant Lin28B-GFP protein lacking both NLS showed cellular localization similar to that of GFP alone suggesting that both NLS elements are important for nuclear and nucleolar localization of Lin28B (Figure 2E). To further determine whether these sequences represent functional NLS, we examined the localization of the NLS#1-GFP and NLS#2-GFP (Figure 2E). When exogenously expressed in Hela cells, NLS#1-GFP localized nearly entirely throughout the nucleus including the nucleoli. This is in contrast to the control GFP construct that is broadly distributed throughout the cell. Localization of NLS#2-GFP the signal was nearly entirely localized to the nucleoli (Figure 2E). Together these results identify that NLS#1 amino acid sequence represents a bona fide NLS, and that NLS#2 is a functional nucleolar localization signal (NoLS). NoLS properties are less well known and have only recently been studied at the amino acid sequence level (Scott et al., 2010). Several reports suggest that proteins with a NoLS also contain an NLS that allows them to cross the nuclear membrane before localizing to the nucleoli. These results were confirmed by biochemical fractionation of Hela cells transiently expressing either Flag-Lin28A, full-length-, truncated-, or double mutant-Flag-Lin28B. As expected Flag-Lin28A was predominantly present in the cytoplasmic fraction, Flag-Lin28B was mostly nuclear, NLS#1 mutant only showed marginal increase of signal in the cytoplasmic fraction, whereas the double mutant Flag-Lin28B showed a more significant increase in cytoplasmic signal (Figure 2F).
Lin28B localizes to nucleoli where Microprocessor is absent
We next compared the specific localization of Lin28B with that of the nuclear miRNA processing machinery. We found that the Microprocessor components, DGCR8 and Drosha, co-localize in the nucleoplasm but are excluded from nucleoli (Figure 3A). To further confirm that localization of Lin28B is distinct and non-overlapping with the Microprocessor, we examined the co-localization of mCherry-DGCR8 and GFP-Lin28A/B proteins in transfected cells (Figure 3B). Lin28A localized mostly to the cytoplasm and therefore showed no overlap with the nuclear DGCR8. Lin28B localized to nucleoli and did not overlap with DGCR8 either. In contrast, the localization of the Lin28B NLS/NoLS mutant showed a broadly dispersed localization throughout the nucleus and cytoplasm (similar to GFP control) and displayed co-localization with DGCR8 in the nucleoplasm. We confirmed this finding that Lin28B and Microprocessor normally occupy distinct compartments in the nucleus by performing large-scale biochemical fractionation and Western blot of a stable Hela cell line expressing Flag-Lin28B. Lin28B was specifically present in the nucleolar-enriched fractions whereas DGCR8 was only detectable in the nuclear fraction and not in the nucleolar fractions (Figure 3C). Overall, these findings suggest a possible mechanism by which Lin28B blocks let-7 processing in the nucleus by sequestering pri-let-7 miRNAs in the nucleoli away from the Microprocessor.
Figure 3. Lin28B localizes to nucleoli where Microprocessor is absent.
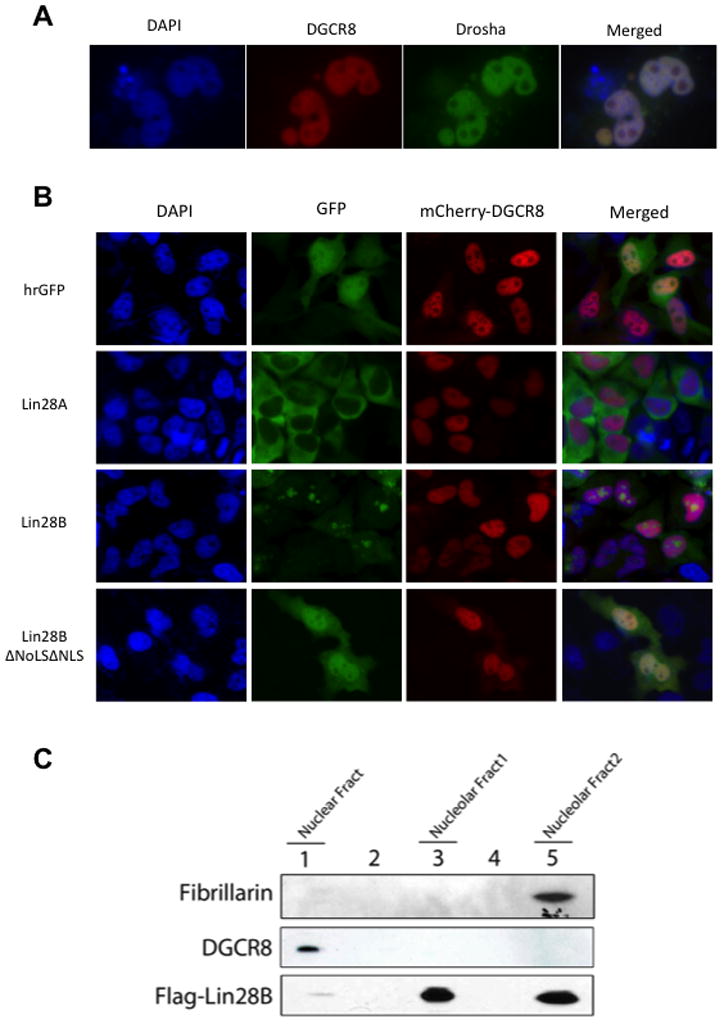
(A) Co-localization of Microprocessor components GFP-Drosha and mCherry-DGCR8 in Hela cells reveals their distribution throughout the nucleus and exclusion from nucleoli. (B) Localization of GFP-Lin28A, Lin28B, or mutant Lin28B proteins with mCherry-DGCR8 in Hela cells reveals non-overlapping localization of Lin28B and DGCR8. (B) Fractionation of a Flag-Lin28B Hela stable cell line. Flag-Lin28B and endogenous DGCR8 were detected western blot and shows a non-overlapping subcellular localization of Lin28B and the Microprocessor. Fibrillarin was used as a control for nucleolar localization.
Lin28B directly binds and sequesters pri-let-7
To further dissect the mechanism of the Lin28B-mediated let-7 processing block we first compared the relative abilities of recombinant human Lin28A and Lin28B proteins to bind pre-let-7 (Figure 4A, B, and Supplementary Figure 2). We performed EMSA with pre-let-7g to analyze the relative binding affinities of the two recombinant proteins. We found Lin28A and Lin28B have apparent Kds of approximately 0.6 nM and 0.5 nM, respectively (Figure 4B). Both these estimated Kds are much lower than a previously reported for recombinant mouse Lin28A. This difference is likely due to the omission here of nonspecific yeast tRNA competitor used previously in the binding buffer (Piskounova et al., 2008). We also performed EMSA with pri-let-7g and demonstrated that both recombinant Lin28A and Lin28B are able to bind pri-let-7g with similar affinities (Figure 4C). Collectively, these assays reveal that both Lin28 proteins can directly bind to let-7 precursors with high-affinity in vitro.
Figure 4. Lin28B directly binds and sequesters pri-let-7.
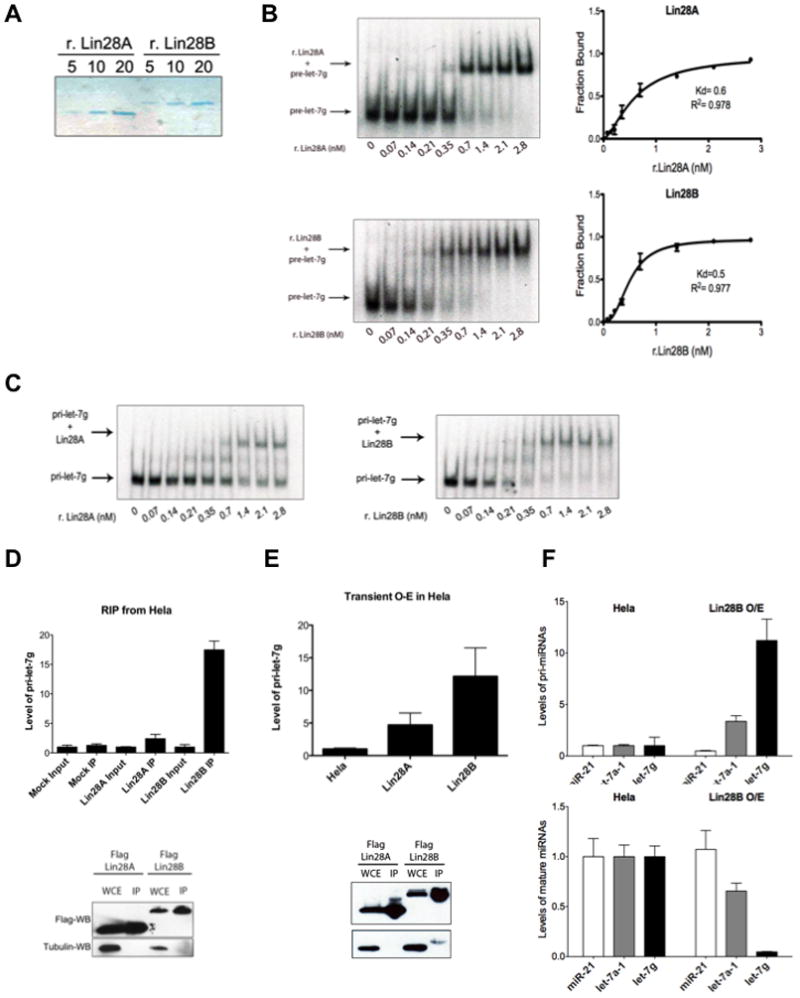
(A) Colloidal Blue staining of purified recombinant His-Lin28A and His-Lin28B proteins. (B) Binding of r.Lin28A and r.Lin28B to pre-let-7g was assessed by EMSA performed with 0.5 nM 5′-end labeled pre-let-7g RNA and the indicated concentration of recombinant protein. Band intensities were quantitated from three independent experiments and represented as the fraction of bound pre-let-7g RNA in the plots. Values are given as average ±S.E.M. (n=3). See also Figure S2. (C) EMSA performed indicated concentration of r.Lin28A and r.Lin28B with in vitro transcribed uniformly labeled pri-let-7g. (D) RNA-Immunoprecipitation (RIP) analysis of RNA associated with immunopurified Flag-Lin28A and Flag-Lin28B from Hela cells. RNA was extracted from IP material and analyzed by q.RT-PCR. Error bars ±S.E.M. (n=3). Lower panel indicates relative Lin28A and Lin28B expression levels by Flag-Western blot. (E) Accumulation of pri-let-7 by transient Lin28B expression in Hela cell detected by q.RT-PCR. Error bars ±S.E.M. (n=3). Lower panel indicates relative expression levels of Lin28A and Lin28B proteins detected by Flag-Western blot in transfected cells. (F) pri-let-7 accumulates (top panel) and mature let-7 levels decrease (bottom panel) in Hela cells stably overexpressing Lin28B. Error bars ±S.E.M. (n=3)
To gain further support for our model in which Lin28B binds and sequesters pri-let-7 in the nucleus to inhibit the Microprocessor we next examined the RNA associated with Lin28B. We individually expressed and purified Flag-Lin28A and Flag-Lin28B, extracted the associated RNA and analyzed relative levels of pri-let-7g by q.RT-PCR. This RNA immunoprecipitation (RIP) analysis revealed that Lin28B directly associates with pri-let-7g RNA (Figure 4D). We detected an ~18-fold enrichment of pri-let-7 associated with Lin28B. Furthermore, we found substantially more pri-let-7 associated with Lin28B than with Lin28A, which is consistent with the differential subcellular localization of these proteins. Taken together these results indicate that this preferential association of pri-let-7g with Lin28B likely reflects the distinct mechanism by which Lin28B represses let-7 expression rather than any possible intrinsic differences in the relative RNA-binding affinities of Lin28A and Lin28B proteins. Next, we examined the effect of transient Lin28B overexpression on pri-let-7g levels by q.RT-PCR. Transient Lin28B overexpression led to ~12-fold accumulation of pri-let-7g levels (Figure 4E). In contrast, overexpression of Lin28A only had a more modest effect on pri-let-7g levels consistent with its predominantly cytoplasmic localization. In order to further assess the effects of Lin28B overexpression on both pri– and mature miRNA levels, we utilized a Flag-Lin28B expressing Hela stable cell line. Analysis of several pri-miRNAs by q.RT-PCR in this cell line demonstrated a substantial accumulation of pri-let-7 miRNAs, >10-fold for pri-let-7g and >3-fold for pri-let-7a-1. There was however no effect on levels of pri-miR-21 (Figure 4F). We detected a corresponding decrease in the levels of mature let-7, with >90% decrease for mature let-7g, and ~40% decrease for mature let-7a. Again no effect was observed on levels of mature miR-21 (Figure 4F). Together, these data support our model whereby nuclear Lin28B directly associates with pri-let-7 sequestering it from cleavage by the Microprocessor to selectively inhibit let-7 maturation, and underscore our findings that the paralogous RNA-biding proteins, Lin28A and Lin28B, operate by distinct mechanisms to selectively repress let-7 expression.
Zcchc11 inhibition blocks the tumorigenicity and invasiveness of Lin28A- but not Lin28B-expressing breast cancer cells in vitro and in vivo
We were motivated to further explore the relevance of our findings that Lin28A and Lin28B block let-7 processing through distinct mechanisms and to examine the effect of Zcchc11 inhibition on the tumorigenicity and invasiveness of human Lin28A/B-expressing cancer cells. Initially, we tested the consequences of Zcchc11 inhibition in the MCF10A ER-Src inducible model of cellular transformation, where MCF10A immortalized breast epithelial cells become transformed 36h post tamoxifen (TAM) treatment (Iliopoulos et al., 2009). We previously reported that Lin28B expression is activated and required for induction and maintenance of the transformed phenotype in this model (Suppl. Figure 3A, 3B). In contrast, Zcchc11 depletion did not affect transformation or colony formation (Suppl. Figure 3B–D) and did not affect the expression of let-7a and its direct downstream target IL6 (Suppl. Figure 3D–F). Accordingly, Zcchc11 inhibition did not affect the tumorigenicity of MCF10A ER-Src transformed cells in xenograft assays (Suppl. Figure 3G). Overall, these data suggest that inhibition of Zcchc11 does not have an inhibitory effect on the tumorigenicity and invasiveness of Lin28B-expressing MCF10A ER-Src cells.
To further explore the distinct requirements for Zcchc11 in Lin28A- and Lin28B-expressing cancer, we compared the effects of Zcchc11 inhibition on the tumorigenicity and invasiveness of MDA-MB-231 breast cancer cells (Lin28B-expressing cells) relative to T47D breast cancer cells (Lin28A-expressing cells). We found that suppression of Zcchc11 expression did not affect let-7a expression in MDA-MB-231 cells, however led to 7-fold increase in mature let-7a levels in T47D cells (Figure 5A, 5B). Furthermore, Zcchc11 inhibition did not affect the tumorigenicity and invasiveness of MDA-MB-231 cells, while it suppressed both the colony formation ability and invasiveness of T47D cells (Figures 5C, D). Zcchc11 inhibition had similar effects on the tumor growth of these cell lines in xenografts. Specifically, Zcchc11 knockdown did not affect tumor growth of MDA-MB-231 while it suppressed T47D tumor growth (Figure 5E). Synthetic let-7a miRNA suppressed both MDA-MB-231 and T47D tumor growth (Figure 5E). Also, in the tumors derived from MDA-MB-231 xenografts (day 30), let-7a expression was not affected by inhibition of Zcchc11, while Lin28B suppression increased let-7a levels about 5-fold (Figure 5F, G). On the other hand, both Zcchc11 and Lin28A inhibition resulted in up-regulation of let-7a expression to similar levels in T47D-derived tumors (day 30) (Figure 5F, G).
Figure 5. Zcchc11 inhibition blocks tumorigenicity and invasiveness of Lin28A-expressing breast cancer cells.
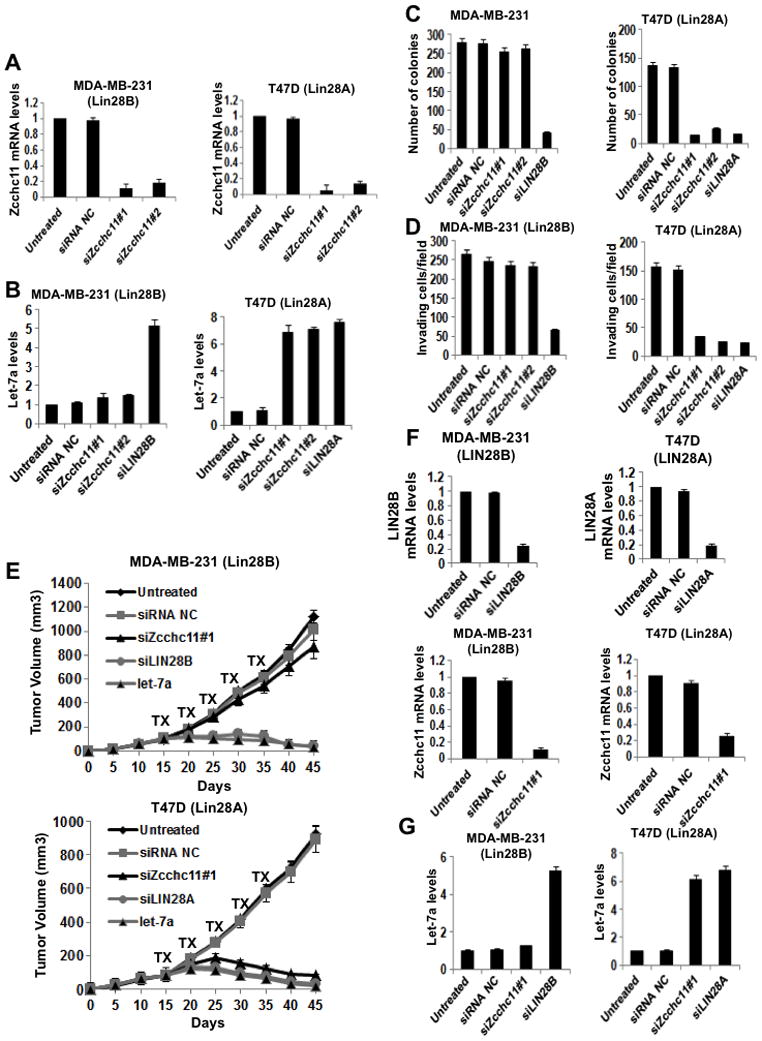
(A) q.RT-PCR analysis of Zcchc11 knockdown in MDA-MB-231 and T47D breast cancer cells. Error bars ±S.E.M. (n=3) (B) Inhibition of Zcchc11 expression does not affect let-7a expression in Lin28B-expressing cells (MDA-MB-231), while it up-regulates let-7a expression in Lin28A-expressing cells (T47D). Let-7a expression levels measured by q.RT-PCR in cells treated with siRNAs for 48hr. Error bars ±S.E.M. (n=3) (C) Inhibition of Zcchc11 expression did not affect the colony formation ability of MDA-MB-231 cells, but suppressed the colony formation ability of T47D cells. The number of colonies was evaluated 20 days post plating in soft agar. The experiment was repeated thrice and the statistical significance was calculated using Student’s t test. (D) Inhibition of Zcchc11 expression did not affect the invasiveness of MDA-MB-231 cells, but suppressed the invasive ability of T47D cells. The number of invasive cells was measured 16h post transfection with indicated siRNAs. In all assays, 10 fields per insert were scored and SD was measured. The experiment was repeated thrice and the statistical significance was calculated using Student’s t test. (E) Inhibition of Zcchc11 expression did not suppress tumor growth of MDA-MB-231 cells in xenografts, however it inhibited tumor growth of T47D cells in xenografts. The treatments with indicated siRNA were performed intraperitoneally (i.p.) for 5 cycles starting on day 15. Each treatment group consisted of 5 mice. (F) q.RT-PCR analysis of siRNA inhibition of Lin28B, Lin28A, and Zcchc11 in xenograft tumors (day 30) derived from MDA-MB-231 and T47D cells. Error bars ±S.E.M. (n=3). (G) Inhibition of Lin28B but not of Zcchc11 allows up-regulation of let-7a expression levels in MDA-MB-231 xenograft tumors (day 30). However, inhibition of Lin28A or Zcchc11 results in let-7a up-regulation in T47D xenograft tumors. Let-7a expression levels measured by q.RT-PCR on tumors untreated or treated with indicated siRNA. Error bars ±S.E.M. (n=3). See also Figure S3.
In addition to the breast cancer cells, we tested the effects of Zcchc11 inhibition on tumor growth of several other (liver, lung, ovarian, melanoma, colon) cancer cell types (Figure 6A). As above, we found that Zcchc11 inhibition (Figure 6B) blocked the growth of LIN28A-expressing tumors (Igrov1) and did not affect the growth of Lin28B-expressing tumors (HepG2, H1299, SK_MEL_28, CaCO2) (Figure 6A). Lin28A and Lin28B inhibition suppressed the growth of the corresponding tumors (Figure 6C, 6D). Taken together, these data suggest that Zcchc11 plays a role in the tumorigenicity and invasiveness of Lin28A-expressing cancer cells but depletion of Zcchc11 in Lin28B-expressing cancer cell lines has no effect on cancer growth.
Figure 6. Inhibition of Zcchc11 expression suppresses tumor growth of Lin28A- but not Lin28B-expressing xenografts.
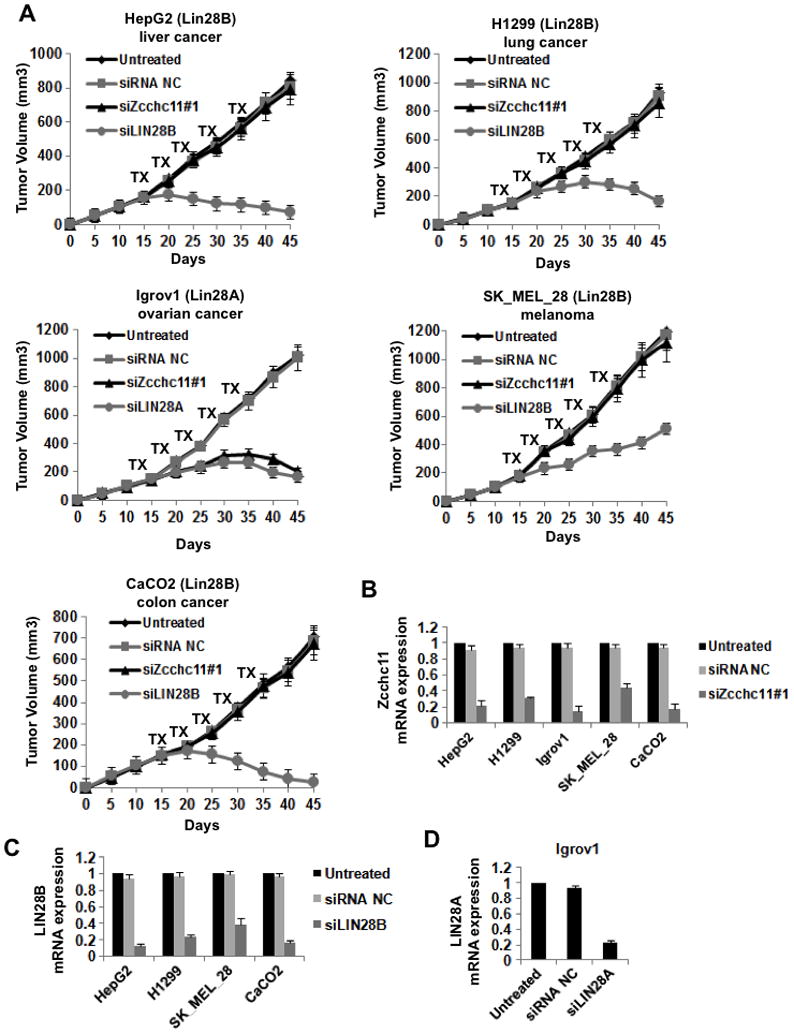
(A) Xenograft experiments were performed with a variety of different human cancer cell lines. Mice were treated with the indicated siRNA for 5 cycles starting on day 15. For all cells lines tested each treatment group consisted of 5 mice. While inhibition of Lin28A or Lin28B suppressed tumor growth in the relevant xenografts, inhibition of Zcchc11 inhibited growth only of Lin28A- but not Lin28B-expressing tumors. Error bars ±S.E.M. (n=3) (B) Analysis of siRNA inhibition of Zcchc11 in xenograft tumors (day 30) derived from the indicated cells. (C) Analysis of siRNA inhibition of Lin28B in xenograft tumors (day 30) derived from the indicated cells. (D) Analysis of siRNA inhibition of Lin28A in xenograft tumors (day 30) derived from IGROV1 cells. mRNA expression levels were measured by q.RT-PCR on tumors untreated or treated with the indicated siRNA. Error bars ±S.E.M. (n=3)
Lin28A and Lin28B expression levels in human colon and breast tissues
To further study the disease relevance of our findings we measured Lin28A and Lin28B expression in human colon and breast tissues. We found that Lin28A or Lin28B are upregulated while let-7a was downregulated in colon adenocarcinomas relative to normal colon tissues (Figure 7A). Specifically, we identified that Lin28A was upregulated in 19/45 colon adenocarcinomas while Lin28B was upregulated in 14/45 colon adenocarcinomas. We found that the colon tumors with Lin28A overexpression had very low levels of Lin28B and vice versa. Furthermore, immunohistochemistry and in situ hybridization analyses in normal and colon cancer tissues revealed that Lin28A or Lin28B proteins are upregulated while let-7a is downregulated in colon carcinomas relative to normal colon tissues (Figure 7B; Suppl. Figure 4). Similar to the mRNA data, immunohistochemistry revealed that tumors that expressed high Lin28A protein levels had low levels for Lin28B and vice versa. This is consistent with our analysis of human cancer cell lines where we did not find cells that express both Lin28A and Lin28B (Figure 1B and data not shown).
Figure 7. Lin28A and Lin28B expression in primary human cancers.
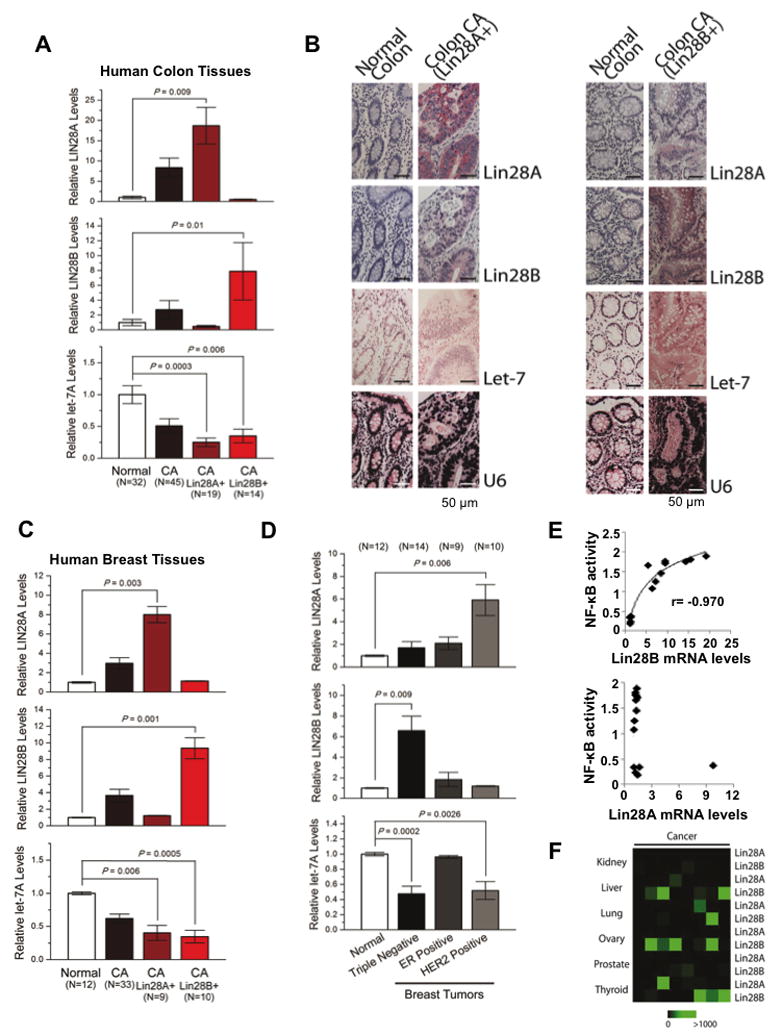
(A) q.RT-PCR analysis of Lin28A, Lin28B and let-7a expression levels in normal and colon cancer tissues. Tumor samples were further classified into two groups expressing either high Lin28A or Lin28B. Data expressed as mean ± SE. n=3. (B) Immunohistochemistry for Lin28A, Lin28B and in situ hybridization for let-7a and U6 in normal colon tissues and colon adenocarcinomas. See also Figure S4. (C) q.RT-PCR analysis of Lin28A, Lin28B and let-7a in human normal and breast cancer tissues. Tumor samples were further classified into two groups expressing either high Lin28A or Lin28B. Data expressed as mean ±SE. n=3. (D) Lin28A, Lin28B and let-7a expression levels in different breast cancer subtypes. (E) Correlation between Lin28A and Lin28B mRNA levels assessed by q.RT-PCR with NF-κB phosphorylation status assessed by ELISA assay. (E) Heatmap representation of Lin28A and Lin28B in carcinomas of different origin measured by q.RT-PCR.
We also found that Lin28A or Lin28B were upregulated while let-7a was downregulated in breast cancer relative to normal breast tissues (Figure 7C). Specifically, we identified that Lin28A was upregulated in 9/33 breast carcinomas while Lin28B was upregulated in 10/33 breast carcinomas. Similar to the colon tissues, we detected that the breast tumors that overexpressed Lin28A had very low levels of Lin28B and vice versa. In addition, we detected that Lin28A was significantly upregulated in HER2-overexpressing breast tumors while Lin28B was significantly upregulated in triple-negative (ER-, PR-, HER2-) breast tumors (Figure 7D). Furthermore, according to our previous studies Lin28B expression is a part of an inflammatory circuit and is regulated by NF-κB transcription factor (Iliopoulos et al., Cell, 2009). Thus, we tested how NF-κB activity correlates with Lin28A or Lin28B mRNA levels in human breast tumors. We found a statistically significant correlation between NF-κB nuclear levels and Lin28B but not Lin28A expression levels (Figure 7E). These data suggest that NF-κB regulates Lin28B but not Lin28A pathway. Also, in order to have a broader view of Lin28A and Lin28B expression levels in human cancer tissues, we tested their expression levels in kidney, liver, lung, ovarian, prostate, and thyroid cancer (Figure 7F). These data reveal that Lin28B is upregulated in liver, ovarian and thyroid carcinomas.
DISCUSSSION
Lin28A and Lin28B inhibit let-7 miRNA biogenesis in ESCs and cancer and it has been assumed that both proteins block let-7 expression through the same mechanism, by recruiting the TUTase Zcchc11 (TUT4) in the cell cytoplasm to uridylate pre-let-7 and target it for degradation. Moreover these paralogous proteins have been used interchangeably in several in vitro assays (Heo et al., 2009). Or results provide the first evidence that despite their high degree of homology, Lin28A and Lin28B function through distinct mechanisms to block let-7 processing, a finding that has broad implications for the development of new cancer therapies and the potential use of Zcchc11 inhibitors in Lin28A-expressing tumors but not in Lin28B-expressing tumors. This distinction derives from the differential subcellular localization of these two proteins: Lin28A localizes primarily to the cytoplasm, whereas Lin28B contains functional nuclear localization signals and specifically localizes to nucleoli.
Due to the differential subcellular localization of the two proteins we find that in human cancer cell lines Lin28A and Lin28B block let-7 processing at different steps of the miRNA-processing pathway. However, the steps at which let-7 processing is blocked by Lin28 in various different organisms is controversial. A recently published report by Van Wynsberghe et al., proposes that Lin28 binds pri-let-7 and blocks let-7 expression co-transcriptionally in C. elegans and disputes earlier conclusions that Lin28 functions at steps further downstream in the let-7 biogenesis pathway (Lehrbach et al., 2009; Van Wynsberghe et al., 2011). They also report that a very small fraction of Lin28A in hESCs localizes to the nucleus and binds pri-let-7 miRNAs, although pre-let-7 is bound abundantly (Van Wynsberghe et al., 2011). This is consistent with our results that demonstrate a small fraction of Lin28A localizes to the nucleus in Igrov1 cell line. We also find that Lin28A binds pri-let-7 miRNA, however not as much as Lin28B, and earlier reports have demonstrated that purified Lin28A can inhibit the Microprocessor in vitro (Newman et al., 2008; Viswanathan et al., 2008).
Our demonstration that the Microprocessor is excluded from nucleoli raises the possibility that sequestration of certain pri-miRNAs to nucleoli by specific RNA-binding proteins may prove to be a more general strategy for the posttranscriptional control of miRNA biogenesis. Previous reports have demonstrated that nucleoli contain machinery responsible for modifying small nucleolar RNAs, for example through RNA methylation or 3′ uridylation. Whether these nucleolar mechanisms play a role in pri-miRNA regulation remains to be determined (Boisvert et al., 2007; Matera et al., 2007). It is possible that additional new factors may be involved in sequestering and possibly degrading and/or modifying pri-let-7 miRNAs in the nucleoli in a Lin28B-dependent manner. The identification of such factors could reveal new potential therapeutic targets aimed at restoring let-7 expression in Lin28B-expressing cancers.
Our proof-of-concept studies with human breast and ovarian cancer cell lines demonstrate that inhibition of Zccch11 may represent a possible new therapeutic target for cancer. Indeed knockdown of Zcchc11 effectively inhibits the tumorigenic capacity and metastatic potential of human breast and ovarian cancer cells and xenografts. Importantly however, our data also predict that therapeutic potential of Zcchc11 inhibition will be limited to Lin28A-expressing cancers. Although Lin28A expression is relatively uncommon in several human cancer cell lines, our analysis of primary human colon and breast cancer indicate that upregulation of Lin28A or Lin28B occurs in a large proportion of tumors with approximately equally frequency for each protein. Furthermore, expression of Lin28A or Lin28B seems to distinguish different classes of breast cancers (Figure 7). Therefore, the identification of small molecule inhibitors of Zcchc11 may lead to the development of novel chemotherapies for Lin28A-expressing cancers.
EXPERIMENTAL PROCEDURES
Cloning
Myc-Lin28A and Lin28B were cloned into pBK-EF1. Lin28A, Lin28B, Lin28BΔNLS#1, were cloned into pFLAG-CMV2 vector (Sigma). Lin28BΔNoLSΔNLS#1 was generated by site-directed mutagenesis using the QuickChange kit (Stratagene). Lin28A, Lin28B, Lin28BΔNLS#1, and Lin28BΔNoLSΔNLS#1 were cloned into CT-GFP-Topo (Invitrogen). NLS#1 and NLS#2 oligos were annealed before ligating into CT-GFP-Topo. N-terminal Cherry-DGCR8 fusion construct was generated by subcloning Cherry cDNA into p3xFLAG-CMV14-DGCR8 (Gregory et al., 2004). Lin28A and Lin28B were subcloned into Pet21 for His-tagged recombinant protein expression. Pri-let-7g was previously reported (Viswanathan et al., 2008), Cloning primers are listed in Supplementary Table1.
Transfection, knockdowns and stable cell line generation
All transfections were preformed with Lipofectamine (Invitrogen) per manufacturer’s instructions. shRNA constructs were generated using pLKO.1 Puro (Addgene #8453) and pLKO.1 Hygro (Addgene #24150) vectors. The sequences of the shRNA hairpins and siRNAs are listed in Supplementary Table 2. Lentivirus production, infection, and stable cell line selection are as described (Viswanathan et al., 2009). siRNAs used in this study: siRNA negative control (100nM), (cat no. AM4611, Ambion Inc); siRNA Zcchc11#1 (100nM) (cat no. s23551, Ambion Inc); siRNA Zcchc11#2 (100nM) (cat no. s23553, Ambion Inc); siRNA Lin28B (100nM) (cat no. s52477, Ambion Inc); siRNA Lin28A (100nM) (cat no. s36195, Ambion Inc).
Western blotting
The following antibodies were used: Lin28A (Cell Signaling, 3978), Lin28B (Cell Signaling, 4196), Zcchc11 (Protein Tech Group, 18980-1-AP), Zcchc11 (Imgenex, IMX-3587), DGCR8 (Protein Tech Group, 10996-1-AP), Fibrillarin (Abcam, ab18380), and β-Tubulin (Abcam AB6046).
Subcellular fractionation
Cellular fractionation was done using NE-PER Nuclear and Cytoplasmic Extraction Kits (Pierce) per manufacturer’s instructions. Large-scale fractionation was performed as described in supplementary experimental procedures.
Electromobility shift assay (EMSA)
EMSA with purified His-Lin28A/B was performed as described but without competitor yeast tRNA (Piskounova et al., 2008). Complexes were resolved on native 3.5% or 5% polyacrylamide gels and visualized by autoradiography. Band intensities of scanned gels were quantified using ImageJ software and used to calculate percentage of probe bound. Graph-Pad Prism was used to plot data. Percent active protein was determined using stoichiometric binding reactions as described in (Ryder et al., 2008). Hills equation for specific binding with one site was, Y=Bmax*X^h/(Kd^h + X^h), was used to calculate Kd.
Colony Formation Assay
MDA-MB-231 cells and T47D cells were transfected with different siRNAs for 48h. Triplicate samples of 105 cells from each cell line were mixed 4:1 (v/v) with 2.0% agarose in growth medium for a final concentration of 0.4% agarose. The cell mixture was plated on top of a solidified layer of 0.5% agarose in growth medium. Cells were fed every 6 to 7 days with growth medium containing 0.4% agarose. The number of colonies was counted after 20 days.
Invasion Assays
We performed invasion assays in MDA-MB-231 and T47D breast cancer cells were transfected under with different siRNAs for 16h. Invasion of matrigel was conducted by using standardized conditions with BDBioCoat growth factor reduced MATRIGEL invasion chambers (PharMingen). Assays were conducted per manufacturer’s protocol, using 10% FBS as chemoattractant. Non-invading cells on the top-side of the membrane were removed while invading cells were fixed and stained with 4′-6-diamidino-2-phenylindole (DAPI, Vector Laboratories Inc.), 16h post seeding.
Mouse experiments
Xenograft experiments with human cancer cell lines are described in detail the supplementary experimental procedures. Briefly, cells were injected subcutaneously in the right flank of athymic nude mice. Tumor growth was monitored every five days. In Vivo Ready siRNAs (Ambion Inc.) were mixed with Invivofectamine 2.0 liposomes (Ambion Inc) and injected intra-peritoneal in a volume of 100ul at a dose of 5mg/kg.
Human Tissues and RNAs
Thirty normal colon tissues and 45 colon adenocarcinomas were collected from the translational pathology core laboratory of the Department of Pathology at UCLA. All subjects gave informed consent and the study was approved by the UCLA Institutional Review Board. RNAs from 12 normal mammary tissues and 33 breast cancer tissues were purchased from Origene Inc. The ER, PR and HER2 status for each of these breast carcinomas was known. Additional RNAs were purchased from Origene (8 renal cell carcinomas, 8 hepatocellular carcinomas, 8 squamous cell lung carcinomas, 8 ovarial adenocarcinomas, 8 prostate adenocarcinomas, 8 papillary thyroid carcinomas).
In situ hybridization and immunohistochemistry
Sections of the colon adenocarcinomas and adjacent uninvolved tissues were analyzed by in situ hybridization as described (Iliopoulos et al., 2009) with modification (supplementary experimental procedures). Double-DIG labeled Mircury LNA Detection probe for the detection of hsa-let-7 (1800-15, Exiqon) was used. Protocol for immunohistochemistry is described in detail the supplementary experimental procedures using the following antibodies: Lin28A (3978, Cell Signaling Technology) and Lin28B (LS-B3423, LSBio).
Supplementary Material
HIGHLIGHTS.
The TUTase Zcchc11 represses expression of the miRNA let-7 via Lin28A but not Lin28B
Lin28B localizes to the cell nucleus and blocks pri-let-7 processing
Zcchc11 depletion selectively inhibits the tumorigenicity of Lin28A-expressing cancer
The majority of human colon and breast tumors express either Lin28A or Lin28B
Acknowledgments
We thank Dr. Akihiko Yoshimura, Keio University, Japan for the human Flag-Zcchc11 plasmid, Dr. Na Liu, Memorial Sloan Kettering Cancer Center, for the Myc-Ago2 plasmid, Dr. Bharat Ramratnam, Brown University, for the GFP-Drosha plasmid, and Dr. Ramin Shiekhattar, Wistar Institute, for the Flag-Eif6 expression plasmid. We thank Chiara Giacomelli, Dr. Saurabh Patel, and Dr. Maria Hatziapostolou for technical assistance. R.I.G was supported by grants from the US National Institute of General Medical Sciences (NIGMS) (1R01GM086386-01A1), The Harvard Stem Cell Institute, and The American Cancer Society. R.I.G is a Pew Research Scholar. D.I was supported by start-up funds from the Dept of Cancer Immunology & AIDS at Dana-Farber Cancer Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barh D, Malhotra R, Ravi B, Sindhurani P. Microrna let-7: an emerging next-generation cancer therapeutic. Curr Oncol. 2010;17:70–80. doi: 10.3747/co.v17i1.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert FM, van Koningsbruggen S, Navascues J, Lamond AI. The multifunctional nucleolus. Nat Rev Mol Cell Biol. 2007;8:574–585. doi: 10.1038/nrm2184. [DOI] [PubMed] [Google Scholar]
- Boyerinas B, Park SM, Hau A, Murmann AE, Peter ME. The role of let-7 in cell differentiation and cancer. Endocr Relat Cancer. 2010;17:F19–36. doi: 10.1677/ERC-09-0184. [DOI] [PubMed] [Google Scholar]
- Bussing I, Slack FJ, Grosshans H. let-7 microRNAs in development, stem cells and cancer. Trends Mol Med. 2008;14:400–409. doi: 10.1016/j.molmed.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Chang TC, Zeitels LR, Hwang HW, Chivukula RR, Wentzel EA, Dews M, Jung J, Gao P, Dang CV, Beer MA, et al. Lin-28B transactivation is necessary for Myc-mediated let-7 repression and proliferation. Proc Natl Acad Sci U S A. 2009;106:3384–3389. doi: 10.1073/pnas.0808300106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangi-Garimella S, Yun J, Eves EM, Newman M, Erkeland SJ, Hammond SM, Minn AJ, Rosner MR. Raf kinase inhibitory protein suppresses a metastasis signalling cascade involving LIN28 and let-7. EMBO J. 2009;28:347–358. doi: 10.1038/emboj.2008.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ. Processing of primary microRNAs by the Microprocessor complex. Nature. 2004;432:231–235. doi: 10.1038/nature03049. [DOI] [PubMed] [Google Scholar]
- Di Leva G, Croce CM. Roles of small RNAs in tumor formation. Trends Mol Med. 2010;16:257–267. doi: 10.1016/j.molmed.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esquela-Kerscher A, Trang P, Wiggins JF, Patrawala L, Cheng A, Ford L, Weidhaas JB, Brown D, Bader AG, Slack FJ. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle. 2008;7:759–764. doi: 10.4161/cc.7.6.5834. [DOI] [PubMed] [Google Scholar]
- Gregory RI, Chendrimada TP, Cooch N, Shiekhattar R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell. 2005;123:631–640. doi: 10.1016/j.cell.2005.10.022. [DOI] [PubMed] [Google Scholar]
- Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- Guo Y, Chen Y, Ito H, Watanabe A, Ge X, Kodama T, Aburatani H. Identification and characterization of lin-28 homolog B (LIN28B) in human hepatocellular carcinoma. Gene. 2006;384:51–61. doi: 10.1016/j.gene.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Hagan JP, Piskounova E, Gregory RI. Lin28 recruits the TUTase Zcchc11 to inhibit let-7 maturation in mouse embryonic stem cells. Nat Struct Mol Biol. 2009;16:1021–1025. doi: 10.1038/nsmb.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo I, Joo C, Cho J, Ha M, Han J, Kim VN. Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Mol Cell. 2008;32:276–284. doi: 10.1016/j.molcel.2008.09.014. [DOI] [PubMed] [Google Scholar]
- Heo I, Joo C, Kim YK, Ha M, Yoon MJ, Cho J, Yeom KH, Han J, Kim VN. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell. 2009;138:696–708. doi: 10.1016/j.cell.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Hutvagner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, Zamore PD. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001;293:834–838. doi: 10.1126/science.1062961. [DOI] [PubMed] [Google Scholar]
- Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693–706. doi: 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji J, Wang XW. A Yin-Yang balancing act of the lin28/let-7 link in tumorigenesis. J Hepatol. 2010;53:974–975. doi: 10.1016/j.jhep.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King CE, Cuatrecasas M, Castells A, Sepulveda AR, Lee JS, Rustgi AK. LIN28B Promotes Colon Cancer Progression and Metastasis. Cancer Res. 2011;71:4260–4268. doi: 10.1158/0008-5472.CAN-10-4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- Lehrbach NJ, Armisen J, Lightfoot HL, Murfitt KJ, Bugaut A, Balasubramanian S, Miska EA. LIN-28 and the poly(U) polymerase PUP-2 regulate let-7 microRNA processing in Caenorhabditis elegans. Nat Struct Mol Biol. 2009;16:1016–1020. doi: 10.1038/nsmb.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, Wong CM, Ying Q, Fan DN, Huang S, Ding J, Yao J, Yan M, Li J, Yao M, et al. MicroRNA-125b suppressesed human liver cancer cell proliferation and metastasis by directly targeting oncogene LIN28B2. Hepatology. 2010;52:1731–1740. doi: 10.1002/hep.23904. [DOI] [PubMed] [Google Scholar]
- Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song JJ, Hammond SM, Joshua-Tor L, Hannon GJ. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004;305:1437–1441. doi: 10.1126/science.1102513. [DOI] [PubMed] [Google Scholar]
- Lu L, Katsaros D, Shaverdashvili K, Qian B, Wu Y, de la Longrais IA, Preti M, Menato G, Yu H. Pluripotent factor lin-28 and its homologue lin-28b in epithelial ovarian cancer and their associations with disease outcomes and expression of let-7a and IGF-II. Eur J Cancer. 2009;45:2212–2218. doi: 10.1016/j.ejca.2009.05.003. [DOI] [PubMed] [Google Scholar]
- Martinez NJ, Gregory RI. MicroRNA gene regulatory pathways in the establishment and maintenance of ESC identity. Cell Stem Cell. 2010;7:31–35. doi: 10.1016/j.stem.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera AG, Terns RM, Terns MP. Non-coding RNAs: lessons from the small nuclear and small nucleolar RNAs. Nat Rev Mol Cell Biol. 2007;8:209–220. doi: 10.1038/nrm2124. [DOI] [PubMed] [Google Scholar]
- Melton C, Judson RL, Blelloch R. Opposing microRNA families regulate self-renewal in mouse embryonic stem cells. Nature. 2010;463:621–626. doi: 10.1038/nature08725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss EG, Lee RC, Ambros V. The cold shock domain protein LIN-28 controls developmental timing in C. elegans and is regulated by the lin-4 RNA. Cell. 1997;88:637–646. doi: 10.1016/s0092-8674(00)81906-6. [DOI] [PubMed] [Google Scholar]
- Newman MA, Thomson JM, Hammond SM. Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. RNA. 2008;14:1539–1549. doi: 10.1261/rna.1155108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh JS, Kim JJ, Byun JY, Kim IA. Lin28-let7 modulates radiosensitivity of human cancer cells with activation of K-Ras. Int J Radiat Oncol Biol Phys. 76:5–8. doi: 10.1016/j.ijrobp.2009.08.028. [DOI] [PubMed] [Google Scholar]
- Peng S, Maihle NJ, Huang Y. Pluripotency factors Lin28 and Oct4 identify a sub-population of stem cell-like cells in ovarian cancer. Oncogene. 2010;29:2153–2159. doi: 10.1038/onc.2009.500. [DOI] [PubMed] [Google Scholar]
- Piskounova E, Viswanathan SR, Janas M, LaPierre RJ, Daley GQ, Sliz P, Gregory RI. Determinants of microRNA processing inhibition by the developmentally regulated RNA-binding protein Lin28. J Biol Chem. 2008;283:21310–21314. doi: 10.1074/jbc.C800108200. [DOI] [PubMed] [Google Scholar]
- Rybak A, Fuchs H, Smirnova L, Brandt C, Pohl EE, Nitsch R, Wulczyn FG. A feedback loop comprising lin-28 and let-7 controls pre-let-7 maturation during neural stem-cell commitment. Nat Cell Biol. 2008;10:987–993. doi: 10.1038/ncb1759. [DOI] [PubMed] [Google Scholar]
- Ryder SP, Recht MI, Williamson JR. Quantitative analysis of protein-RNA interactions by gel mobility shift. Methods Mol Biol. 2008;488:99–115. doi: 10.1007/978-1-60327-475-3_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott MS, Boisvert FM, McDowall MD, Lamond AI, Barton GJ. Characterization and prediction of protein nucleolar localization sequences. Nucleic acids research. 2010;38:7388–7399. doi: 10.1093/nar/gkq653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shell S, Park SM, Radjabi AR, Schickel R, Kistner EO, Jewell DA, Feig C, Lengyel E, Peter ME. Let-7 expression defines two differentiation stages of cancer. Proc Natl Acad Sci U S A. 2007;104:11400–11405. doi: 10.1073/pnas.0704372104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack F. let-7 microRNA reduces tumor growth. Cell Cycle. 2009;8:1823. doi: 10.4161/cc.8.12.8639. [DOI] [PubMed] [Google Scholar]
- Small EM, Olson EN. Pervasive roles of microRNAs in cardiovascular biology. Nature. 2011;469:336–342. doi: 10.1038/nature09783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64:3753–3756. doi: 10.1158/0008-5472.CAN-04-0637. [DOI] [PubMed] [Google Scholar]
- Trang P, Medina PP, Wiggins JF, Ruffino L, Kelnar K, Omotola M, Homer R, Brown D, Bader AG, Weidhaas JB, et al. Regression of murine lung tumors by the let-7 microRNA. Oncogene. 2010;29:1580–1587. doi: 10.1038/onc.2009.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wynsberghe PM, Kai ZS, Massirer KB, Burton VH, Yeo GW, Pasquinelli AE. LIN-28 co-transcriptionally binds primary let-7 to regulate miRNA maturation in Caenorhabditis elegans. Nat Struct Mol Biol. 2011;18:302–308. doi: 10.1038/nsmb.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan SR, Daley GQ. Lin28: A microRNA regulator with a macro role. Cell. 2010;140:445–449. doi: 10.1016/j.cell.2010.02.007. [DOI] [PubMed] [Google Scholar]
- Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of microRNA processing by Lin28. Science. 2008;320:97–100. doi: 10.1126/science.1154040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S, O’Sullivan M, Lu J, Phillips LA, Lockhart VL, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet. 2009;41:843–848. doi: 10.1038/ng.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YC, Chen YL, Yuan RH, Pan HW, Yang WC, Hsu HC, Jeng YM. Lin-28B expression promotes transformation and invasion in human hepatocellular carcinoma. Carcinogenesis. 2010;31:1516–1522. doi: 10.1093/carcin/bgq107. [DOI] [PubMed] [Google Scholar]
- West JA, Viswanathan SR, Yabuuchi A, Cunniff K, Takeuchi A, Park IH, Sero JE, Zhu H, Perez-Atayde A, Frazier AL, et al. A role for Lin28 in primordial germ-cell development and germ-cell malignancy. Nature. 2009;460:909–913. doi: 10.1038/nature08210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Lin X, Zhong X, Kaur S, Li N, Liang S, Lassus H, Wang L, Katsaros D, Montone K, et al. Double-negative feedback loop between reprogramming factor LIN28 and microRNA let-7 regulates aldehyde dehydrogenase 1-positive cancer stem cells. Cancer Res. 2010;70:9463–9472. doi: 10.1158/0008-5472.CAN-10-2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, Huang Y, Hu X, Su F, Lieberman J, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007a;131:1109–1123. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007b;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Zhu H, Shah S, Shyh-Chang N, Shinoda G, Einhorn WS, Viswanathan SR, Takeuchi A, Grasemann C, Rinn JL, Lopez MF, et al. Lin28a transgenic mice manifest size and puberty phenotypes identified in human genetic association studies. Nat Genet. 2010;42:626–630. doi: 10.1038/ng.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.